Genome-Scale Metabolic Modeling Reveals Metabolic Alterations of Multidrug-Resistant Acinetobacter baumannii in a Murine Bloodstream Infection Model

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Growth Condition
2.2. Animals
2.3. Non-Neutropenic Murine Bacteremia Model
2.4. Construction of the Genome-Scale Metabolic Model (GSMM) iAB5075
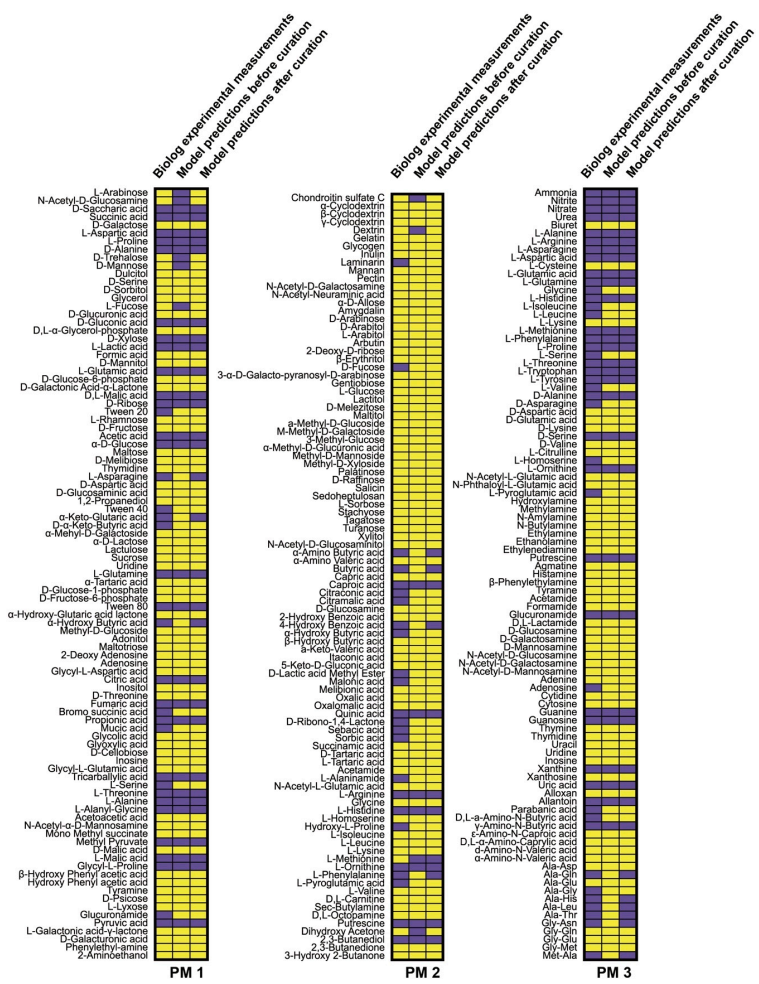
2.5. Biolog Assay and Prediction of Nutrient Utilizations
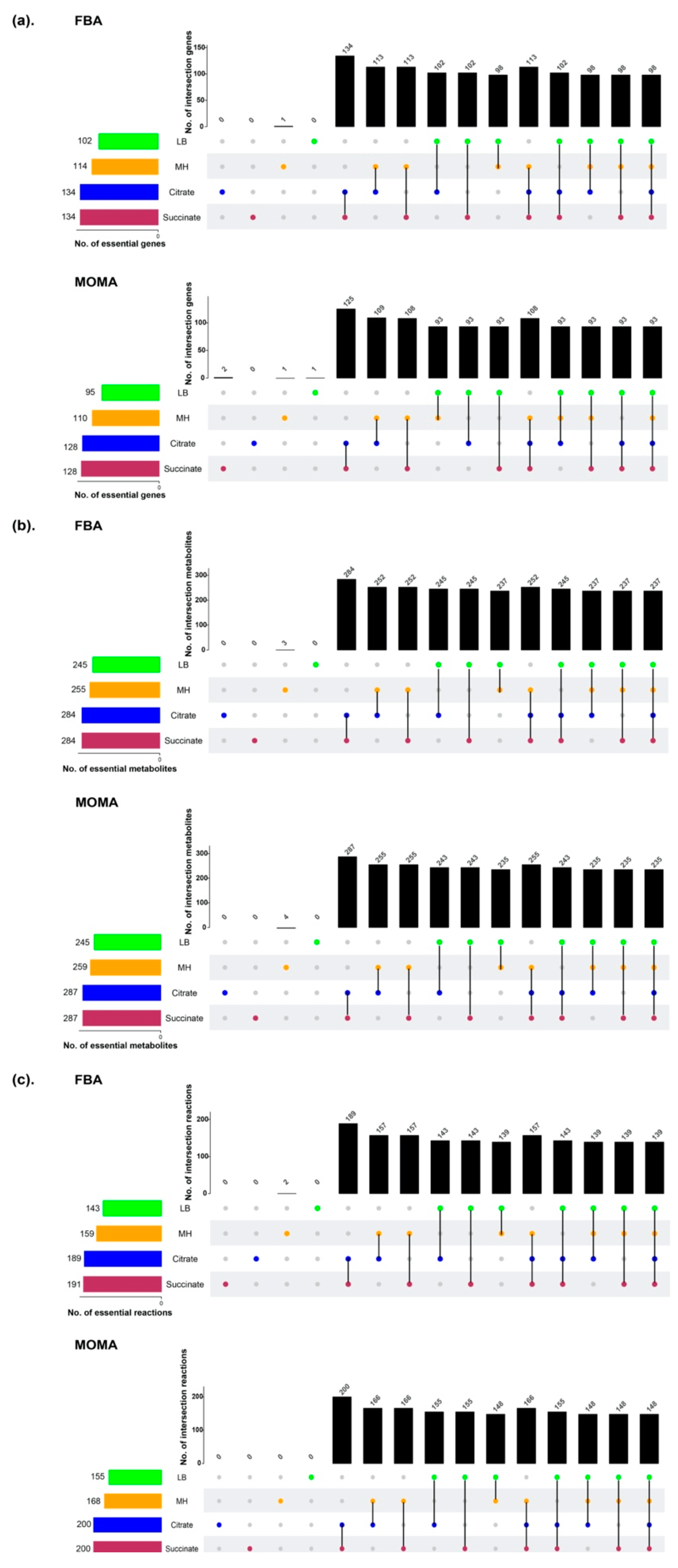
2.6. Gene Essentiality Analysis
2.7. Calculating Metabolic Fluxes with Transcriptomics Constraints
3. Results
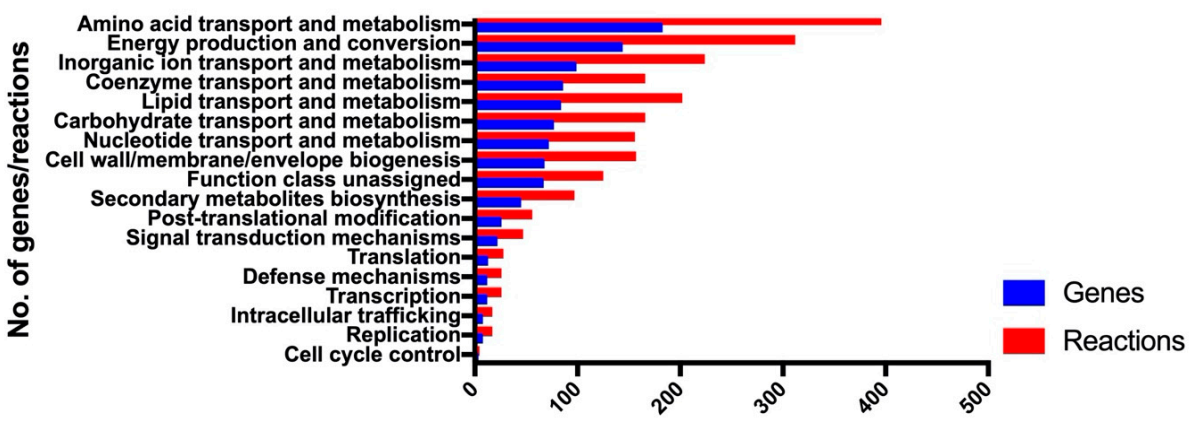
3.1. Construction of the Genome-Scale Metabolic Model iAB5075
3.2. Prediction of Bacterial Growth on Various Nutrients
3.3. Prediction of Essential Genes, Reactions, and Metabolites for Bacterial Growth
3.4. Modeling Metabolic Changes of Strain AB5075 in a Murine Bacteremia Model with Transcriptomic Constraints
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: A review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Dijkshoorn, L.; Nemec, A.; Seifert, H. An increasing threat in hospitals: Multidrug-resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 2007, 5, 939–951. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Monserrat-Martinez, A.; Gambin, Y.; Sierecki, E. Thinking outside the bug: Molecular targets and strategies to overcome antibiotic resistance. Int. J. Mol. Sci. 2019, 20, 1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.C.; Frawley, E.R.; Tapscott, T.; Vázquez-Torres, A. Bacterial