Targeted Next-Generation Sequencing and Informatics as an Effective Tool to Establish the Composition of Bovine Piroplasm Populations in Endemic Regions

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
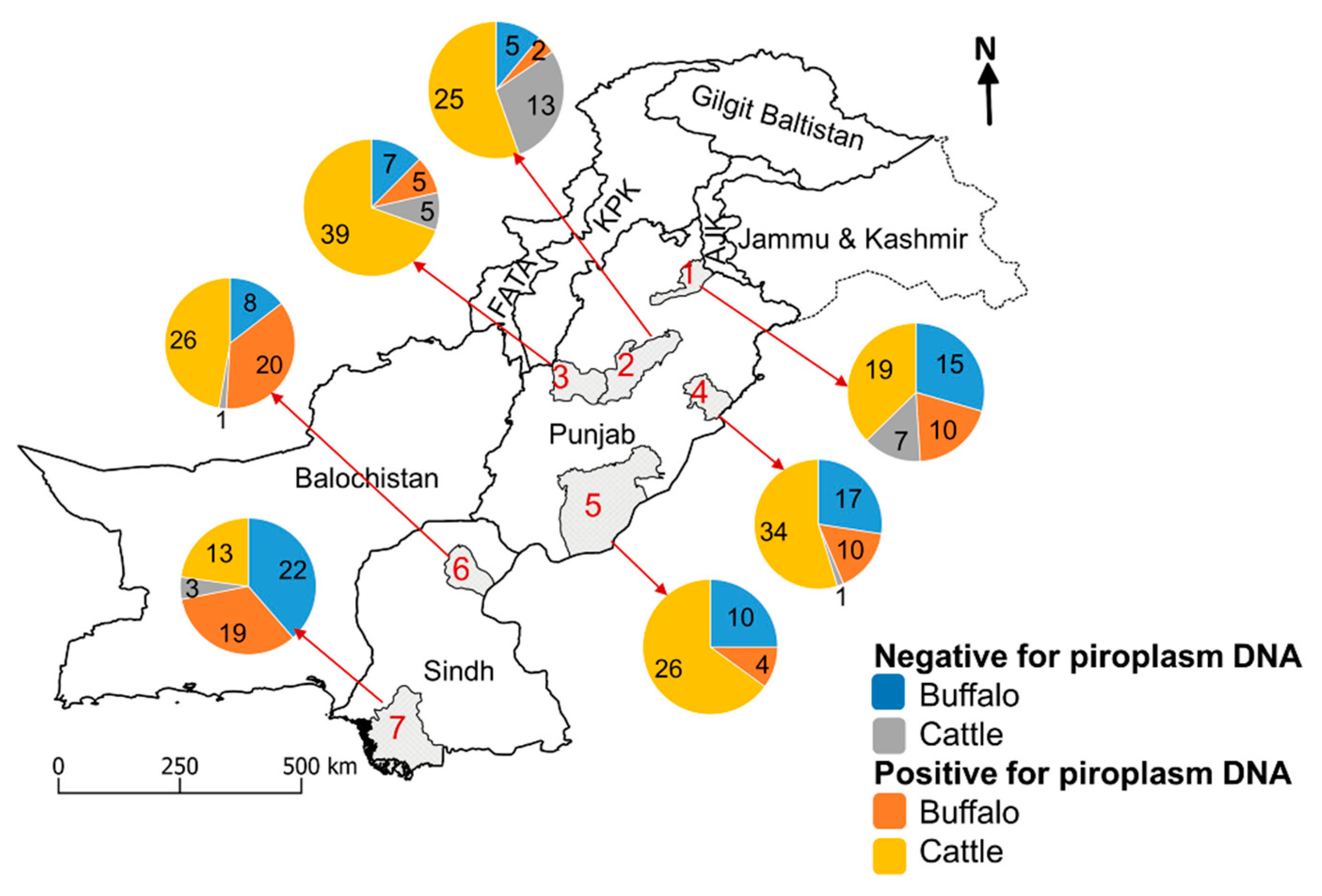
2.1. Collection of Blood Samples and DNA Extraction
2.2. PCR-Based Next-Generation Sequencing
2.3. Pre-Processing and Analysis of Sequence Data, and Taxonomic Assignment
2.4. Phylogenetic Analyses
2.5. Statistical Analyses
2.6. Data Availability Statement
3. Results
3.1. Sequence Data Sets and Definition of ASVs
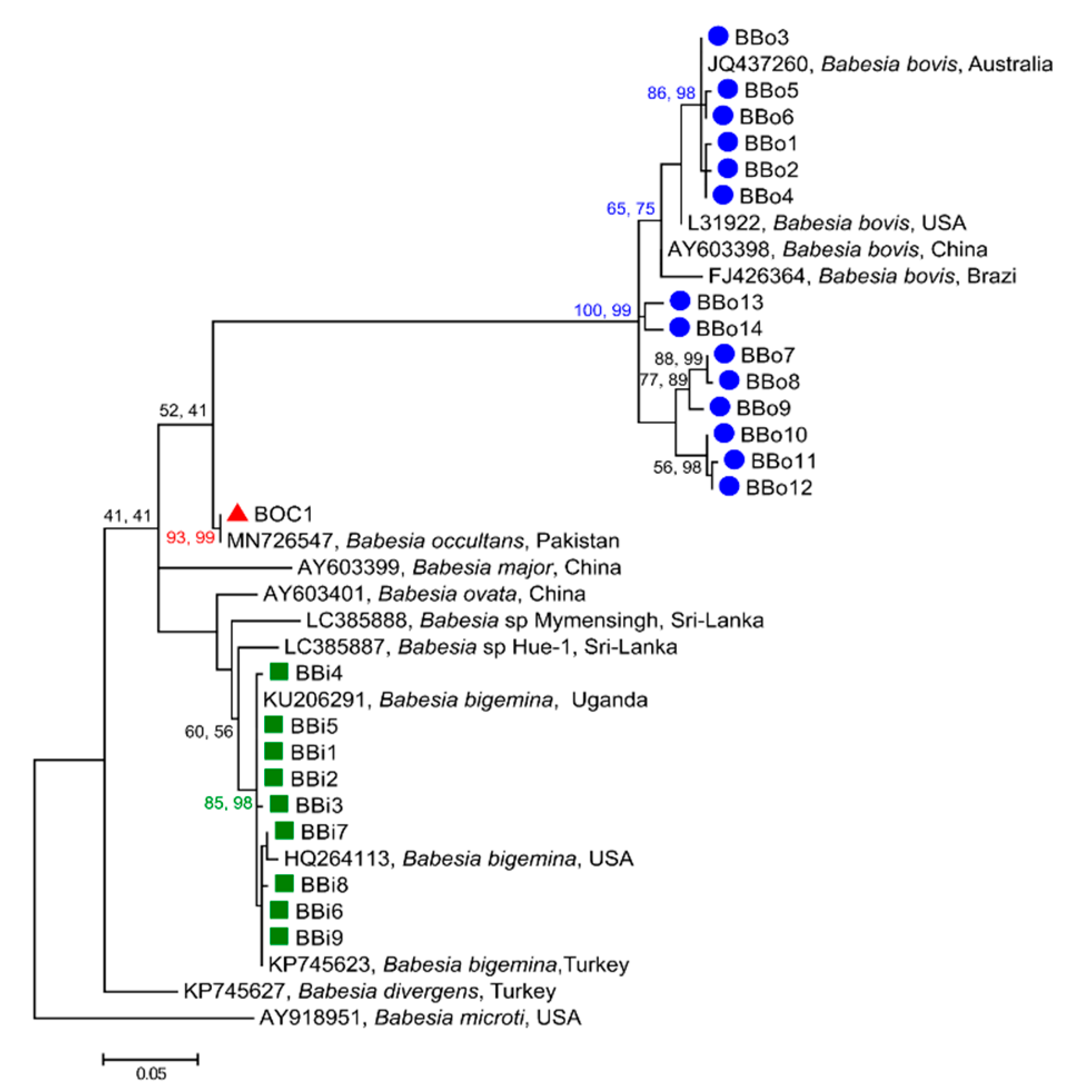
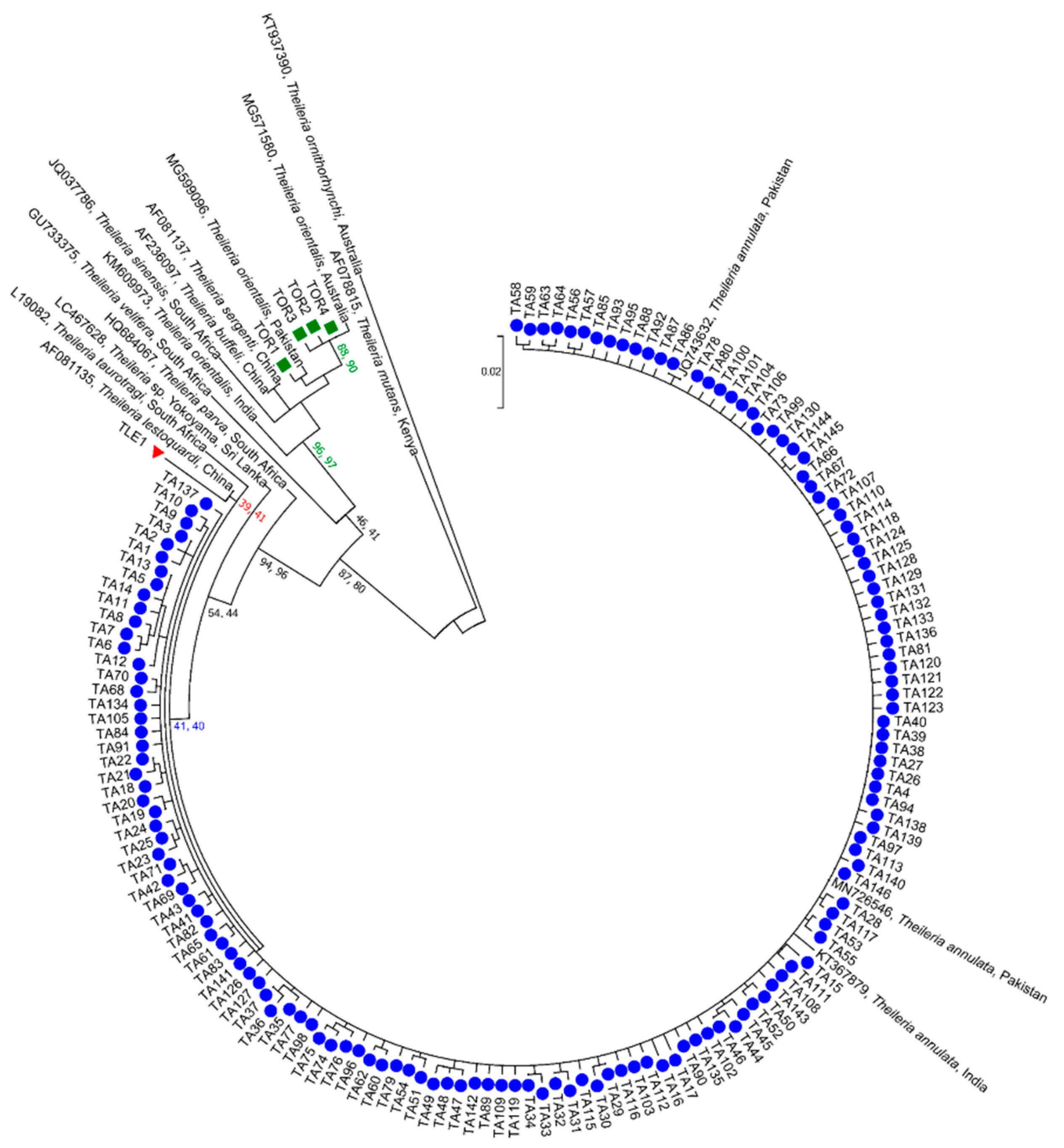
3.2. Phylogenetic Relationships of ASVs
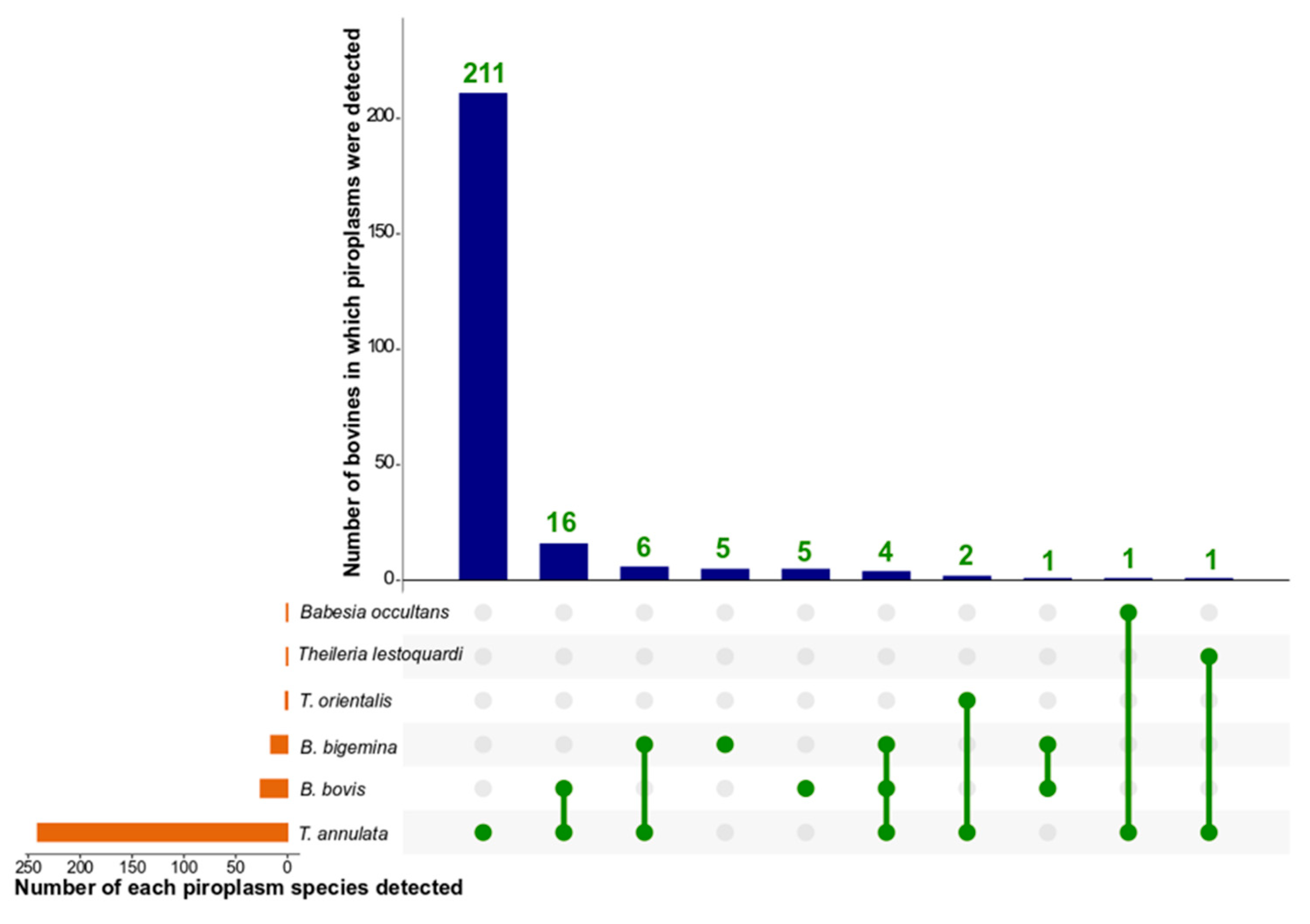
3.3. Composition of Piroplasm Populations in Individual Bovines
4. Discussions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, C.G.D. Dynamics and impact of tick-borne diseases of cattle. Trop. Anim. Health Prod. 1997, 29, 1S–3S. [Google Scholar] [CrossRef] [PubMed]
- Uilenberg, G. International collaborative research: Significance of tick-borne hemoparasitic diseases to world animal health. Vet. Parasitol. 1995, 57, 19–41. [Google Scholar] [CrossRef]
- De Castro, J.J. Sustainable tick and tickborne disease control in livestock improvement in developing countries. Vet. Parasitol. 1997, 71, 77–97. [Google Scholar] [CrossRef]
- Bock, R.; Jackson, L.; De Vos, A.; Jorgensen, W. Babesiosis of cattle. Parasitology 2004, 129, S247–S269. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, A.; Moreau, E.; Bonnet, S.; Plantard, O.; Malandrin, L. Babesia and its hosts: Adaptation to long-lasting interactions as a way to achieve efficient transmission. Vet. Res. 2009, 40, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zintl, A.; Mulcahy, G.; Skerrett, H.E.; Taylor, S.M.; Gray, J.S. Babesia divergens, a bovine blood parasite of veterinary and zoonotic importance. Clin. Microbiol. Rev. 2003, 16, 622–636. [Google Scholar] [CrossRef] [Green Version]
- Földvári, G.; Široký, P.; Szekeres, S.; Majoros, G.; Sprong, H. Dermacentor reticulatus: A vector on the rise. Parasit. Vectors 2016, 9, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.C.; Palmer, G. Designing blood-stage vaccines against Babesia bovis and B. bigemina. Parasitol. Today 1999, 15, 275–281. [Google Scholar] [CrossRef]
- Suarez, C.E.; Noh, S. Emerging perspectives in the research of bovine babesiosis and anaplasmosis. Vet. Parasitol. 2011, 180, 109–125. [Google Scholar] [CrossRef]
- Bishop, R.P.; Musoke, A.; Morzaria, S.; Gardner, M.; Nene, V. Theileria: Intracellular protozoan parasites of wild and domestic ruminants transmitted by ixodid ticks. Parasitology 2004, 129, S271–S283. [Google Scholar] [CrossRef]
