
Adaptation and Codon-Usage Preference of Apple and Pear-Infecting Apple Stem Grooving Viruses

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
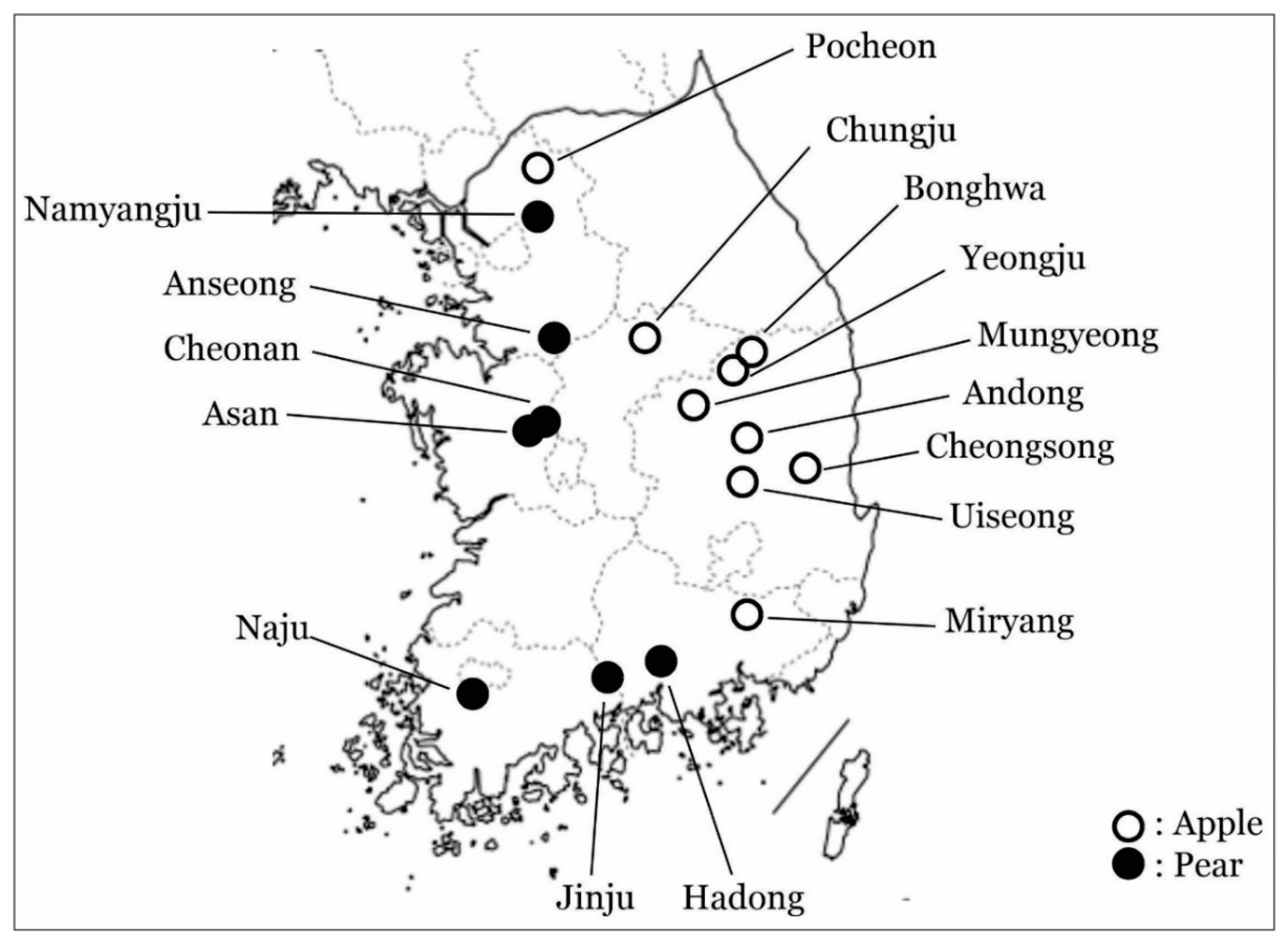
2.1. Sample Collection
2.2. cDNA Amplification
2.3. Phylogenetic Analysis
2.4. Protein Structure Prediction
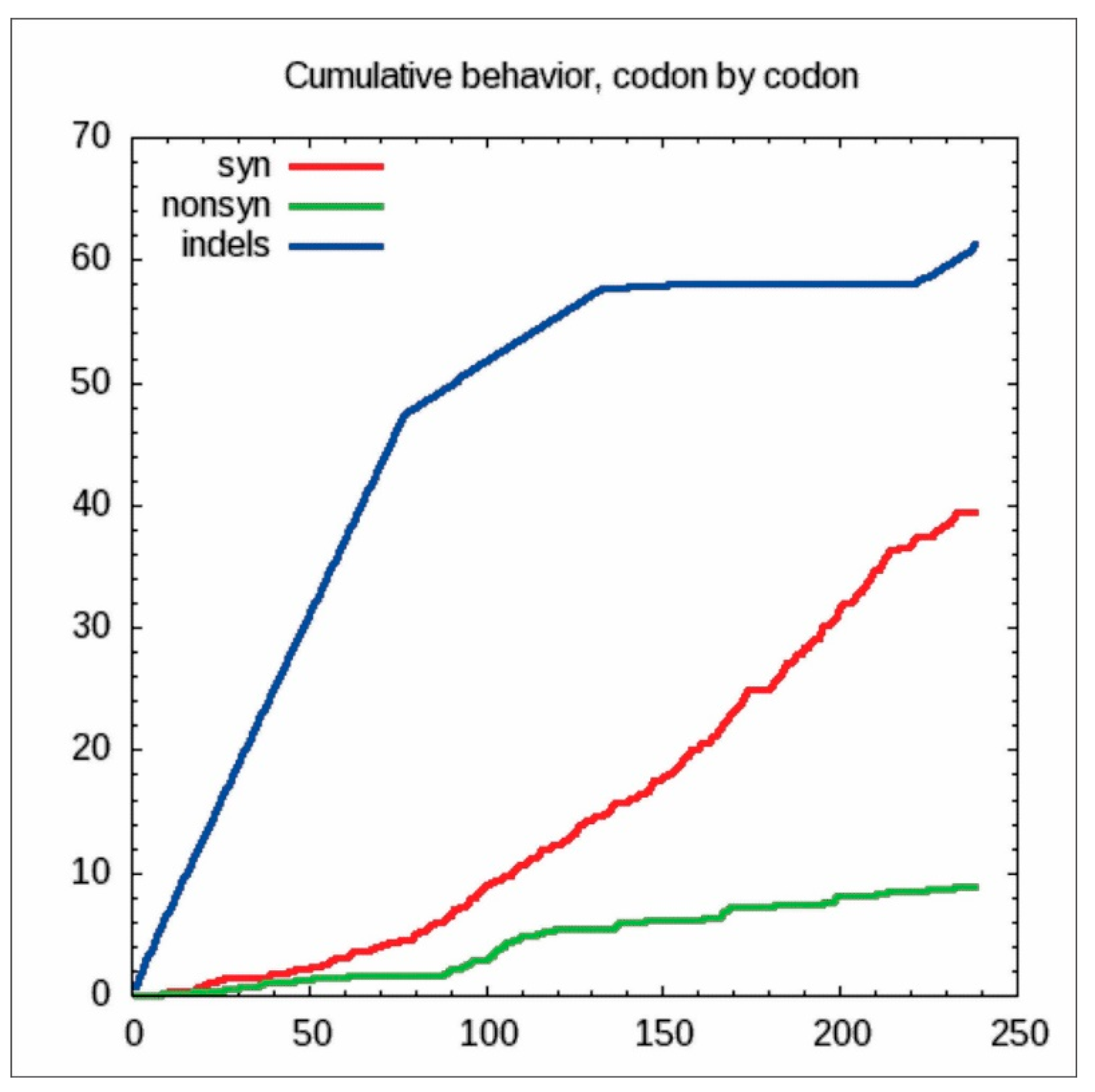
2.5. Codon-by-Codon Behavior Analysis for Synonymous and Non-Synonymous Substitutions
2.6. Codon-by-Codon Maximum Likelihood Analysis of Natural Selection
2.7. Preferred Codon Usage of ASGV CP Gene
3. Results
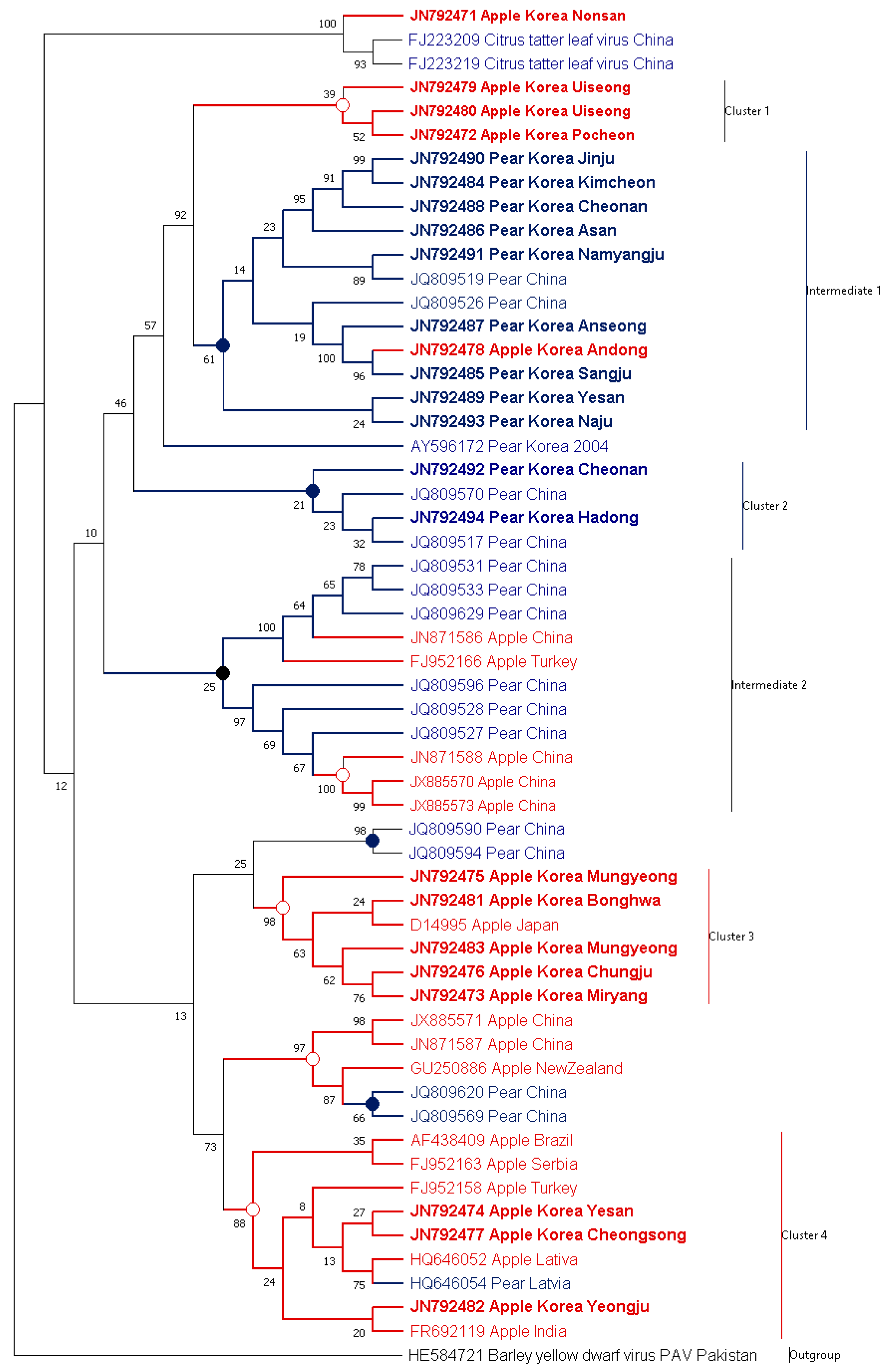
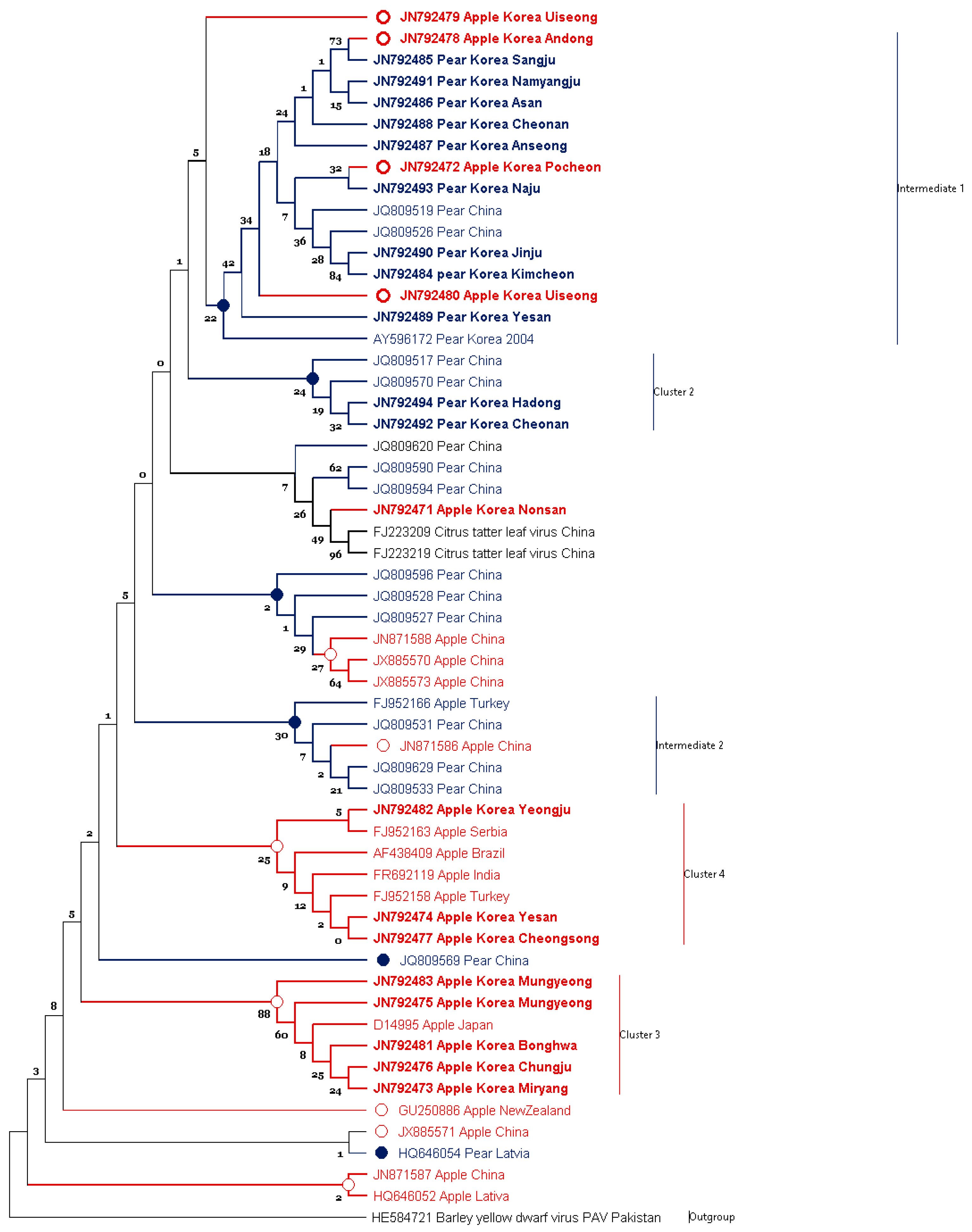
3.1. Phylogenetic Analysis of ASGV Showed Host-Specificity
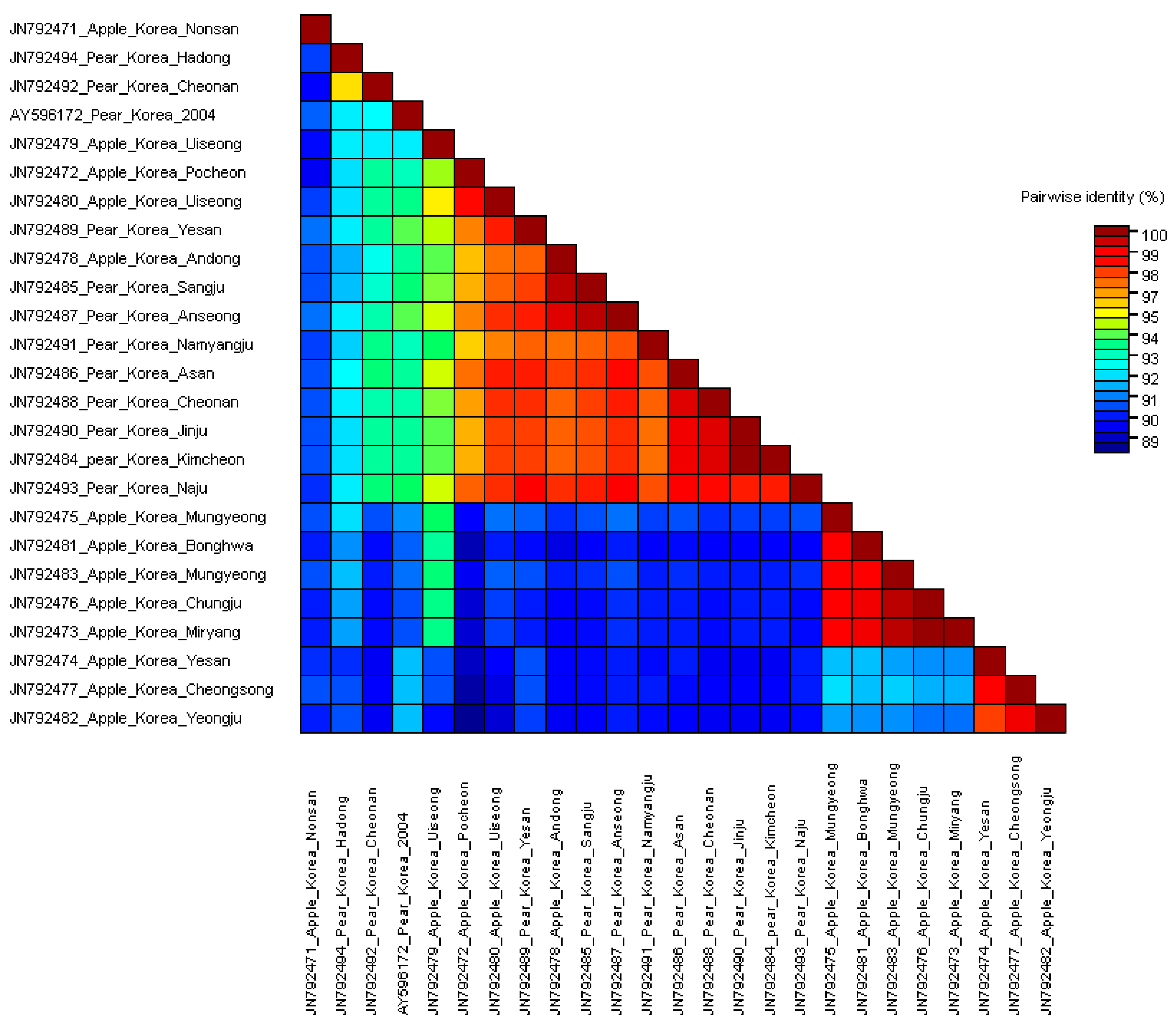
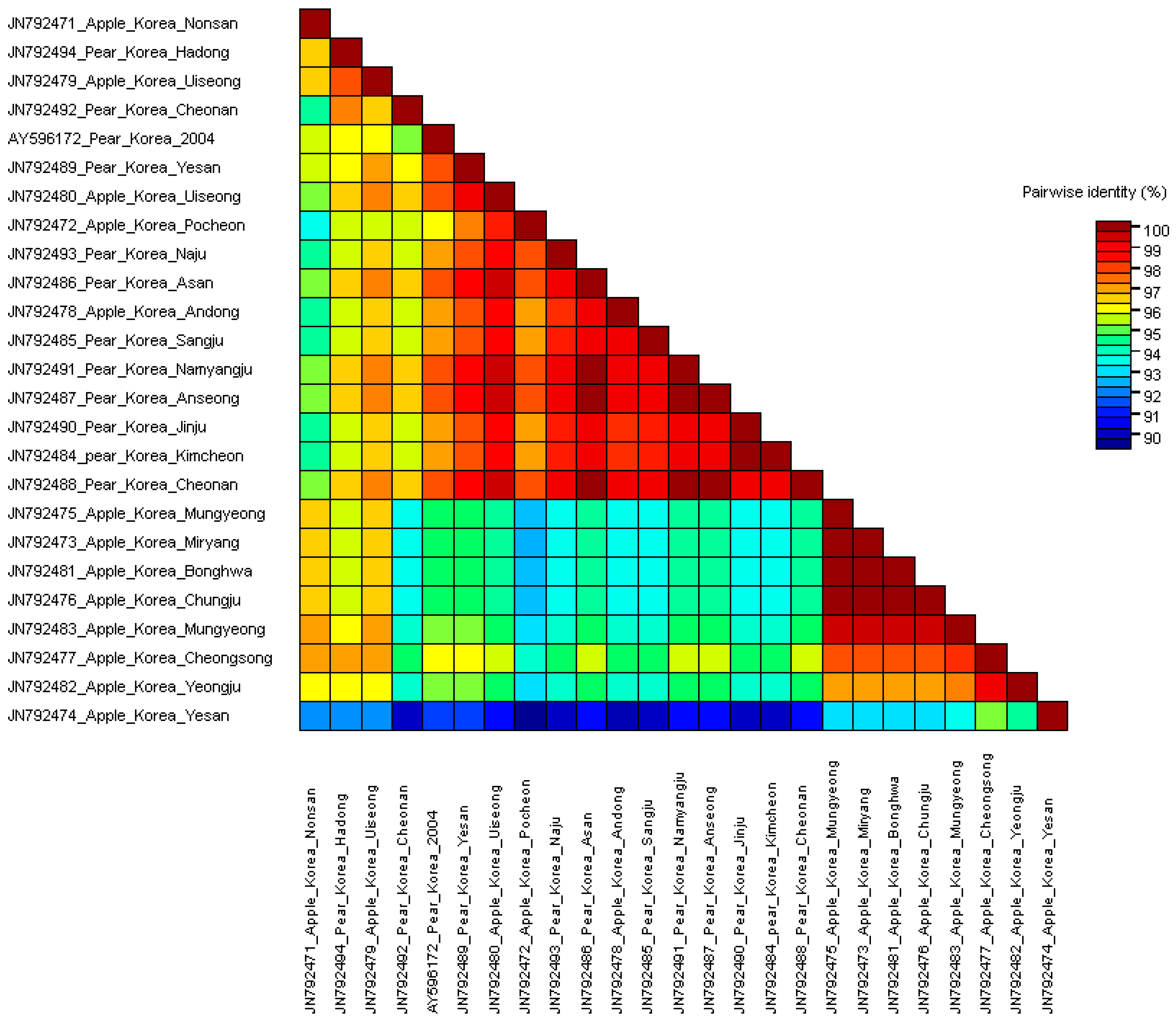
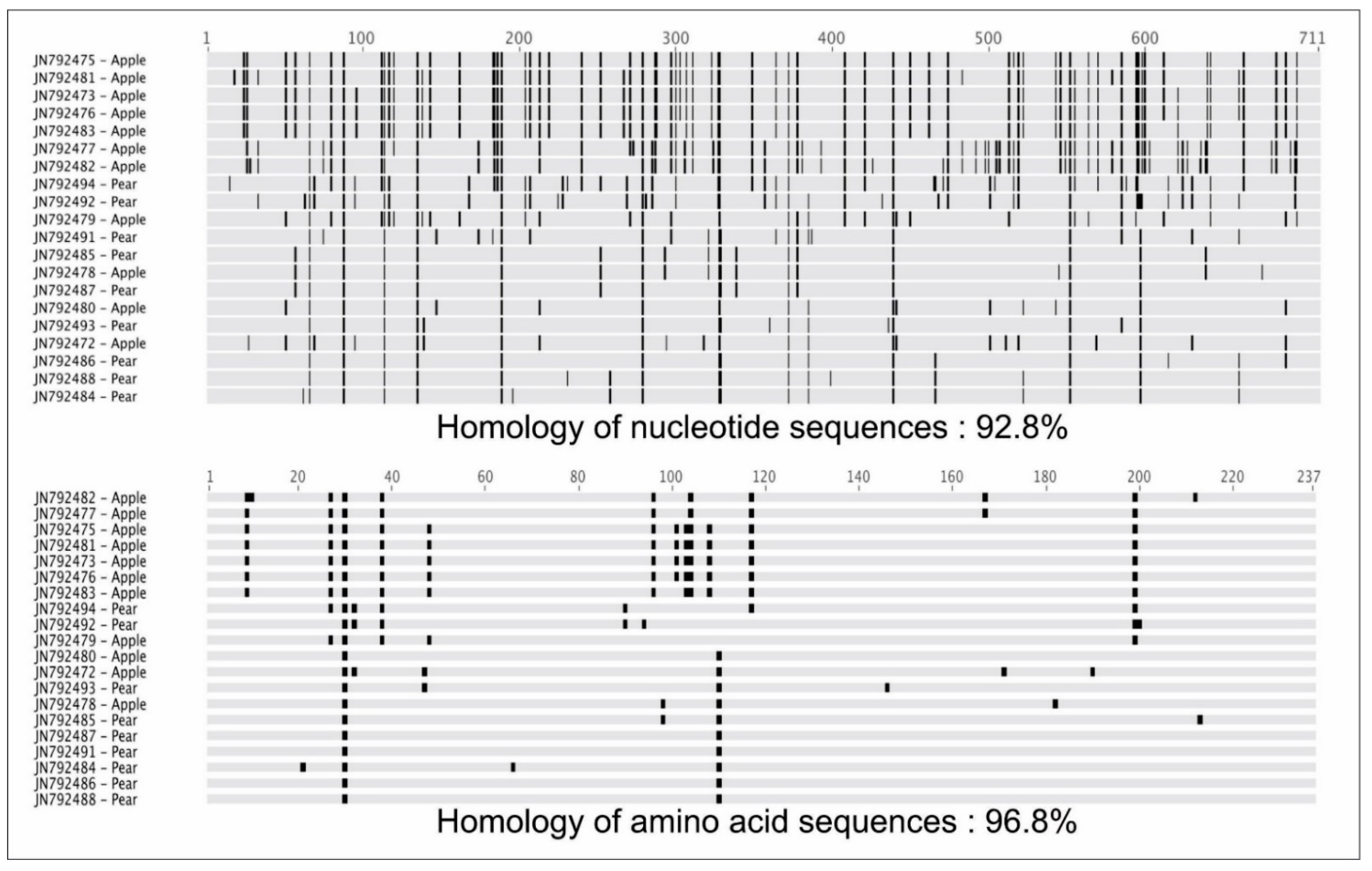
3.2. Comparisons of Nucleotide and Protein Sequences
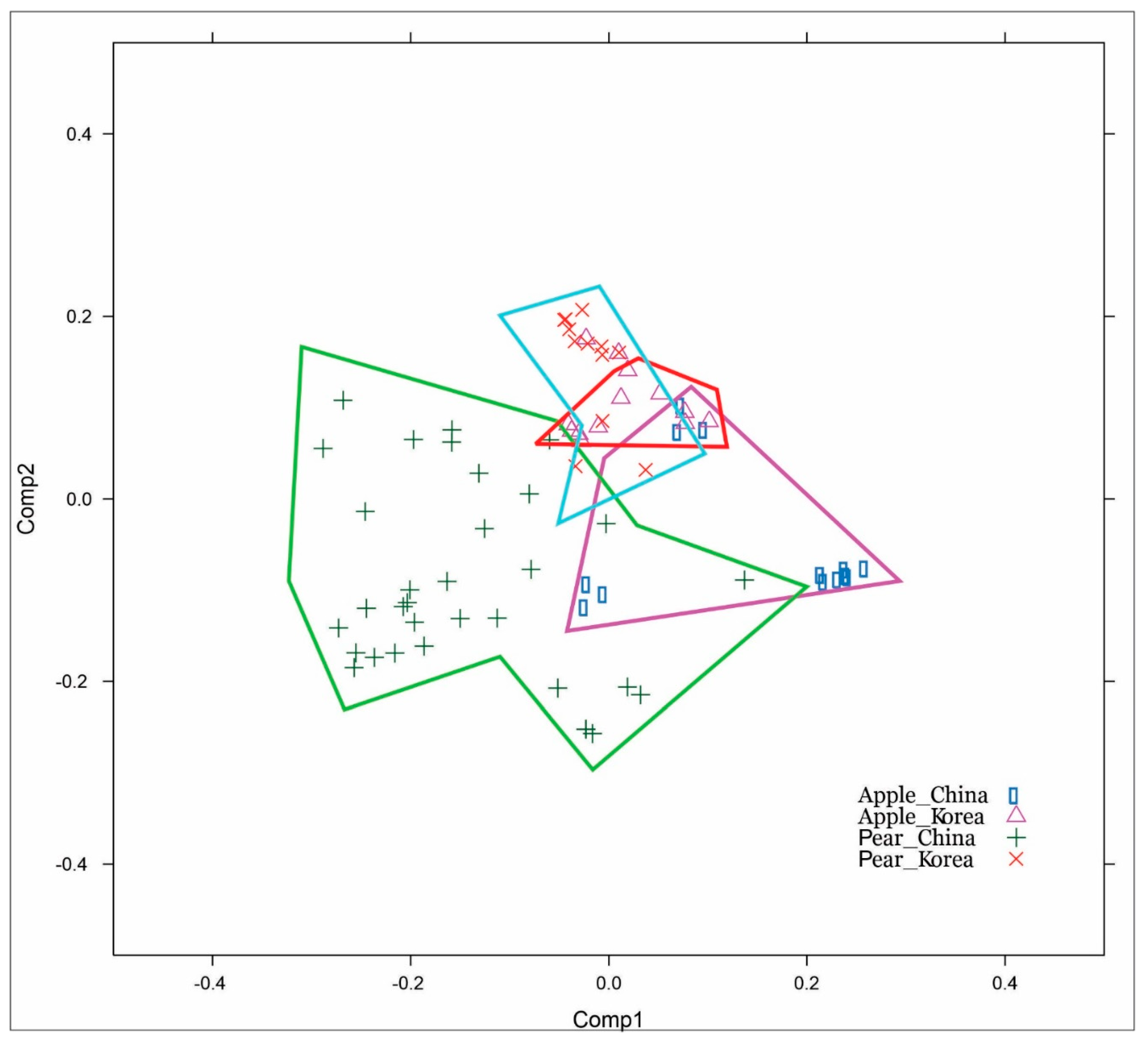
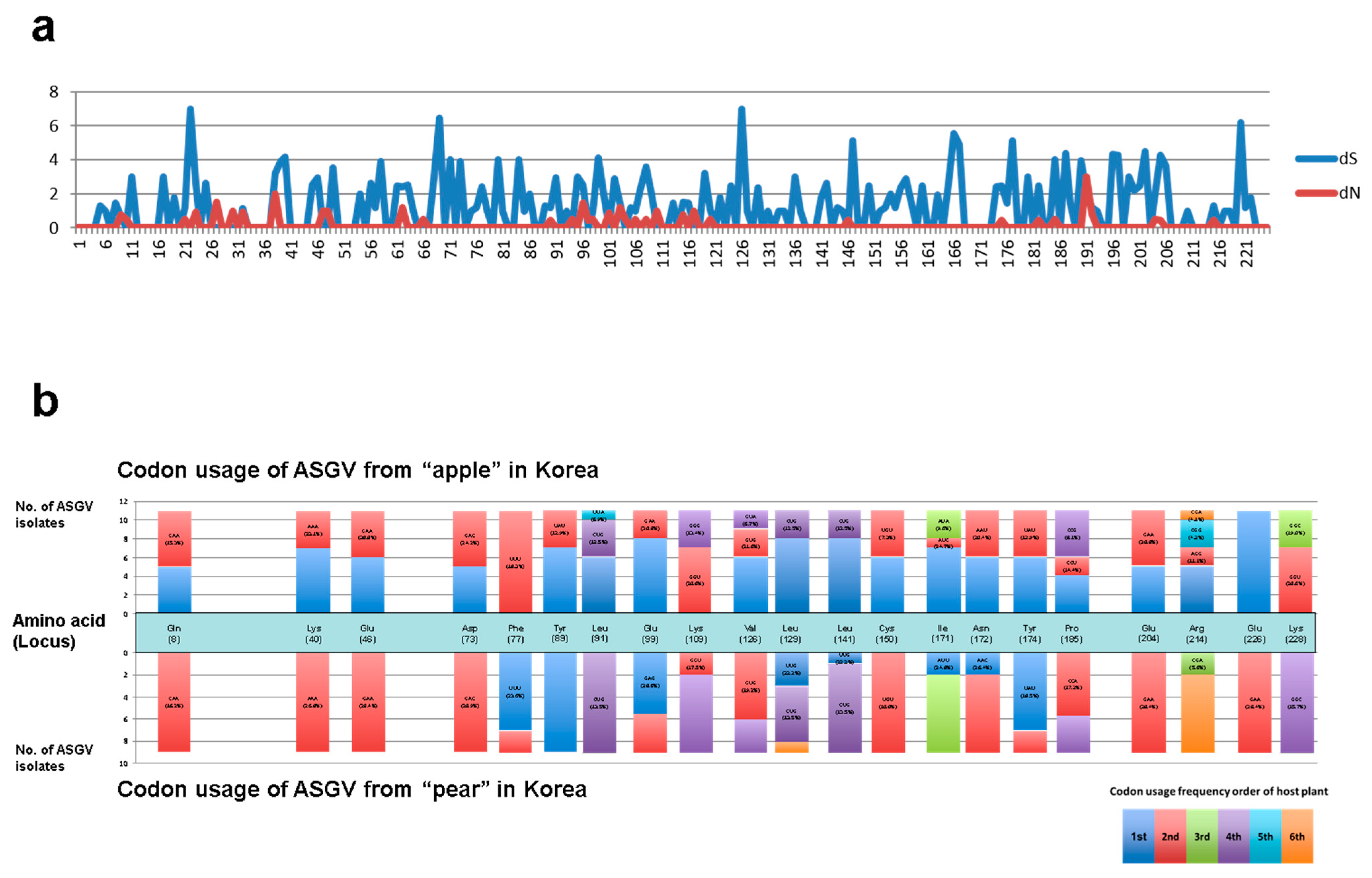
3.3. The CP Gene Corresponds with the Host Codon Usage Frequency
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ohshima, K.; Yamaguchi, Y.; Hirota, R.; Hamamoto, T.; Tomimura, K.; Tan, Z.; Sano, T.; Azuhata, F.; Walsh, J.A.; Fletcher, J.; et al. Molecular evolution of Turnip mosaic virus: Evidence of host adaptation, genetic recombination and geographical spread. J. Gen. Virol. 