
Genomic Diversity among Actinomyces naeslundii Strains and Closely Related Species
 , , ,
, , ,
Abstract
:1. Introduction
2. Material and Methods
2.1. DNA Extraction from Actinomyces
2.1.1. Bacterial Strains and Growth Conditions
2.1.2. Lysis, Purification, and Isolation of Genomic DNA
2.2. Library Preparation for Illumina Paired-End Sequencing
2.3. Single-End Sequencing Using the Roche 454 GS (FLX) Titanium Platform
2.4. De Novo Sequence Assembly and RAST Annotation
2.5. Genomic Comparisons among Actinomyces Strains
2.5.1. Comparative Analysis of Whole Genomes using Mauve
2.5.2. Comparative Analysis using Progressive Mauve (ClonalFrame)
Whole Genomes Analysis


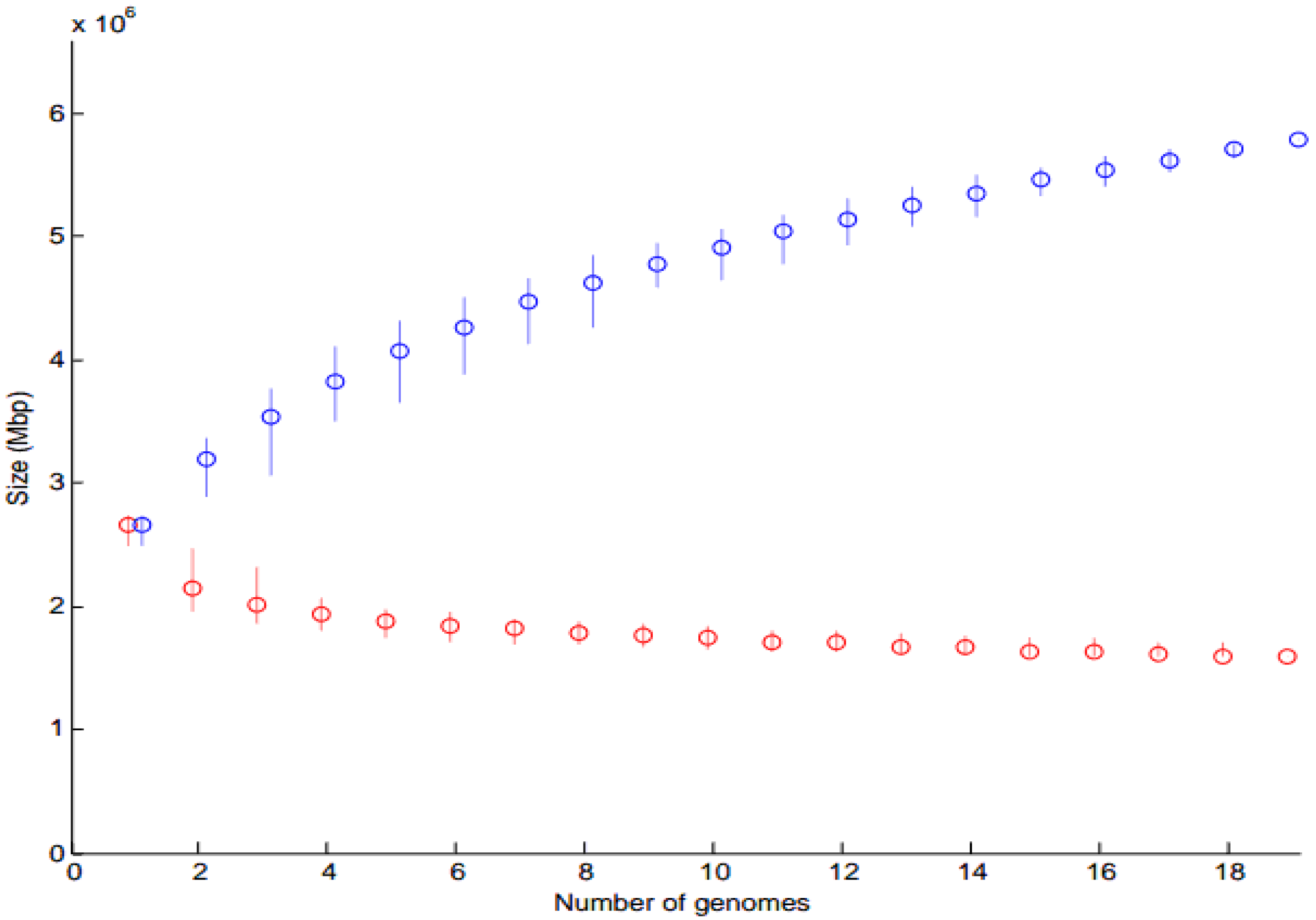
Core and Pan-Genome Analysis