- Gill, B.S.; Bhattacharyulu, Y.; Kaur, D. Symptoms and pathology of experimental bovine tropical theileriosis (Theileria annulata infection). Ann. Parasitol. Hum. Comparée 1977, 52, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nietz, W. Theileriosis, gonderioses and cytauxzoonoses. Onderstepoort J. Vet. Res. 1956, 27, 275–430. [Google Scholar]
- Martínez, J.A.A.; Rojas-Martínez, C.; Figueroa-Millán, J.V. Diagnostic tools for the identification of Babesia sp. in persistently infected cattle. Pathogens 2019, 8, 143. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.; Cruz, D.; Holman, P.; Waghela, S.; Perrone, J.; Shompole, S.; Rurangirwa, F. Non-immunologic methods of diagnosis of babesiosis. Memórias Inst. Oswaldo Cruz 1992, 87, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubbels, J.M.; De Vos, A.P.; Van Der Weide, M.; Viseras, J.; Schouls, L.M.; De Vries, E.; Jongejan, F. Simultaneous detection of bovine Theileria and Babesia species by reverse line blot hybridization. J. Clin. Microbiol. 1999, 37, 1782–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bursakov, S.A.; Kovalchuk, S.N. Co-infection with tick-borne disease agents in cattle in Russia. Ticks Tick Borne Dis. 2019, 10, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Perera, P.K.; Gasser, R.B.; Firestone, S.M.; Smith, L.; Roeber, F.; Jabbar, A. Semiquantitative multiplexed tandem PCR for detection and differentiation of four Theileria orientalis genotypes in cattle. J. Clin. Microbiol. 2015, 53, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Carr, I.M.; Robinson, J.I.; Dimitriou, R.; Markham, A.F.; Morgan, A.W.; Bonthron, D.T. Inferring relative proportions of DNA variants from sequencing electropherograms. Bioinformatics 2009, 25, 3244–3250. [Google Scholar] [CrossRef]
- Gandhi, K.; Thera, M.A.; Coulibaly, I.; Traoré, K.; Guindo, A.B.; Doumbo, O.K.; Takala-Harrison, S.; Plowe, C.V. Next generation sequencing to detect variation in the Plasmodium falciparum circumsporozoite protein. Am. J. Trop. Med. Hyg. 2012, 86, 775–781. [Google Scholar] [CrossRef]
- Glidden, C.K.; Koehler, A.V.; Hall, R.S.; Saeed, M.A.; Coppo, M.; Beechler, B.R.; Charleston, B.; Gasser, R.B.; Jolles, A.E.; Jabbar, A. Elucidating cryptic dynamics of Theileria communities in African buffalo using a high-throughput sequencing informatics approach. Ecol. Evol. 2019, 10, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, J.M.; Nickerson, D.A.; Reiner, A.P. Massively parallel sequencing: The new frontier of hematologic genomics. Blood 2013, 122, 3268–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, S.; Michelet, L.; Moutailler, S.; Cheval, J.; Hébert, C.; Vayssier-Taussat, M.; Eloit, M. Identification