stress responses during host infection. Cell Host Microbe 2016, 20, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totsika, M. Benefits and challenges of antivirulence antimicrobials at the dawn of the post-antibiotic era. Drug Deliv. Lett. 2016, 6, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Damron, F.H.; Oglesby-Sherrouse, A.G.; Wilks, A.; Barbier, M. Dual-seq transcriptomics reveals the battle for iron during Pseudomonas aeruginosa acute murine pneumonia. Sci. Rep. 2016, 6, 39172. [Google Scholar] [CrossRef]
- Subashchandrabose, S.; Smith, S.; DeOrnellas, V.; Crepin, S.; Kole, M.; Zahdeh, C.; Mobley, H.L. Acinetobacter baumannii genes required for bacterial survival during bloodstream infection. mSphere 2016, 1, 00013–00015. [Google Scholar] [CrossRef] [Green Version]
- Westermann, A.J.; Barquist, L.; Vogel, J. Resolving host–pathogen interactions by dual RNA-seq. PLoS Pathog. 2017, 13, e1006033. [Google Scholar] [CrossRef] [Green Version]
- Pisu, D.; Huang, L.; Grenier, J.K.; Russell, D.G. Dual RNA-Seq of Mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell Rep. 2020, 30, 335–350. [Google Scholar] [CrossRef]
- Bhuiyan, M.S.; Ellett, F.; Murray, G.L.; Kostoulias, X.; Cerqueira, G.M.; Schulze, K.E.; Maifiah, M.H.M.; Li, J.; Creek, D.J.; Lieschke, G.J. Acinetobacter baumannii phenylacetic acid metabolism influences infection outcome through a direct effect on neutrophil chemotaxis. Proc. Natl. Acad. Sci. USA 2016, 113, 9599–9604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, G.L.; Tsyganov, K.; Kostoulias, X.P.; Bulach, D.M.; Powell, D.; Creek, D.J.; Boyce, J.D.; Paulsen, I.T.; Peleg, A.Y. Global gene expression profile of Acinetobacter baumannii during bacteremia. J. Infect. Dis. 2017, 215, S52–S57. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Lu, J.; Zhao, J.; Zhang, X.; Heidi, H.Y.; Velkov, T.; Li, J. Complete genome sequence and genome-scale metabolic modelling of Acinetobacter baumannii type strain ATCC 19606. Int. J. Med. Microbiol. 2020, 151412. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhao, J.; Maifiah, M.H.M.; Velkov, T.; Schreiber, F.; Li, J. Metabolic responses to polymyxin treatment in Acinetobacter baumannii ATCC 19606: Integrating transcriptomics and metabolomics with genome-scale metabolic modeling. mSystems 2019, 4, e00157-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presta, L.; Bosi, E.; Mansouri, L.; Dijkshoorn, L.; Fani, R.; Fondi, M. Constraint-based modeling identifies new putative targets to fight colistin-resistant A. baumannii infections. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hadadi, N.; Pandey, V.; Chiappino-Pepe, A.; Morales, M.; Gallart-Ayala, H.; Mehl, F.; Ivanisevic, J.; Sentchilo, V.; van der Meer, J.R. Mechanistic insights into bacterial metabolic reprogramming from omics-integrated genome-scale models. NPJ Syst. Biol. Appl. 2020, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Health and Medical Research Council (NHMRC). Australian Code of Practice for the Care and Use of Animals for Scientific Purposes; National Health and Medical Research Council: Canberra, Australia, 2004.