2.5.3. Mapping and Alignment of Whole Genomes (Core Genomes) of Actinomyces Using Gene-to-Gene Analysis
2.5.4. Digital DNA–DNA Hybridization
3. Results
3.1. Comparative Analysis of Whole Genomes Using Mauve
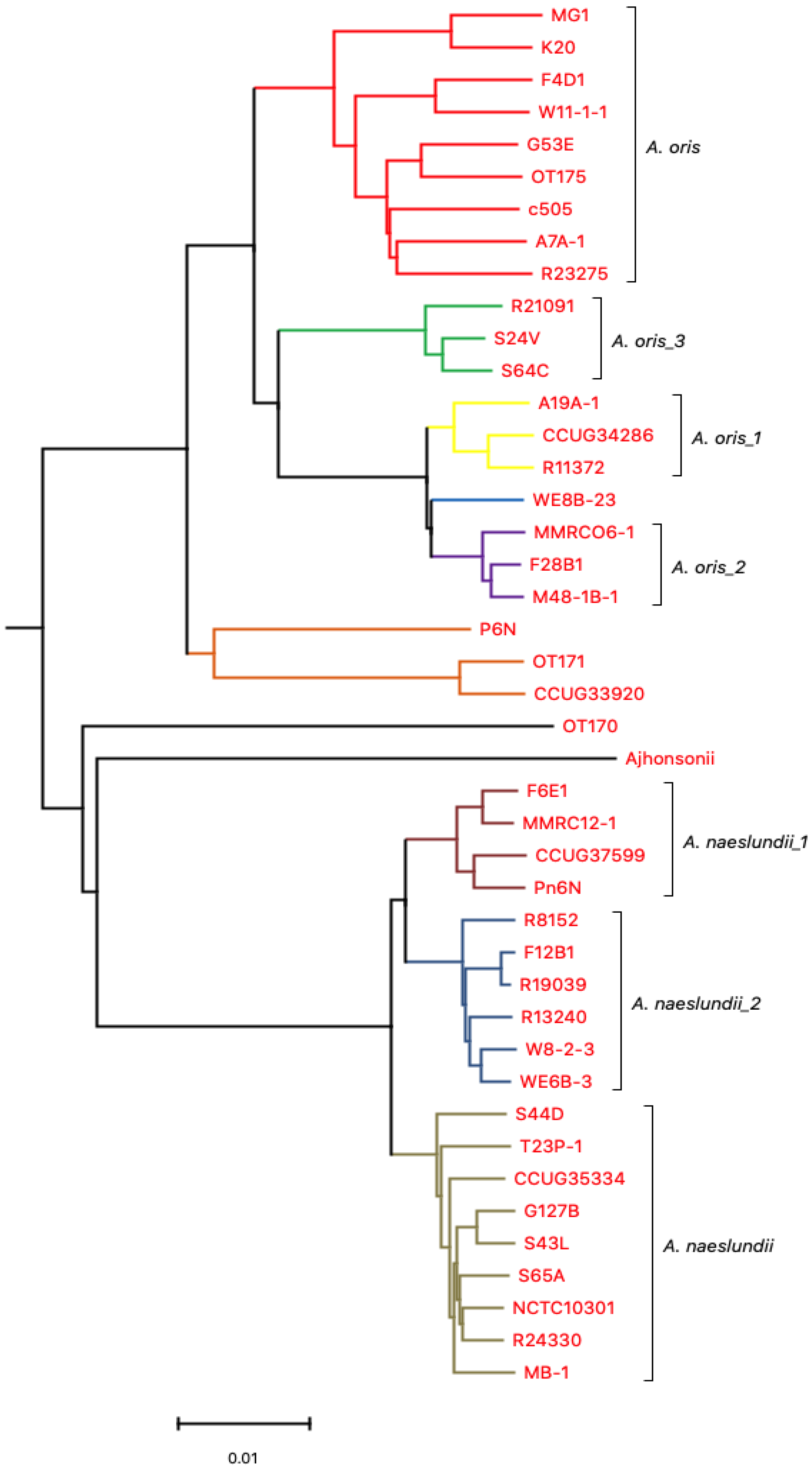
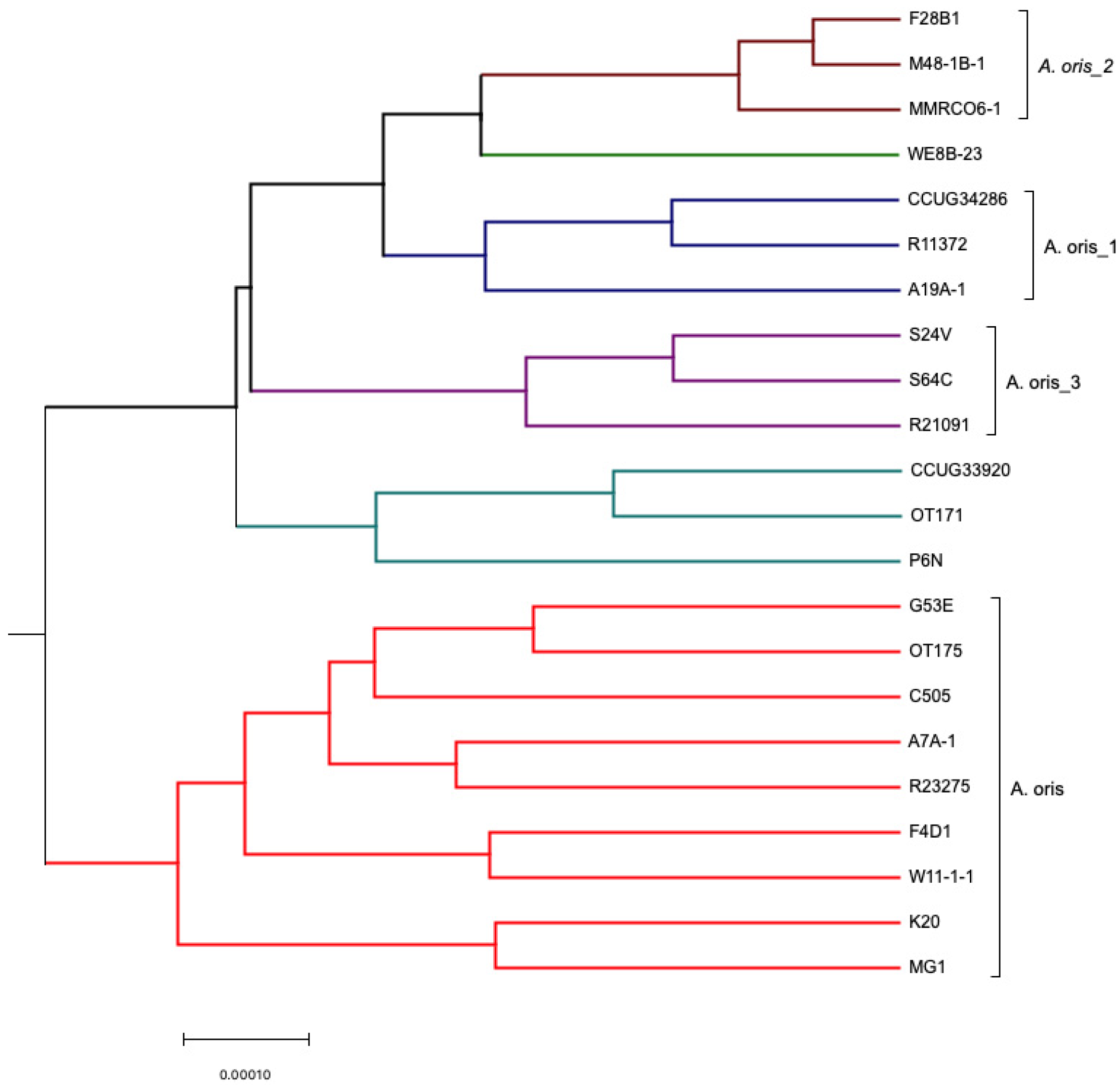
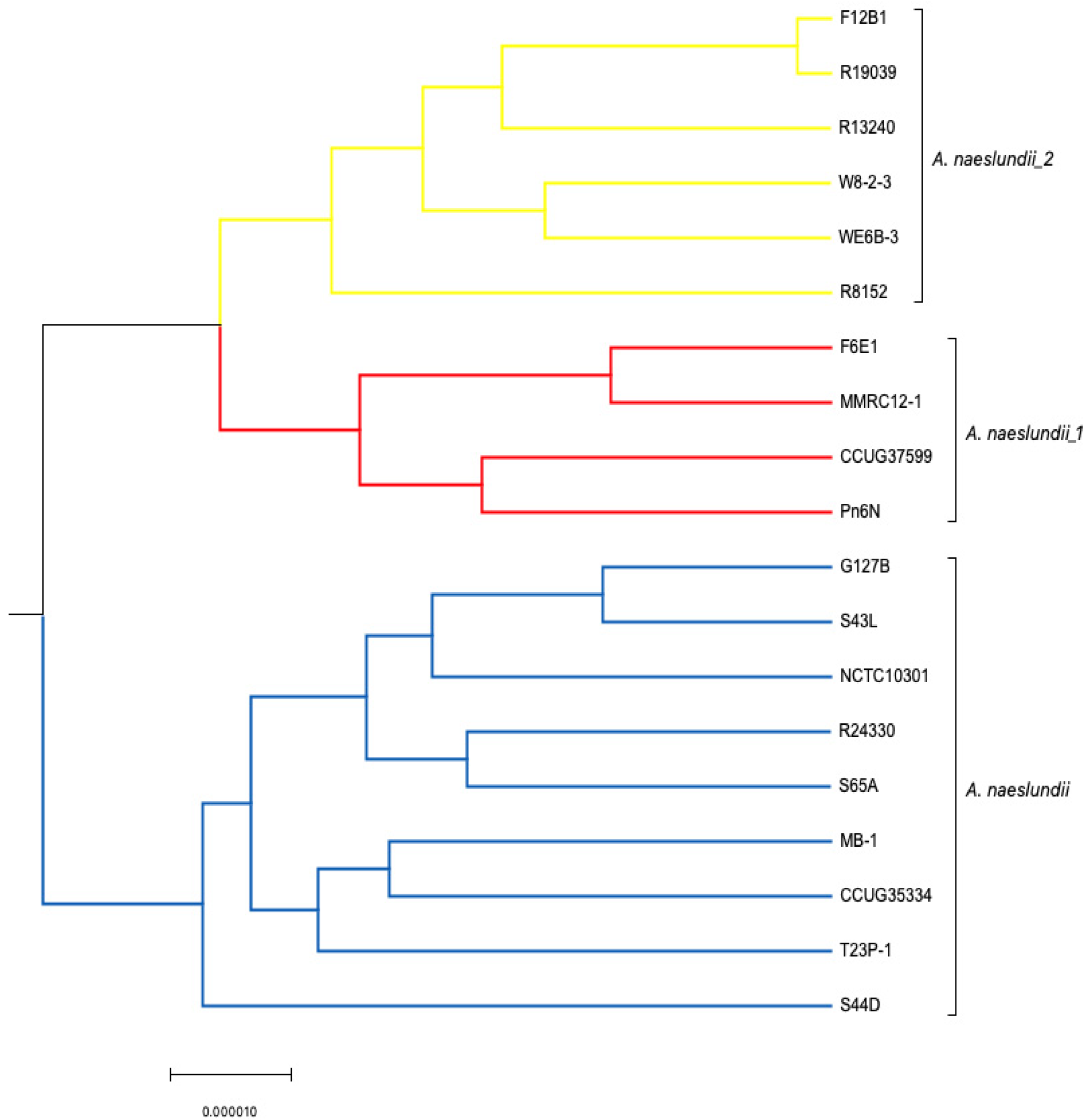
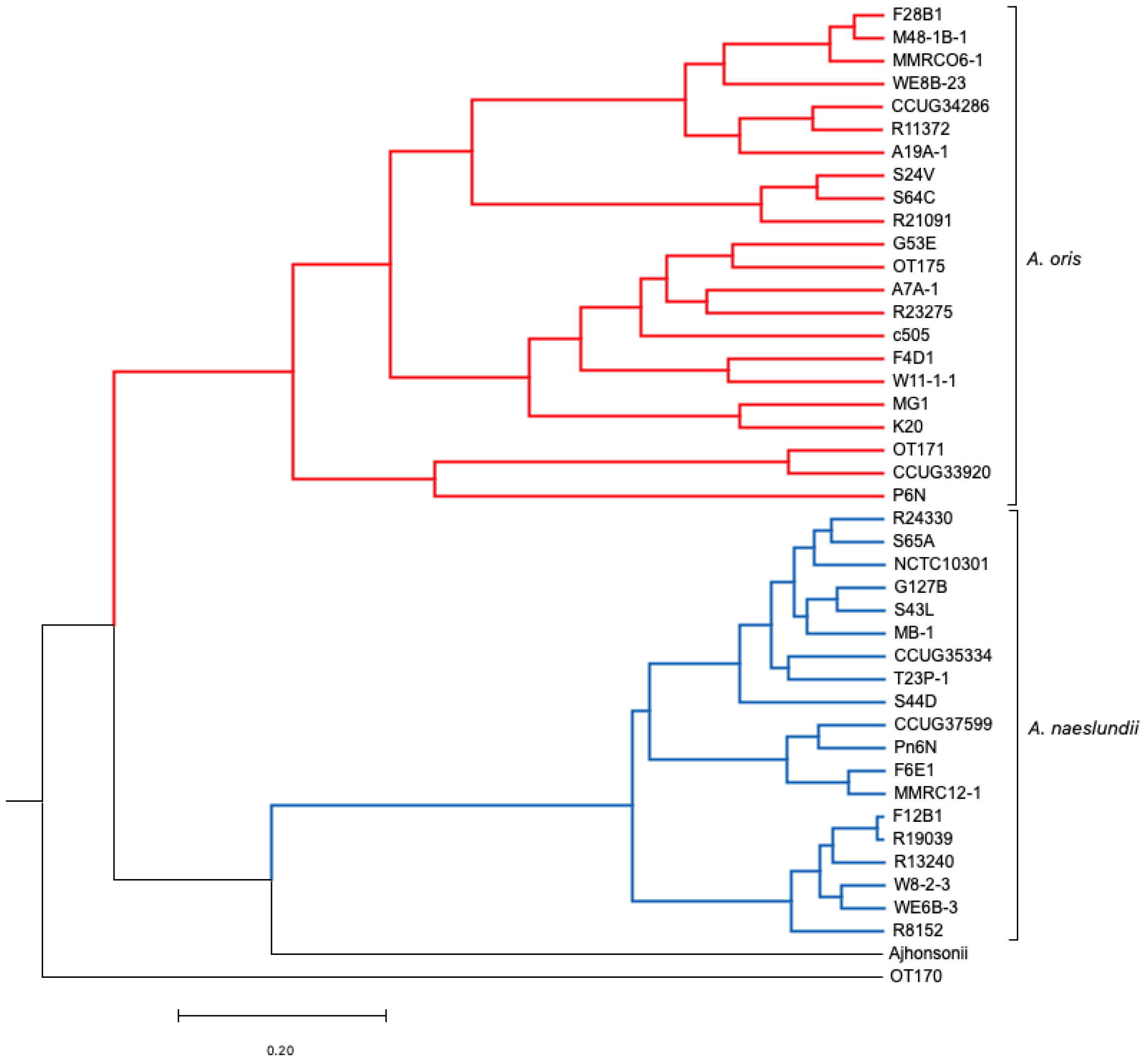