of parasitic communities within European ticks using next-generation sequencing. PLoS Negl. Trop. Dis. 2014, 8, e2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naveed, U.; Ali, Q.; Rashid, I.; Shabbir, M.Z.; Ijaz, M.; Abbas, M.; Evans, M.; Ashraf, K.; Morrison, I.; Morrison, L.; et al. Development of a deep amplicon sequencing method to determine the species composition of piroplasm haemoprotozoa. Ticks Tick Borne Dis. 2019, 10, 101276. [Google Scholar] [CrossRef]
- Hinsu, A.T.; Thakkar, J.R.; Koringa, P.G.; Vrba, V.; Jakhesara, S.J.; Psifidi, A.; Guitian, J.; Tomley, F.; Rank, D.N.; Raman, M.; et al. Illumina Next Generation Sequencing for the Analysis of Eimeria Populations in Commercial Broilers and Indigenous Chickens. Front. Vet. Sci. 2018, 5, 176. [Google Scholar] [CrossRef]
- Huggins, L.G.; Koehler, A.V.; Ng-Nguyen, D.; Wilcox, S.; Schunack, B.; Inpankaew, T.; Traub, R.J. A novel metabarcoding diagnostic tool to explore protozoan haemoparasite diversity in mammals: A proof-of-concept study using canines from the tropics. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Koehler, A.V.; Jabbar, A.; Hall, R.S.; Gasser, R.B. A Targeted “Next-Generation” Sequencing-Informatic approach to define genetic diversity in Theileria orientalis populations within individual cattle: Proof-of-principle. Pathogens 2020, 9, 448. [Google Scholar] [CrossRef]
- Šlapeta, J.; Saverimuttu, S.; Vogelnest, L.; Sangster, C.; Hulst, F.; Rose, K.; Thompson, P.; Whittington, R. Deep-sequencing to resolve complex diversity of apicomplexan parasites in platypuses and echidnas: Proof of principle for wildlife disease investigation. Infect. Genet. Evol. 2017, 55, 218–227. [Google Scholar] [CrossRef]
- Jabbar, A.; Abbas, T.; Sandhu, Z.-U.-D.; Saddiqi, H.A.; Qamar, M.F.; Gasser, R.B. Tick-borne diseases of bovines in Pakistan: Major scope for future research and improved control. Parasit. Vectors 2015, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ali, Z.; Maqbool, A.; Muhammad, K.; Khan, M.; Younis, M. Prevalence of Theileria annulata infected hard ticks of cattle and buffalo in Punjab, Pakistan. Pak. Vet. J. 2013, 23, 20–26. [Google Scholar]
- Ashraf, Q.U.; Khan, A.U.; Khattak, R.M.; Ali, M.; Shaikh, R.S.; Iqbal, F. A report on the high prevalence of Anaplasma sp. in buffaloes from two provinces in Pakistan. Ticks Tick Borne Dis. 2013, 4, 395–398. [Google Scholar] [CrossRef]
- Atif, F.A. Prevalence of Anaplasma marginale, Babesia bigemina and Theileria annulata infections among cattle in Sargodha district, Pakistan. Afr. J. Agric. Res. 2012, 7, 3302–3307. [Google Scholar] [CrossRef]