- Zhao, J.; Li, M.; Wang, J.; Velkov, T.; Li, J. Dual RNA-seq revealed complex host-pathogen-polymyxin interactions in Acinetobacter baumannii bacteraemia. In Proceedings of the 29th European Congress of Clinical Microbiology & Infectious Diseases, Amsterdam, The Netherlands, 13 April 2019. [Google Scholar]
- Brittnacher, M.J.; Fong, C.; Hayden, H.; Jacobs, M.; Radey, M.; Rohmer, L. PGAT: A multistrain analysis resource for microbial genomes. Bioinformatics 2011, 27, 2429–2430. [Google Scholar] [CrossRef]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef]
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish-Bowden, A. The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v. 3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Czauderna, T.; Zhao, J.; Klapperstueck, M.; Maifiah, M.H.M.; Han, M.-L.; Lu, J.; Sommer, B.; Velkov, T.; Lithgow, T. Genome-scale metabolic modeling of responses to polymyxins in Pseudomonas aeruginosa. GigaScience 2018, 7, giy021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Gallagher, L.A.; Ramage, E.; Weiss, E.J.; Radey, M.; Hayden, H.S.; Held, K.G.; Huse, H.K.; Zurawski, D.V.; Brittnacher, M.J.; Manoil, C. Resources for genetic and genomic analysis of emerging pathogen Acinetobacter baumannii. J. Bacteriol. 2015, 197, 2027–2035. [Google Scholar] [CrossRef] [Green Version]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Jeske, L.; Placzek, S.; Schomburg, I.; Chang, A.; Schomburg, D. BRENDA in 2019: A European ELIXIR core data resource. Nucleic Acids Res. 2019, 47, D542–D549. [Google Scholar] [CrossRef]
- Wang, C.-H.; Zhang, C.; Xing, X.-H. Xanthine dehydrogenase: An old enzyme with new knowledge and prospects. Bioengineered 2016, 7, 395–405. [Google Scholar] [CrossRef]
- Yuan, H.; Chai, S.C.; Lam, C.K.; Xu, H.H.; Ye, Q.-Z. Two methionine aminopeptidases from Acinetobacter baumannii are functional enzymes. Bioorg. Med. Chem. Lett. 2011, 21, 3395–3398. [Google Scholar] [CrossRef] [Green Version]
- Huynh, K.-H.; Hong, M.-k.; Lee, C.; Tran, H.-T.; Lee, S.H.; Ahn, Y.-J.; Cha, S.-S.; Kang, L.-W. The crystal structure of the D-alanine-D-alanine ligase from Acinetobacter baumannii suggests a flexible conformational change in the central domain before nucleotide binding. J. Microbiol. 2015, 53, 776–782. [Google Scholar] [CrossRef]
- Sorci, L.; Blaby, I.; De Ingeniis, J.; Gerdes, S.; Raffaelli, N.; de Crécy Lagard, V.; Osterman, A. Genomics-driven reconstruction of Acinetobacter NAD metabolism insights for antibacterial target selection. J. Biol. Chem. 2010, 285, 39490–39499. [Google Scholar] [CrossRef] [Green Version]
- Mo, M.L.; Palsson, B.Ø.; Herrgård, M.J. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst. Biol. 2009, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Saa, P.A.; Nielsen, L.K. ll-ACHRB: A scalable algorithm for sampling the feasible solution space of metabolic networks. Bioinformatics 2016, 32, 2330–2337. [Google Scholar] [CrossRef]
- Runci, F.; Gentile, V.; Frangipani, E.; Rampioni, G.; Leoni, L.; Lucidi, M.; Visaggio, D.; Harris, G.; Chen, W.; Stahl, J. Contribution of active iron uptake to Acinetobacter baumannii pathogenicity. Infect. Immun. 2019, 87, e00755-18. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, B.L.; Skaar, E.P. Host–microbe interactions that shape the pathogenesis of A cinetobacter baumannii infection. Cell. Microbiol. 2012, 14, 1336–1344. [Google Scholar] [CrossRef] [Green Version]