2002, 83, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Mechanisms of plant virus evolution. Annu. Rev. Phytopathol. 1997, 35, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Akaishi, S.; Kajiyama, H.; Koga, R.; Gibbs, A.J. Evolutionary trajectory of turnip mosaic virus populations adapting to a new host. J. Gen. Virol. 2010, 91, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Wichman, H.A. Different Trajectories of Parallel Evolution during Viral Adaptation. Science 1999, 285, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Novella, I.S.; Zárate, S.; Metzgar, D.; Ebendick-Corpus, B.E. Positive selection of synonymous mutations in vesicular stomatitis virus. J. Mol. Biol. 2004, 342, 1415–1421. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Viboud, C.; Holmes, E.C.; Koonin, E.V.; Lipman, D.J. Long intervals of stasis punctuated by bursts of positive selection in the seasonal evolution of influenza A virus. Biol. Direct. 2006, 1, 34. [Google Scholar] [CrossRef] [Green Version]
- Woods, R.; Schneider, D.; Winkworth, C.L.; Riley, M.A.; Lenski, R.E. Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc. Natl. Acad. Sci. USA 2006, 103, 9107–9112. [Google Scholar] [CrossRef] [Green Version]
- Bazykin, G.A.; Kondrashov, F.A.; Brudno, M.; Poliakov, A.; Dubchak, I.; Kondrashov, A.S. Extensive parallelism in protein evolution. Biol. Direct. 2007, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musset, L.; Le Bras, J.; Clain, J. Parallel Evolution of Adaptive Mutations in Plasmodium falciparum Mitochondrial DNA during Atovaquone-Proguanil Treatment. Mol. Biol. Evol. 2007, 24, 1582–1585. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wang, H.; Huang, L.F.; Naylor, M.; Clifford, P. Heterogeneity in codon usages of sobemovirus genes. Arch. Virol. 2005, 150, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Pinel-Galzi, A.; Rakotomalala, M.; Sangu, E.; Sorho, F.; Kanyeka, Z.; Traoré, O.; Sérémé, D.; Poulicard, N.; Rabenantoandro, Y.; Séré, Y.; et al. Theme and variations in the evolutionary pathways to virulence of an RNA plant virus species. PLoS Pathog. 2007, 3, e180. [Google Scholar] [CrossRef] [Green Version]