- Farooqi, S.H.; Ijaz, M.; Rashid, M.I.; Aqib, A.I.; Ahmad, Z.; Saleem, M.H.; Hussain, K.; Islam, S.; Naeem, H.; Khan, A. Molecular epidemiology of Babesia bovis in bovine of Khyber Pakhtunkhwa, Pakistan. Pak. Vet. J. 2017, 37, 275–280. [Google Scholar]
- Farooqi, S.H.; Ijaz, M.; Saleem, M.H.; Rashid, M.; Ahmad, S.S.; Islam, S.; Aqib, A.I.; Khan, A.; Hussain, K.; Khan, N.U. Prevalence and molecular diagnosis of Theileria annulata in bovine from three distinct zones of Khyber Pakhtunkhwa province, Pakistan. JAPS J. Anim. Plant Sci. 2017, 27, 1836–1841. [Google Scholar]
- Hassan, M.A.; Liu, J.; Rashid, M.; Iqbal, N.; Guan, G.; Yin, H.; Luo, J.-X. Molecular survey of piroplasm species from selected areas of China and Pakistan. Parasit. Vectors 2018, 11, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.K.; He, L.; Hussain, A.; Azam, S.; Zhang, W.-J.; Wang, L.-X.; Zhang, Q.-L.; Hu, M.; Zhou, Y.-Q.; Zhao, J. Molecular epidemiology of Theileria annulata and identification of 18S rRNA gene and ITS regions sequences variants in apparently healthy buffaloes and cattle in Pakistan. Infect. Genet. Evol. 2013, 13, 124–132. [Google Scholar] [CrossRef]
- Qayyum, M.; Farooq, U.; Samad, H.; Chauhdry, H. Prevalence, clinicotherapeutic and prophylactic studies on theileriosis in district Sahiwal (Pakistan). J. Anim. Plant Sci. 2010, 20, 266–270. [Google Scholar]
- Zeb, J.; Shams, S.; Din, I.U.; Ayaz, S.; Khan, A.; Nasreen, N.; Khan, H.; Khan, M.A.; Senbill, H. Molecular epidemiology and associated risk factors of Anaplasma marginale and Theileria annulata in cattle from North-western Pakistan. Vet. Parasitol. 2020, 279, 109044. [Google Scholar] [CrossRef]
- Khan, A.G. Technical Report on the Characterization of the Agro Ecological Context in which FAnGR (Farm Animal Genetic Resource) Are Found; ILRI: Nairobi, Kenya, 2004. [Google Scholar]
- Zia, U.E.; Mahmood, T.; Ali, M.R. Dairy Development in Pakistan; Food and Agriculture Organization of the United Nations: Rome, Italy, 2011. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: https://www.R-project.org/ (accessed on 11 August 2018).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, C.; Keatley, S.; Northover, A.; Gofton, A.W.; Brigg, F.; Lymbery, A.J.; Pallant, L.; Clode, P.L.; Thompson, R.A. Next generation sequencing reveals widespread trypanosome diversity and polyparasitism in marsupials from Western Australia. Int. J. Parasitol. Parasites Wildl. 2018, 7, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Government of Pakistan. Pakistan Economic Survey 2019–2020; Ministry of Finance: Islamabad, Pakistan, 2020; pp. 17–41.