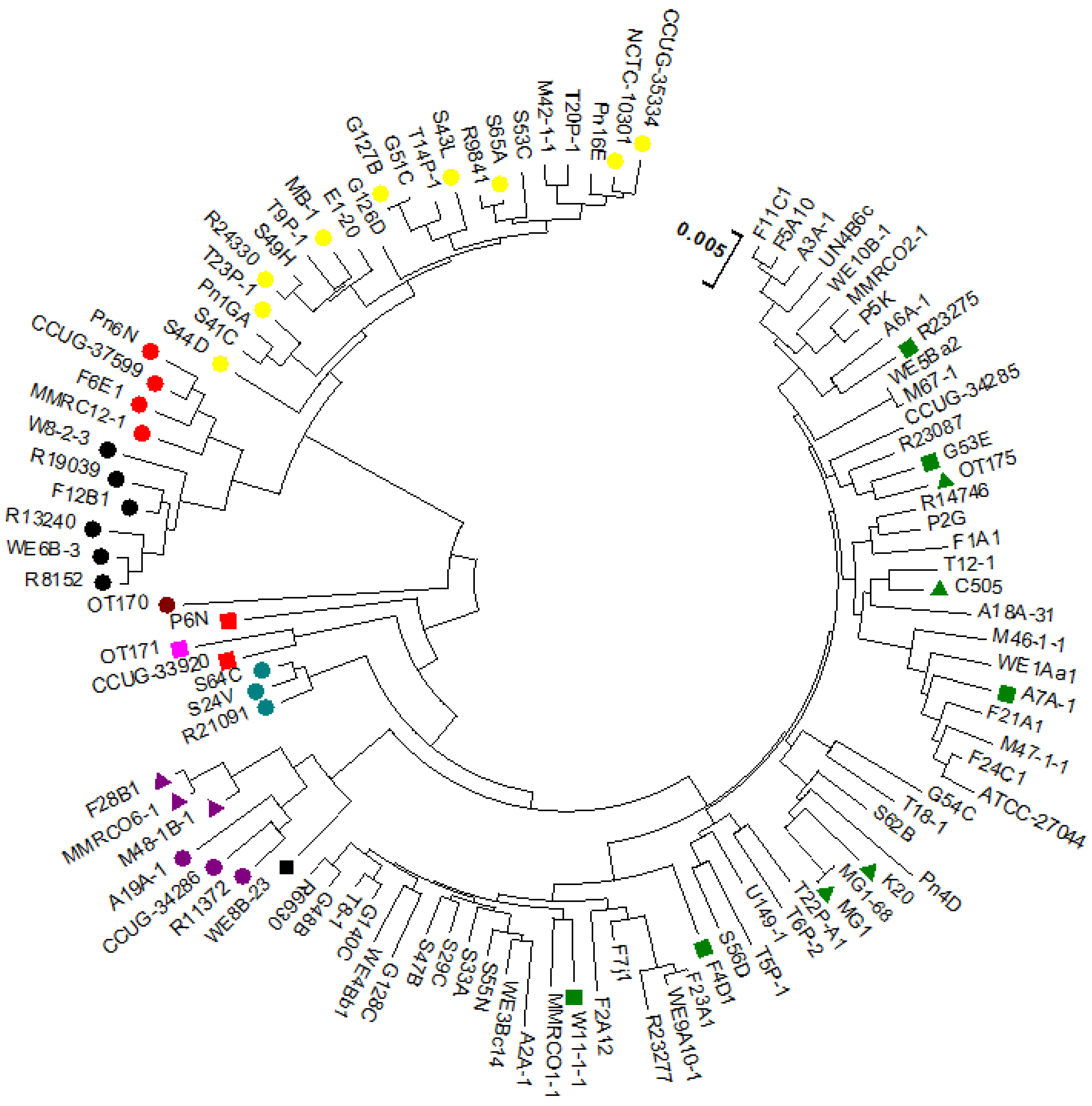
3.2. Comparative Analysis Using Progressive Mauve (ClonalFrame)
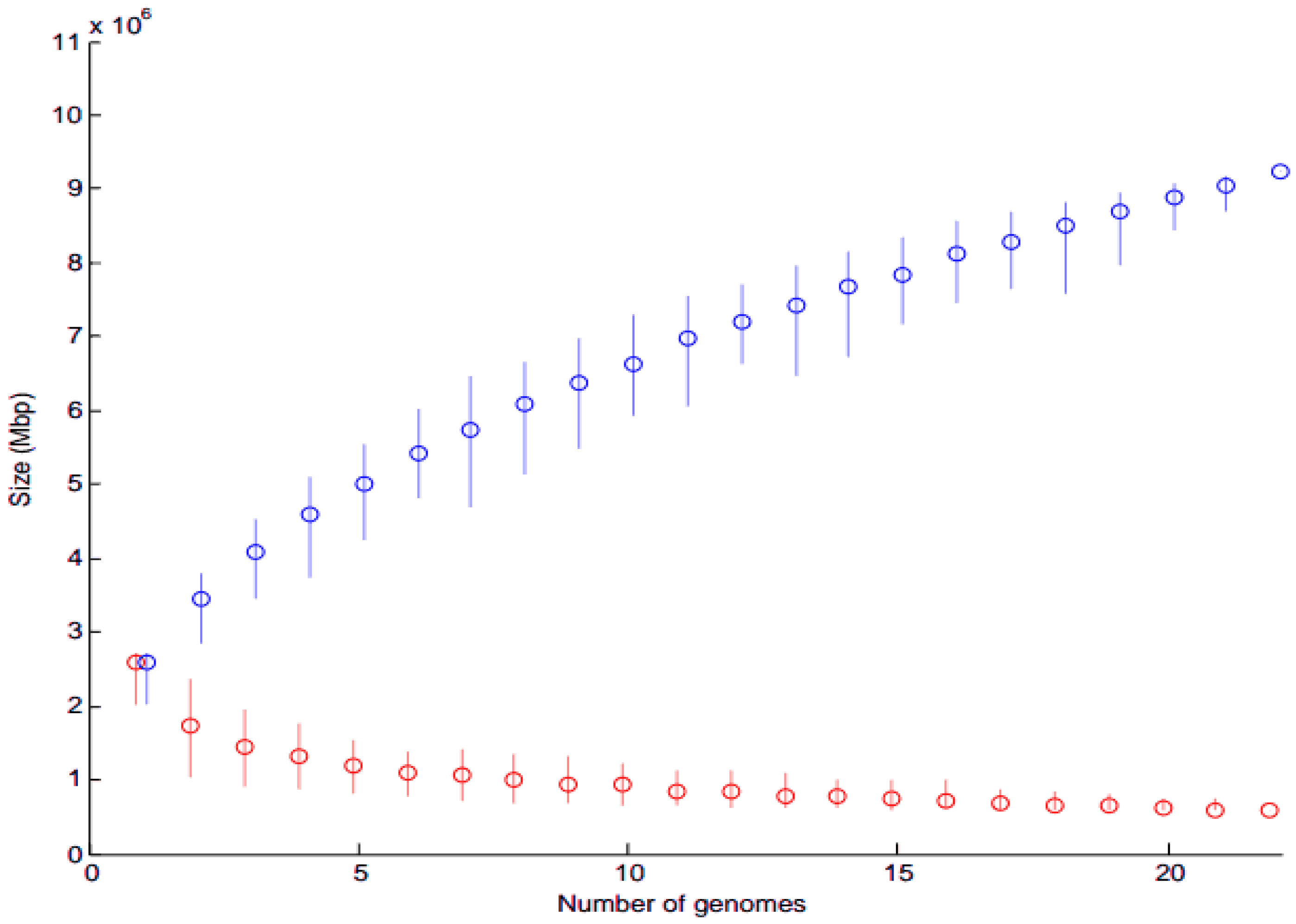
3.2.1. Core and Pan-Genome Analysis


3.2.2. Core Genome Analysis (Evolutionary Analysis)
3.3. Digital DNA–DNA Hybridization
4. Discussion
4.1. Clonal Frame Analysis
4.2. Core Gene Analysis
4.3. Digital DNA–DNA Hybridization Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1
 ; A. oris_1:
; A. oris_1:  ; A. oris_2:
; A. oris_2:  ; A. oris_3:
; A. oris_3:  ; A. naeslundii:
; A. naeslundii:  ; A. naeslundii_1:
; A. naeslundii_1:  ; A. naeslundii_2:
; A. naeslundii_2:  ; c505, MG1, k20, OT175 (A. oris):
; c505, MG1, k20, OT175 (A. oris):  ; OT170:
; OT170:  ; OT171:
; OT171:  .