- Magome, H.; Yoshikawa, N.; Takahashi, T.; Ito, T.; Miyakawa, T. Molecular variability of the genomes of capilloviruses from apple, Japanese pear, European pear, and citrus trees. Phytopathology 1997, 87, 389–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanese, H.; Mink, G.I.; Sawamura, K.; Yamaguchi, A. Apple topworking disease. In Compendium of Apple and Pear Diseases; Jones, A.L., Aldwinckle, H.S., Eds.; APS Press: St. Paul, MN, USA, 1990; pp. 74–75. [Google Scholar]
- Shim, H.; Min, Y.; Hong, S.; Kwon, M.; Kim, D.; Kim, H.; Choi, Y.; Lee, S.; Yang, J. Nucleotide sequences of a Korean isolate of apple stem grooving virus associated with black necrotic leaf spot disease on pear (Pyrus pyrifolia). Mol. Cells 2004, 18, 192–199. [Google Scholar] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, B.; Leitner, T.; Apetrei, C.; Hahn, B.; Mizrachi, I.; Mullins, J.; Rambaut, A.; Wolinsky, S.; Korber, B. (Eds.) HIV Sequence Compendium 2018; LA-UR 18-25673; Theoretical Biology and Biophysics Group, Los Alamos National Laboratory: Santa Fe, NM, USA, 2018. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Gojobori, T.; Ikemura, T. Codon usage tabulated from international DNA sequence databases: Status for the year 2000. Nucleic Acids Res. 2000, 28, 292. [Google Scholar] [CrossRef] [Green Version]