- Ghafar, A.; Gasser, R.B.; Rashid, I.; Ghafoor, A.; Jabbar, A. Exploring the prevalence and diversity of bovine ticks in five agro-ecological zones of Pakistan using phenetic and genetic tools. Ticks Tick Borne Dis. 2020, 11, 101472. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Nijhof, A.M.; Sauter-Louis, C.; Schauer, B.; Staubach, C.; Conraths, F.J. Distribution of ticks infesting ruminants and risk factors associated with high tick prevalence in livestock farms in the semi-arid and arid agro-ecological zones of Pakistan. Parasit. Vectors 2017, 10, 1–15. [Google Scholar] [CrossRef]
- Pipano, E.; Shkap, V. Vaccination against tropical theileriosis. Ann. N. Y. Acad. Sci. 2000, 916, 484–500. [Google Scholar] [CrossRef]
- Sajid, M.S.; Iqbal, Z.; Khan, M.N.; Muhammad, G.; Khan, M.K. Prevalence and associated risk factors for bovine tick infestation in two districts of lower Punjab, Pakistan. Prev. Vet. Med. 2009, 92, 386–391. [Google Scholar] [CrossRef]
- Bock, R.E.; De Vos, A.J.; Kingston, T.G.; McLellan, D.J. Effect of breed of cattle on innate resistance to infection with Babesia bovis, B. bigemina and Anaplasma marginale. Aust. Vet. J. 1997, 75, 337–340. [Google Scholar] [CrossRef]
- Glass, E.J.; Preston, P.M.; Springbett, A.; Craigmile, S.; Kirvar, E.; Wilkie, G.; Brown, C.D. Bos taurus and Bos indicus (Sahiwal) calves respond differently to infection with Theileria annulata and produce markedly different levels of acute phase proteins. Int. J. Parasitol. 2005, 35, 337–347. [Google Scholar] [CrossRef]
- Larcombe, S.; Kolte, S.; Ponnudurai, G.; Kurkure, N.; Magar, S.; Velusamy, R.; Rani, N.; Rubinibala, B.; Rekha, B.; Alagesan, A.; et al. The impact of tick-borne pathogen infection in Indian bovines is determined by host type but not the genotype of Theileria annulata. Infect. Genet. Evol. 2019, 75, 103972. [Google Scholar] [CrossRef]
- Calleja-Bueno, L.; Sainz, A.; García-Sancho, M.; Rodríguez-Franco, F.; González-Martín, J.V.; Villaescusa, A. Molecular, epidemiological, haematological and biochemical evaluation in asymptomatic Theileria annulata infected cattle from an endemic region in Spain. Ticks Tick Borne Dis. 2017, 8, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Aktas, M.; Altay, K.; Dumanli, N. A molecular survey of bovine Theileria parasites among apparently healthy cattle and with a note on the distribution of ticks in eastern Turkey. Vet. Parasitol. 2006, 138, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Suarez, C.J.; Banaei, N.; Pinsky, B.A. Next-generation sequencing for infectious disease diagnosis and management: A report of the Association for Molecular Pathology. J. Mol. Diagn. 2015, 17, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, R.; Xiao, S.; Lin, R.; Wang, Y.; Wang, T. Technical validation of a high-sensitivity target capture NGS assay using unique molecular identifier approach. J. Clin. Oncol. 2020, 38, e13657. [Google Scholar] [CrossRef]