; A. oris_1: ; A. oris_2: ; A. oris_3: ; A. naeslundii: ; A. naeslundii_1: ; A. naeslundii_2: ; c505, MG1, k20, OT175 (A. oris): ; OT170: ; OT171: .
.
; A. oris_1: ; A. oris_2: ; A. oris_3: ; A. naeslundii: ; A. naeslundii_1: ; A. naeslundii_2: ; c505, MG1, k20, OT175 (A. oris): ; OT170: ; OT171: .
Appendix A.2
Appendix A.3
Appendix A.4
References
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paster, B.J.; Olsen, I.; Aas, J.A.; Dewhirst, F.E. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontology 2000 2006, 42, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Escapa, I.F.; Chen, T.; Huang, Y.; Gajare, P.; Dewhirst, F.E.; Lemon, K.P. New Insights into Human Nostril Microbiome from the Expanded Human Oral Microbiome Database (eHOMD): A Resource for the Microbiome of the Human Aerodigestive Tract. mSystems 2018, 3, e00187-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auch, A.F.; von Jan, M.; Klenk, H.P.; Göker, M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2010, 2, 117–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aas, J.A.; Paster, B.J.; Stokes, L.N.; Olsen, I.; Dewhirst, F.E. Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol. 2005, 43, 5721–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bik, E.M.; Long, C.D.; Armitage, G.C.; Loomer, P.; Emerson, J.; Mongodin, E.F.; Nelson, K.E.; Gill, S.R.; Fraser-Liggett, C.M.; Relman, D.A. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 2010, 4, 962–974. [Google Scholar] [CrossRef]
- Slots, J.; Möenbo, D.; Langebaek, J.; Frandsen, A. Microbiota of gingivitis in man. Scand. J. Dent. Res. 1978, 86, 174–181. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Zhou, Y.; Wang, C.; Wu, B.; Wan, J. Actinomyces and Alimentary Tract Diseases: A Review of Its Biological Functions and Pathology. Biomed. Res. Int. 2018, 2018, 3820215. [Google Scholar]
- Nyvad, B.; Kilian, M. Microbiology of the early colonization of human enamel and root surfaces in vivo. Scand. J. Dent. Res. 1987, 95, 369–380. [Google Scholar] [CrossRef]
- Nyvad, B.; Kilian, M. Comparison of the initial streptococcal microflora on dental enamel in caries-active and in caries-inactive individuals. Caries Res. 1990, 24, 267–272. [Google Scholar] [CrossRef]
- Li, J.; Helmerhorst, E.J.; Leone, C.W.; Troxler, R.F.; Yaskell, T.; Haffajee, A.D.; Socransky, S.S.; Oppenheim, F.G. Identification of early microbial colonizers in human dental biofilm. J. Appl. Microbiol. 2004, 97, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Diaz, P.I.; Chalmers, N.I.; Rickard, A.H.; Kong, C.; Milburn, C.L.; Palmer, R.J.; Kolenbrander, P.E. Molecular characterization of subject-specific oral microflora during initial colonization of enamel. Appl. Environ. Microbiol. 2006, 72, 2837–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periasamy, S.; Kolenbrander, P.E. Central role of the early colonizer Veillonella sp. in establishing multispecies biofilm communities with initial, middle, and late colonizers of enamel. J. Bacteriol. 2010, 192, 2965–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henssge, U.; Do, T.; Radford, D.R.; Gilbert, S.C.; Clark, D.; Beighton, D. Emended description of Actinomyces naeslundii and descriptions of Actinomyces oris sp. nov. and Actinomyces johnsonii sp. nov., previously identified as Actinomyces naeslundii genospecies 1, 2 and WVA 963. Int. J. Syst. Evol. Microbiol. 2009, 59, 509–516. [Google Scholar] [CrossRef]
- Do, T.; Henssge, U.; Gilbert, S.C.; Clark, D.; Beighton, D. Evidence for recombination between a sialidase (nanH) of Actinomyces naeslundii and Actinomyces oris, previously named ‘Actinomyces naeslundii genospecies 1 and 2’. FEMS Microbiology Letters. 2008, 288, 156–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.L.; Moore, L.V.H.; Kaneko, B.; Moore, W.E.C. Actinomyces georgiae sp. nov., Actinomyces gerencseriae sp. nov., Designation of Two Genospecies of Actinomyces naeslundii, and Inclusion of A. naeslundii serotypes II and III and Actinomyces viscosus serotype II in A. naeslundii Genospecies 2. Int. J. Syst. Bacteriol. 1990, 40, 273–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henssge, U.; Do, T.; Gilbert, S.C.; Cox, S.; Clark, D.; Wickström, C.; Ligtenberg, A.J.M.; Radford, D.R.; Beighton, D. Application of MLST and pilus gene sequence comparisons to investigate the population structures of Actinomyces naeslundii and Actinomyces oris. PLoS ONE 2011, 6, e21430. [Google Scholar] [CrossRef] [Green Version]
- Brailsford, S.R.; Lynch, E.; Beighton, D. The isolation of Actinomyces naeslundii from sound root surfaces and root carious lesions. Caries Res. 1998, 32, 100–106. [Google Scholar] [CrossRef]