- Desbiez, C.; Gal-On, A.; Girard, M.; Wipf-Scheibel, C.; Lecoq, H. Increase in Zucchini yellow mosaic virus Symptom Severity in Tolerant Zucchini Cultivars Is Related to a Point Mutation in P3 Protein and Is Associated with a Loss of Relative Fitness on Susceptible Plants. Phytopathology 2003, 93, 1478–1484. [Google Scholar] [CrossRef] [Green Version]
- Simon, A.E.; Bujarski, J.J. RNA-RNA Recombination and Evolution in Virus-Infected Plants. Annu. Rev. Phytopathol. 1994, 32, 337–362. [Google Scholar] [CrossRef]
- Revers, F.; Le Gall, O.; Candresse, T.; Le Romancer, M.; Dunez, J. Frequent occurrence of recombinant potyvirus isolates. J. Gen. Virol. 1996, 77 Pt 8, 1953–1965. [Google Scholar] [CrossRef]
- Ali, M.; Tahir, M.; Hameed, S. Phylogenetic and genome-wide pairwise distance analysis of the genus Luteovirus. Pak. J. Agric. Sci. 2017, 54, 363–371. [Google Scholar] [CrossRef]
- Hajimorad, M.R.; Eggenberger, A.L.; Hill, J.H. Adaptation of Soybean mosaic virus Avirulent Chimeras Containing P3 Sequences from Virulent Strains to Rsv1-Genotype Soybeans Is Mediated by Mutations in HC-Pro. Mol. Plant-Microbe Interact. 