- Gebrekidan, H.; Perera, P.K.; Ghafar, A.; Abbas, T.; Gasser, R.B.; Jabbar, A. An appraisal of oriental theileriosis and the Theileria orientalis complex, with an emphasis on diagnosis and genetic characterisation. Parasitol. Res. 2019, 119, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Aparna, M.; Ravindran, R.; Vimalkumar, M.; Lakshmanan, B.; Rameshkumar, P.; Kumar, K.A.; Promod, K.; Ajithkumar, S.; Ravishankar, C.; Devada, K.; et al. Molecular characterization of Theileria orientalis causing fatal infection in crossbred adult bovines of South India. Parasitol. Int. 2011, 60, 524–529. [Google Scholar] [CrossRef]
- Sivakumar, T.; Kothalawala, H.; Abeyratne, S.A.E.; Vimalakumar, S.C.; Meewewa, A.S.; Hadirampela, D.T.; Puvirajan, T.; Sukumar, S.; Kuleswarakumar, K.; Chandrasiri, A.D.N.; et al. A PCR-based survey of selected Babesia and Theileria parasites in cattle in Sri Lanka. Vet. Parasitol. 2012, 190, 263–267. [Google Scholar] [CrossRef]
- Liu, A.H.; Guan, G.Q.; Liu, J.L.; Liu, Z.J.; Leblanc, N.; Li, Y.Q.; Gao, J.L.; Ma, M.L.; Niu, Q.L.; Ren, Q.Y.; et al. Polymorphism analysis of Chinese Theileria sergenti using allele-specific polymerase chain reaction of the major piroplasm surface protein gene. J. Parasitol. 2011, 97, 116–121. [Google Scholar] [CrossRef]
- Gebrekidan, H.; Abbas, T.; Wajid, M.; Ali, A.; Gasser, R.B.; Jabbar, A. Molecular characterisation of Theileria orientalis in imported and native bovines from Pakistan. Infect. Genet. Evol. 2017, 47, 19–25. [Google Scholar] [CrossRef]
- Gebrekidan, H.; Gasser, R.B.; Perera, P.K.; McGrath, S.; McGrath, S.; Stevenson, M.A.; Jabbar, A. Investigating the first outbreak of oriental theileriosis in cattle in South Australia using multiplexed tandem PCR (MT-PCR). Ticks Tick Borne Dis. 2015, 6, 574–578. [Google Scholar] [CrossRef]
- Perera, P.K.; Gasser, R.B.; Anderson, G.A.; Jeffers, M.; Bell, C.M.; Jabbar, A. Epidemiological survey following oriental theileriosis outbreaks in Victoria, Australia, on selected cattle farms. Vet. Parasitol. 2013, 197, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Benitez, D.; Mesplet, M.; Echaide, I.; De Echaide, S.T.; Schnittger, L.; Florin-Christensen, M. Mitigated clinical disease in water buffaloes experimentally infected with Babesia bovis. Ticks Tick Borne Dis. 2018, 9, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Jaimes-Dueñez, J.; Triana-Chavez, O.; Holguín-Rocha, A.; Tobón-Castaño, A.; Mejia-Jaramillo, A.M. Molecular surveillance and phylogenetic traits of Babesia bigemina and Babesia bovis in cattle (Bos taurus) and water buffaloes (Bubalus bubalis) from Colombia. Parasit. Vectors 2018, 11, 510. [Google Scholar] [CrossRef] [PubMed]