- Peleg, A.Y.; Seifert, H.; Paterson, D.L. Acinetobacter baumannii: Emergence of a successful pathogen. Clin. Microbiol. Rev. 2008, 21, 538–582. [Google Scholar] [CrossRef] [Green Version]
- Norsigian, C.J.; Kavvas, E.; Seif, Y.; Palsson, B.O.; Monk, J.M. iCN718, an updated and improved genome-scale metabolic network reconstruction of Acinetobacter baumannii AYE. Front. Genet. 2018, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Rahim, N.A.; Cheah, S.-E.; Johnson, M.D.; Yu, H.; Sidjabat, H.E.; Boyce, J.; Butler, M.S.; Cooper, M.A.; Fu, J.; Paterson, D.L.; et al. Synergistic killing of NDM-producing MDR Klebsiella pneumoniae by two ‘old’ antibiotics-polymyxin B and chloramphenicol. J. Antimicrob. Chemother. 2015, 70, 2589–2597. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, A.C.; Thompson, M.G.; Black, C.C.; Kessler, J.L.; Clark, L.P.; McQueary, C.N.; Gancz, H.Y.; Corey, B.W.; Moon, J.K.; Si, Y. AB5075, a highly virulent isolate of Acinetobacter baumannii, as a model strain for the evaluation of pathogenesis and antimicrobial treatments. mBio 2014, 5, e01076-14. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Chavez, J.D.; Schweppe, D.K.; Zheng, C.; Weisbrod, C.R.; Eng, J.K.; Murali, A.; Lee, S.A.; Ramage, E.; Gallagher, L.A. In vivo protein interaction network analysis reveals porin-localized antibiotic inactivation in Acinetobacter baumannii strain AB5075. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Tipton, K.A.; Rather, P.N. An ompR-envZ two-component system ortholog regulates phase variation, osmotic tolerance, motility, and virulence in Acinetobacter baumannii strain AB5075. J. Bacteriol. 2017, 199, e00705-16. [Google Scholar] [CrossRef] [Green Version]
- Khaledi, A.; Schniederjans, M.; Pohl, S.; Rainer, R.; Bodenhofer, U.; Xia, B.; Klawonn, F.; Bruchmann, S.; Preusse, M.; Eckweiler, D. Transcriptome profiling of antimicrobial resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 4722–4733. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.P.; Wright, J.R.; Wong, H.T.; Ardasheva, A.; Brumbaugh, J.; McLimans, C.; Lamendella, R. Using bacterial transcriptomics to investigate Targets of Host-Bacterial Interactions in Caenorhabditis elegans. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Lo, N.W.-S.; Loo, J.F.-C.; Lin, X.; Yim, A.K.-Y.; Tsui, S.K.-W.; Lau, T.C.-K.; Ip, M.; Chan, T.-F. Comparative transcriptomics of multidrug-resistant Acinetobacter baumannii in response to antibiotic treatments. Sci. Rep. 2018, 8, 3515. [Google Scholar] [CrossRef]
- Son, M.S.; Matthews, W.J.; Kang, Y.; Nguyen, D.T.; Hoang, T.T. In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect. Immun. 2007, 75, 5313–5324. [Google Scholar] [CrossRef] [Green Version]
- Yimga, M.T.; Leatham, M.P.; Allen, J.H.; Laux, D.C.; Conway, T.; Cohen, P.S. Role of gluconeogenesis and the tricarboxylic acid cycle in the virulence of Salmonella enterica serovar Typhimurium in BALB/c mice. Infect. Immun. 2006, 74, 1130–1140. [Google Scholar] [CrossRef] [Green Version]