2008, 21, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.X.; Rubio, L.; Smythe, A.B.; Falk, B.W. Molecular Population Genetics of Cucumber Mosaic Virus in California: Evidence for Founder Effects and Reassortment. J. Virol. 2004, 78, 6666–6675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, J.; Zhang, H.; Tang, X.; Du, Z. Molecular evidence and sequence analysis of a natural reassortant between Cucumber mosaic virus subgroup IA and II strains. Virus Genes 2007, 35, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Antoniw, J.F.; Bar-Joseph, M.; Brunt, A.A.; Candresse, T.; Foster, G.D.; Martelli, G.P.; Milne, R.G.; Fauquet, C.M. Virology Division News: The new plant virus family Flexiviridae and assessment of molecular criteria for species demarcation. Arch. Virol. 2004, 149, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Martelli, G.P.; Adams, M.J.; Kreuze, J.F.; Dolja, V.V. Family Flexiviridae: A Case Study in Virion and Genome Plasticity. Annu. Rev. Phytopathol. 2007, 45, 73–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moury, B.; Cardin, L.; Onesto, J.P.; Candresse, T.; Poupet, A. Survey of Prunus necrotic ringspot virus in Rose and Its Variability in Rose and Prunus spp. Phytopathology 2001, 91, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sentandreu, V.; Castro, J.A.; Ayllón, M.A.; Rubio, L.; Guerri, J.; González-Candelas, F.; Moreno, P.; Moya, A. Evolutionary analysis of genetic variation observed in citrus tristeza virus (CTV) after host passage. Arch. Virol. 2005, 151, 875–894. [Google Scholar] [CrossRef]
- Rico, P.; Ivars, P.; Elena, S.F.; Hernández, C. Insights into the selective pressures restricting Pelargonium flower break virus genome variability: Evidence for host adaptation. J. Virol. 2006, 80, 8124–8132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moury, B. A new lineage sheds light on the evolutionary history of Potato virus Y. Mol. Plant Pathol. 2010, 11, 161–168. [Google Scholar] [CrossRef]
- Antoniw, J.F.; Adams, M.J. Codon usage bias amongst plant viruses. Arch. Virol. 2003, 149, 113–135. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Pagel, M.; Gould, E.A.; de A Zanotto, P.M.; Holmes, E.C. Evolution of base composition and codon usage bias in the genus Flavivirus. J. Mol. Evol. 2001, 52, 383–390. [Google Scholar] [CrossRef] [PubMed]