- Bhoora, R.; Franssen, L.; Oosthuizen, M.C.; Guthrie, A.J.; Zweygarth, E.; Penzhorn, B.L.; Jongejan, F.; Collins, N.E. Sequence heterogeneity in the 18S rRNA gene within Theileria equi and Babesia caballi from horses in South Africa. Vet. Parasitol. 2009, 159, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, J.-S.; Allsopp, B.A.; Waghela, S.D.; Park, J.-H.; Kakuda, T.; Sugimoto, C.; Allsopp, M.T.E.P.; Wagner, G.G.; Holman, P.J. A study of the systematics of Theileria spp. based upon small-subunit ribosomal RNA gene sequences. Parasitol. Res. 1999, 85, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, M.-J.; Hong, Y.; Van Der Weide, M.; Qi, B.; Nijman, I.J.; Guangyuan, L.; Jongejan, F. Molecular characterisation of the Theileria buffeli/orientalis group. Int. J. Parasitol. 2000, 30, 943–952. [Google Scholar] [CrossRef]
- Mans, B.J.; Pienaar, R.; Latif, A.A.; Potgieter, F.T. Diversity in the 18S SSU rRNA V4 hyper-variable region of Theileria spp. in Cape buffalo (Syncerus caffer) and cattle from southern Africa. Parasitology 2011, 138, 766–779. [Google Scholar] [CrossRef] [Green Version]
- Zakian, A.; Nouri, M.; Barati, F.; Kahroba, H.; Jolodar, A.; Rashidi, F. Vertical transmission of Theileria lestoquardi in sheep. Vet. Parasitol. 2014, 203, 322–325. [Google Scholar] [CrossRef]
- Jalali, S.M.; Jolodar, A.; Rasooli, A.; Darabifard, A. Detection of Theileria lestoquardi cross infection in cattle with clinical theileriosis in Iran. Acta Parasitol. 2016, 61, 756–761. [Google Scholar] [CrossRef]
- Taha, K.; Salih, D.; Ali, A.; Omer, R.; El Hussein, A. Naturally occurring infections of cattle with Theileria lestoquardi and sheep with Theileria annulata in the Sudan. Vet. Parasitol. 2013, 191, 143–145. [Google Scholar] [CrossRef]
- Ahmed, J.S.; Schnittger, L.; Yin, H.; Gubbels, M.J.; Beyer, D.; Jongejan, F.; Niemann, S. Phylogeny of sheep and goat Theileria and Babesia parasites. Parasitol. Res. 2003, 91, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Katzer, F. Phylogenetic analysis of Theileria and Babesia equi in relation to the establishment of parasite populations within novel host species and the development of diagnostic tests. Mol. Biochem. Parasitol. 1998, 95, 33–44. [Google Scholar] [CrossRef]
- Leemans, I.; Hooshmand-Rad, P.; Uggla, A. The indirect fluorescent antibody test based on schizont antigen for study of the sheep parasite Theileria lestoquardi. Vet. Parasitol. 1997, 69, 9–18. [Google Scholar] [CrossRef]