- Wayne, L.G.; Brenner, D.J.; Colwell, R.R.; Grimont, P.A.D.; Kandler, O.; Krichevsky, M.I.; Moore, L.H.; Moore, W.E.C.; Murray, R.G.E.; Stackebrandt, E.; et al. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Bacteriol. 1987, 37, 463–464. [Google Scholar] [CrossRef] [Green Version]
- Grimont, P.A.D.; Popoff, M.Y.; Grimont, F.; Coynault, C.; Lemelin, M. Reproducibility and correlation study of three deoxyribonucleic acid hybridization procedures. Current Microbiol. 1980, 4, 325–330. [Google Scholar] [CrossRef]
- Huss, V.A.R.; Festl, H.; Schleifer, K.H. Studies on the spectrophotometric determination of DNA hybridization from renaturation rates. Syst. Appl. Microbiol. 1983, 4, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Cohan, F.M.; Lawrence, J.G.; Spratt, B.G.; Coenye, T.; Feil, E.J.; Stackebrandt, E.; Van de Peer, Y.; Vandamme, P.; Thompson, F.L.; et al. Re-evaluating prokaryotic species. Nat. Rev. Micro. 2005, 3, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E. The richness of prokaryotic diversity: There must be a species somewhere. Food Technol. Biotechnol. 2003, 41, 17–22. [Google Scholar]
- Cho, J.-C.; Tiedje, J.M. Bacterial Species Determination from DNA-DNA Hybridization by Using Genome Fragments and DNA Microarrays. Appl. Environ. Microbiol. 2001, 67, 3677–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coenye, T.; Gevers, D.; Van de Peer, Y.; Vandamme, P.; Swings, J. Towards a prokaryotic genomic taxonomy. FEMS Microbiol. Rev. 2005, 29, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Deloger, M.; El Karoui, M.; Petit, M.A. A genomic distance based on MUM indicates discontinuity between most bacterial species and genera. J. Bacteriol. 2009, 191, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Goker, M.; Sproer, C.; Klenk, H.P. When should a DDH experiment be mandatory in microbial taxonomy? Arch. Microbiol. 2013, 195, 413–418. [Google Scholar] [CrossRef]
- Auch, A.F.; Klenk, H.P.; Goker, M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genomic Sci. 2010, 2, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Smokvina, T.; Wels, M.; Polka, J.; Chervaux, C.; Brisse, S.; Boekhorst, J.; Vlieg, J.E.; Siezen, R.J. Lactobacillus paracasei Comparative Genomics: Towards Species Pan-Genome Definition and Exploitation of Diversity. PLoS ONE 2013, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.E.; Miklos, I.; Ragan, M.A. Dynamics of genome rearrangement in bacterial populations. PLoS Genet. 2008, 4, e1000128. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Didelot, X.; Falush, D. Inference of Bacterial Microevolution Using Multilocus Sequence Data. Genetics 2007, 175, 1251–1266. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Yu, W.-H.; Izard, J.; Baranova, O.V.; Lakshmanan, A.; Dewhirst, F.E. The Human Oral Microbiome Database: A web accessible resource for investigating oral microbe taxonomic and genomic information. Database 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, Q.; Wang, Y.; Zhang, K.; Zou, J. Comparing dental plaque microbiome diversity of extrinsic black stain in the primary dentition using Illumina MiSeq sequencing technique. BMC Oral Health 2019, 19, 269. [Google Scholar] [CrossRef]
- Hall, V.; Brazier, J.S. Identification of actinomyces—What are the major problems? In Anaerobic Pathogens; Eley, A.R., Bennett, K.W., Eds.; Academic Press: Sheffield, UK, 1997; pp. 187–192. [Google Scholar]
- Ellen, R.P.; Balcerzak-Raczkowski, I.B. Differential medium for detecting dental plaque bacteria resembling Actinomyces viscosus and Actinomyces naeslundii. J. Clin. Microbiol. 1975, 2, 305–310. [Google Scholar] [CrossRef]
- Ellen, R.P. Establishment and distribution of Actinomyces viscosus and Actinomyces naeslundii in the human oral cavity. Infect. Immun. 1976, 14, 1119–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaal, K.P. Genus Actinomyces. In Bergey’s Manual of Systematic Bacteriology; Sneath, P.H.A., Mair, N.S., Sharpe, M.E., Holt, J.G., Eds.; Williams and Wilkins: Baltimore, MD, USA, 1986; pp. 1383–1418. [Google Scholar]
- Stackebrandt, E.; Charfreitag, O. Partial 16S rRNA primary structure of five Actinomyces species: Phylogenetic implications and development of an Actinomyces israelii-specific oligonucleotide probe. J. Gen. Microbiol. 1990, 136, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Arif, N.; Sheehy, E.C.; Do, T.; Beighton, D. Diversity of Veillonella spp. from sound and carious sites in children. J. Dent. Res. 2008, 87, 278–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, H.; Kuhnert, P.; Olsen, J.E.; Bisgaard, M. Comparative phylogenies of the housekeeping genes atpD, infB and rpoB and the 16S rRNA gene within the Pasteurellaceae. Int. J. Syst. Evol. Microbiol. 2004, 54, 1601–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiratisin, P.; Li, L.; Murray, P.R.; Fischer, S.H. Use of housekeeping gene sequencing for species identification of viridans streptococci. Diagn. Microbiol. Infect. Dis. 2005, 51, 297–301. [Google Scholar] [CrossRef]