- van Hemert, F.J.; Berkhout, B.; Lukashov, V.V. Host-related nucleotide composition and codon usage as driving forces in the recent evolution of the Astroviridae. Virology 2007, 361, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahir, I.; Fromer, M.; Prat, Y.; Linial, M. Viral adaptation to host: A proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 2009, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Lal, A.; Kil, E.-J.; Kwak, H.-R.; Yoon, H.-S.; Choi, H.-S.; Kim, M.; Ali, M.; Lee, S. Adaptation and Codon-Usage Preference of Apple and Pear-Infecting Apple Stem Grooving Viruses. Microorganisms 2021, 9, 1111. https://doi.org/10.3390/microorganisms9061111
Kim J, Lal A, Kil E-J, Kwak H-R, Yoon H-S, Choi H-S, Kim M, Ali M, Lee S. Adaptation and Codon-Usage Preference of Apple and Pear-Infecting Apple Stem Grooving Viruses. Microorganisms. 2021; 9(6):1111. https://doi.org/10.3390/microorganisms9061111
Chicago/Turabian StyleKim, Jaedeok, Aamir Lal, Eui-Joon Kil, Hae-Ryun Kwak, Hwan-Su Yoon, Hong-Soo Choi, Mikyeong Kim, Muhammad Ali, and Sukchan Lee. 2021. "Adaptation and Codon-Usage Preference of Apple and Pear-Infecting Apple Stem Grooving Viruses" Microorganisms 9, no. 6: 1111. https://doi.org/10.3390/microorganisms9061111