- Gray, J.S.; De Vos, A.J. Studies on a bovine Babesia transmitted by Hyalomma marginatum rufipes Koch, 1844. Onderstepoort J. Vet. Res. 1981, 48, 215–223. [Google Scholar] [PubMed]
- De Caro, N.; LaRocca, V.; Parisi, A.; Losurdo, M.; Lia, R.P.; Greco, M.F.; Miccolis, A.; Ventrella, G.; Otranto, D.; Buonavoglia, C. Clinical bovine piroplasmosis caused by Babesia occultans in Italy. J. Clin. Microbiol. 2013, 51, 2432–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafar, A.; Cabezas-Cruz, A.; Galon, C.; Obregon, D.; Gasser, R.B.; Moutailler, S.; Jabbar, A. Bovine ticks harbour a diverse array of microorganisms in Pakistan. Parasit. Vectors 2020, 13, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Piroplasm Species | District(s) | Water Buffalo | Cattle | ||
|---|---|---|---|---|---|
| Male | Female | Male | Female | ||
| Theileria annulata | Okara | — | 10/23 | 7/7 | 27/28 |
| Jhelum | 3/7 | 7/18 | — | 19/22 | |
| Bahawalpur | — | 4/11 | 5/5 | 21/21 | |
| Layyah | — | 5/9 | 3/5 | 36/39 | |
| Jhang | — | 2/7 | 4/4 | 21/34 | |
| Sukkur | 5/9 | 9/19 | 8/8 | 18/19 | |
| Thatta | 4/13 | 10/28 | 4/5 | 9/11 | |
| T. lestoquardi-like | Thatta | 1/13 | — | — | — |
| T. orientalis | Okara | — | — | — | 1/28 |
| Jhang | — | — | — | 1/34 | |
| Babesia bovis | Okara | — | — | 1/5 | 1/28 |
| Jhelum | — | — | — | 4/22 | |
| Bahawalpur | — | — | — | 1/21 | |
| Layyah | — | 1/9 | — | 1/39 | |
| Jhang | — | — | — | 1/19 | |
| Sukkur | 1/9 | 4/19 | 1/5 | 2/11 | |
| Thatta | 3/13 | 5/28 | — | — | |
| B. bigemina | Okara | — | — | — | 2/28 |
| Bahawalpur | — | — | 2/5 | — | |
| Layyah | — | — | — | — | |
| Sukkur | 1/9 | 6/19 | 1/8 | — | |
| Thatta | 1/13 | 1/28 | — | 2/11 | |
| B. occultans | Layyah | — | — | — | 1/39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghafar, A.; Koehler, A.V.; Hall, R.S.; Gauci, C.G.; Gasser, R.B.; Jabbar, A. Targeted Next-Generation Sequencing and Informatics as an Effective Tool to Establish the Composition of Bovine Piroplasm Populations in Endemic Regions. Microorganisms 2021, 9, 21. https://doi.org/10.3390/microorganisms9010021
Ghafar A, Koehler AV, Hall RS, Gauci CG, Gasser RB, Jabbar A. Targeted Next-Generation Sequencing and Informatics as an Effective Tool to Establish the Composition of Bovine Piroplasm Populations in Endemic Regions. Microorganisms. 2021; 9(1):21. https://doi.org/10.3390/microorganisms9010021
Chicago/Turabian StyleGhafar, Abdul, Anson V. Koehler, Ross S. Hall, Charles G. Gauci, Robin B. Gasser, and Abdul Jabbar. 2021. "Targeted Next-Generation Sequencing and Informatics as an Effective Tool to Establish the Composition of Bovine Piroplasm Populations in Endemic Regions" Microorganisms 9, no. 1: 21. https://doi.org/10.3390/microorganisms9010021