- Harding, C.M.; Hennon, S.W.; Feldman, M.F. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 2018, 16, 91. [Google Scholar] [CrossRef]
- Auer, G.K.; Weibel, D.B. Bacterial cell mechanics. Biochemistry 2017, 56, 3710–3724. [Google Scholar] [CrossRef]
- Sukhithasri, V.; Nisha, N.; Biswas, L.; Kumar, V.A.; Biswas, R. Innate immune recognition of microbial cell wall components and microbial strategies to evade such recognitions. Microbiol. Res. 2013, 168, 396–406. [Google Scholar] [CrossRef]
- Bojkovic, J.; Richie, D.L.; Six, D.A.; Rath, C.M.; Sawyer, W.S.; Hu, Q.; Dean, C.R. Characterization of an Acinetobacter baumannii lptD deletion strain: Permeability defects and response to inhibition of lipopolysaccharide and fatty acid biosynthesis. J. Bacteriol. 2016, 198, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Hengge, R. Linking bacterial growth, survival, and multicellularity–small signaling molecules as triggers and drivers. Curr. Opin. Microbiol. 2020, 55, 57–66. [Google Scholar] [CrossRef]
- Whiteley, A.T.; Eaglesham, J.B.; de Oliveira Mann, C.C.; Morehouse, B.R.; Lowey, B.; Nieminen, E.A.; Danilchanka, O.; King, D.S.; Lee, A.S.; Mekalanos, J.J. Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature 2019, 567, 194–199. [Google Scholar] [CrossRef]
- Hengge, R.; Gründling, A.; Jenal, U.; Ryan, R.; Yildiz, F. Bacterial signal transduction by cyclic di-GMP and other nucleotide second messengers. J. Bacteriol. 2016, 198, 15–26. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Content | iAB5075draft | iAB5075 | iATCC19606v2 | iCN718 |
|---|---|---|---|---|
| Genome size (Mb) | 3.97 | 3.98 | 3.90 | |
| Assembly status | Complete | Complete | Complete | |
| GC content | 39.1% | 39.2% | 39.0% | |
| No. of genes | 3895 | 3805 | 3694 | |
| No. of CDS | 3771 | 3663 | 3600 | |
| No. of reactions | 2184 | 2229 | 2114 | 1016 |
| No. of metabolites | 1480 | 1530 | 1422 | 890 |
| No. of involved genes | 1010 | 1015 | 1009 | 718 |
| Compartment a | 3 (c, p, e) | 3 (c, p, e) | 3 (c, p, e) | 2(c, e) |
| Prediction accuracy b | 72.8%/74.3% | 86.3%/87.6% | 85.6%/82.1% | 83.7/80% |
| MCC b | 0.58/0.26 | 0.76/0.28 | 0.68/0.33 | 0.72/0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Zhu, Y.; Han, J.; Lin, Y.-W.; Aichem, M.; Wang, J.; Chen, K.; Velkov, T.; Schreiber, F.; Li, J. Genome-Scale Metabolic Modeling Reveals Metabolic Alterations of Multidrug-Resistant Acinetobacter baumannii in a Murine Bloodstream Infection Model. Microorganisms 2020, 8, 1793. https://doi.org/10.3390/microorganisms8111793
Zhao J, Zhu Y, Han J, Lin Y-W, Aichem M, Wang J, Chen K, Velkov T, Schreiber F, Li J. Genome-Scale Metabolic Modeling Reveals Metabolic Alterations of Multidrug-Resistant Acinetobacter baumannii in a Murine Bloodstream Infection Model. Microorganisms. 2020; 8(11):1793. https://doi.org/10.3390/microorganisms8111793
Chicago/Turabian StyleZhao, Jinxin, Yan Zhu, Jiru Han, Yu-Wei Lin, Michael Aichem, Jiping Wang, Ke Chen, Tony Velkov, Falk Schreiber, and Jian Li. 2020. "Genome-Scale Metabolic Modeling Reveals Metabolic Alterations of Multidrug-Resistant Acinetobacter baumannii in a Murine Bloodstream Infection Model" Microorganisms 8, no. 11: 1793. https://doi.org/10.3390/microorganisms8111793