- Poyart, C.; Quesne, G.; Coulon, S.; Berche, P.; Trieu-Cuot, P. Identification of streptococci to species level by sequencing the gene encoding the manganese-dependent superoxide dismutase. J. Clin. Microbiol. 1998, 36, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Wertz, J.E.; Goldstone, C.; Gordon, D.M.; Riley, M.A. A molecular phylogeny of enteric bacteria and implications for a bacterial species concept. J. Evol. Biol. 2003, 16, 1236–1248. [Google Scholar] [CrossRef] [Green Version]
- Bisharat, N.; Cohen, D.I.; Maiden, M.C.; Crook, D.W.; Peto, T.; Harding, R.M. The evolution of genetic structure in the marine pathogen, Vibrio vulnificus. Infect Genet. Evol. 2007, 7, 685–693. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, W.; Zheng, H.; Lan, R.; Wang, H.; Du, P.; Bai, X.; Ji, S.; Meng, Q.; Jin, D.; et al. Minimum Core Genome Sequence Typing of Bacterial Pathogens: A Unified Approach for Clinical and Public Health Microbiology. J. Clin. Microbiol. 2013, 51, 2582–2591. [Google Scholar] [CrossRef] [Green Version]
- Ezaki, T.; Hashimoto, Y.; Yabuuchi, E. Fluorometric Deoxyribonucleic Acid-Deoxyribonucleic Acid Hybridization in Microdilution Wells as an Alternative to Membrane Filter Hybridization in which Radioisotopes Are Used To Determine Genetic Relatedness among Bacterial Strains. Int. J. Syst. Bacteriol. 1989, 39, 224–229. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Rossello-Mora, R. DNA-DNA Reassociation Methods Applied to Microbial Taxonomy and Their Critical Evaluation. In Molecular Identification, Systematics, and Population Structure of Prokaryotes; Springer: Berlin/Heidelberg, Germany, 2006; pp. 23–50. [Google Scholar]
- Rossello-Mora, R.; Amann, R. The species concept for prokaryotes. FEMS Microbiol. Rev. 2001, 25, 39–67. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E.; Goebel, B.M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S rRNA Sequence Analysis in the Present Species Definition in Bacteriology. Int. J. Syst. Bacteriol. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Liesack, W. Nucleic acids and classification. In Handbook of New bacterial Systematics; Goodfellow, M., O’Donnell, A.G., Eds.; Academic Press: London, UK, 1993; pp. 151–194. [Google Scholar]
- Stackebrandt, E.; Frederiksen, W.; Garrity, G.M.; Grimont, P.A.D.; Kämpfer, P.; Maiden, M.C.J.; Nesme, X.; Rosselló-Mora, R.; Swings, J.; Trüper, H.; et al. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 2002, 52, 1043–1047. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aoris | WE8B-23 | AO1 | AO2 | P6N | CCUG-33920 | AO3 | AN | AN1 | AN2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Aoris | 66.29 (53.3–100) | 48.8 (48.7–49.2) | 47.5 (47.0–48.3) | 48.3 (47.8–48.9) | 43.0 (42.5–42.8) | 43.0 (42.6–43.4) | 47.4 (47.7–46.9) | 36 (35.7–36.0) | 35 (35.1–35.4) | 35.5 (35.4–35.9) |
| WE8B-23 | 100 * | 72.7 (71.9–73.9) | 74.8 (74.3–75.5) | 42.8 | 43.2 | 51.2 (51.1–51.23 | 36 (35.8–36.3) | 35.5 (35.5–35.6) | 35.9 (35.8–36.0) | |
| AO1 | 87.8 (79–100) | 72.8 (72.6–73.6) | 42.5 (42.5–42.6) | 43.0 (42.9–43.1) | 51.4 (51.1–51.7) | 35.7 (35.9–36.0) | 36 (35.0–35.9) | 36 (35.7–35.9) | ||
| A02 | 92.4 (86.8–100) | 42.9 (4.8–43.0) | 43 (43.1–43.0) | 51.0 (50.9–51.2) | 36.7 (36.0–38.1) | 36 (36.0–36.2) | 36 (35.0–36.0) | |||
| P6N | 100 * | 47.5 | 44.6 (44.5–44.8) | 38.1 (38–38.3) | 37.2 (37.2–37.2) | 37.2 (37.2–37.4) | ||||
| CCUG 33920 | 100 * | 44.7 (44.6–44.9) | 36.6 (36.5–36.6) | 35.9 (35.9–36) | 36.2 (36.1–36.2) | |||||
| AO3 | 88.2 (80–100) | 36.7 (36.5–37) | 36 (36.0–36.5) | 36 (36.1–36.8) | ||||||
| AN | 86.8 (81.6–100) | 70 (69.1–71.5) | 71 (70.0–72.4) | |||||||
| AN1 | 90.6 (85.4–100) | 75 (72.1–75.1) | ||||||||
| AN2 | 91.6 (87.3–100) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mughal, S.R.; Niazi, S.A.; Do, T.; Gilbert, S.C.; Didelot, X.; Radford, D.R.; Beighton, D. Genomic Diversity among Actinomyces naeslundii Strains and Closely Related Species. Microorganisms 2023, 11, 254. https://doi.org/10.3390/microorganisms11020254
Mughal SR, Niazi SA, Do T, Gilbert SC, Didelot X, Radford DR, Beighton D. Genomic Diversity among Actinomyces naeslundii Strains and Closely Related Species. Microorganisms. 2023; 11(2):254. https://doi.org/10.3390/microorganisms11020254
Chicago/Turabian StyleMughal, Sadaf Rasheed, Sadia Ambreen Niazi, Thuy Do, Steven C. Gilbert, Xavier Didelot, David R. Radford, and David Beighton. 2023. "Genomic Diversity among Actinomyces naeslundii Strains and Closely Related Species" Microorganisms 11, no. 2: 254. https://doi.org/10.3390/microorganisms11020254