
Three New Isoprenylated Flavonoids from the Root Bark of Morus alba

 ,
,  ,
,
Abstract
:1. Introduction
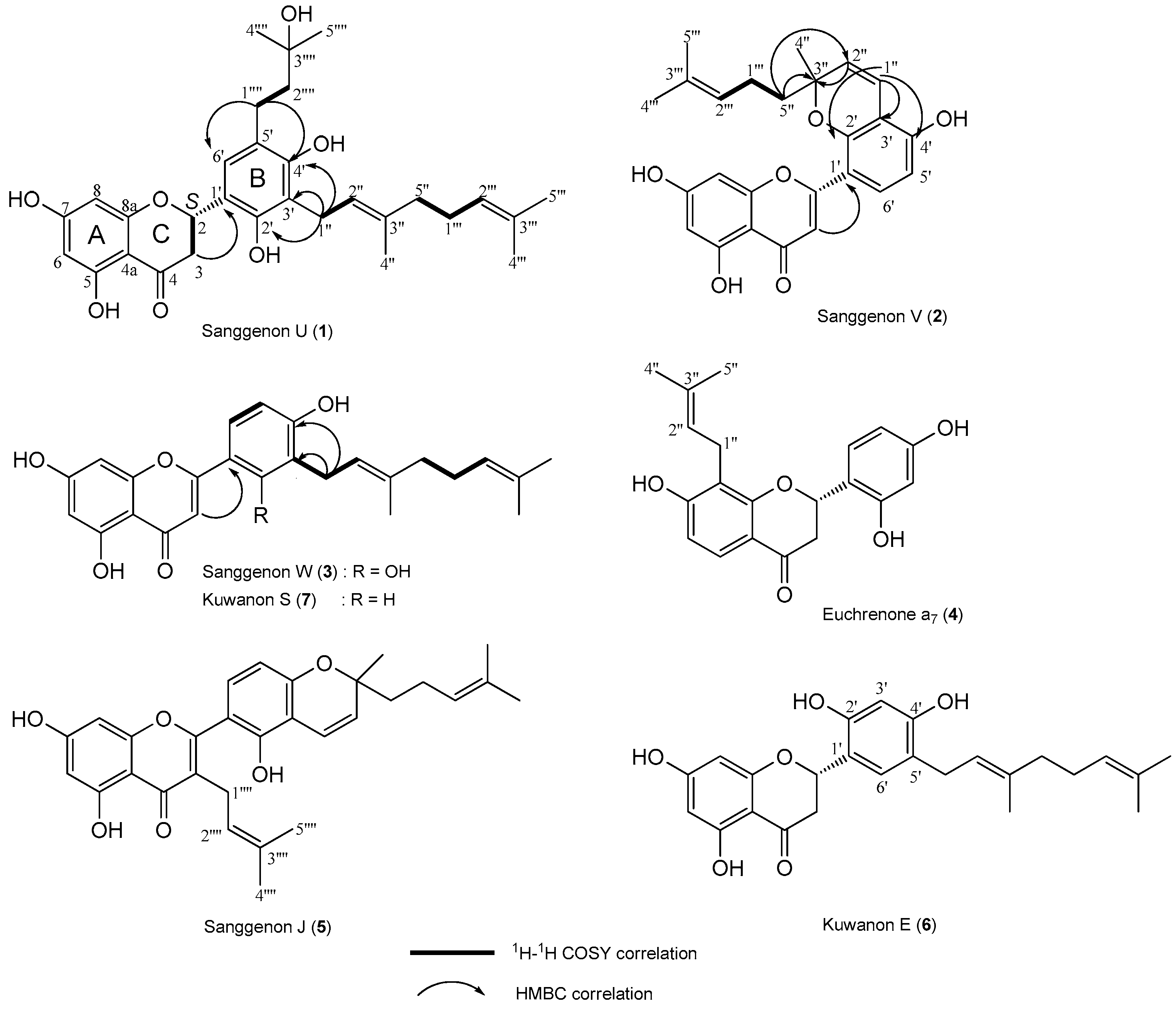
2. Results and Discussion
3. Experimental Section
3.1. Plant Materials
3.2. General
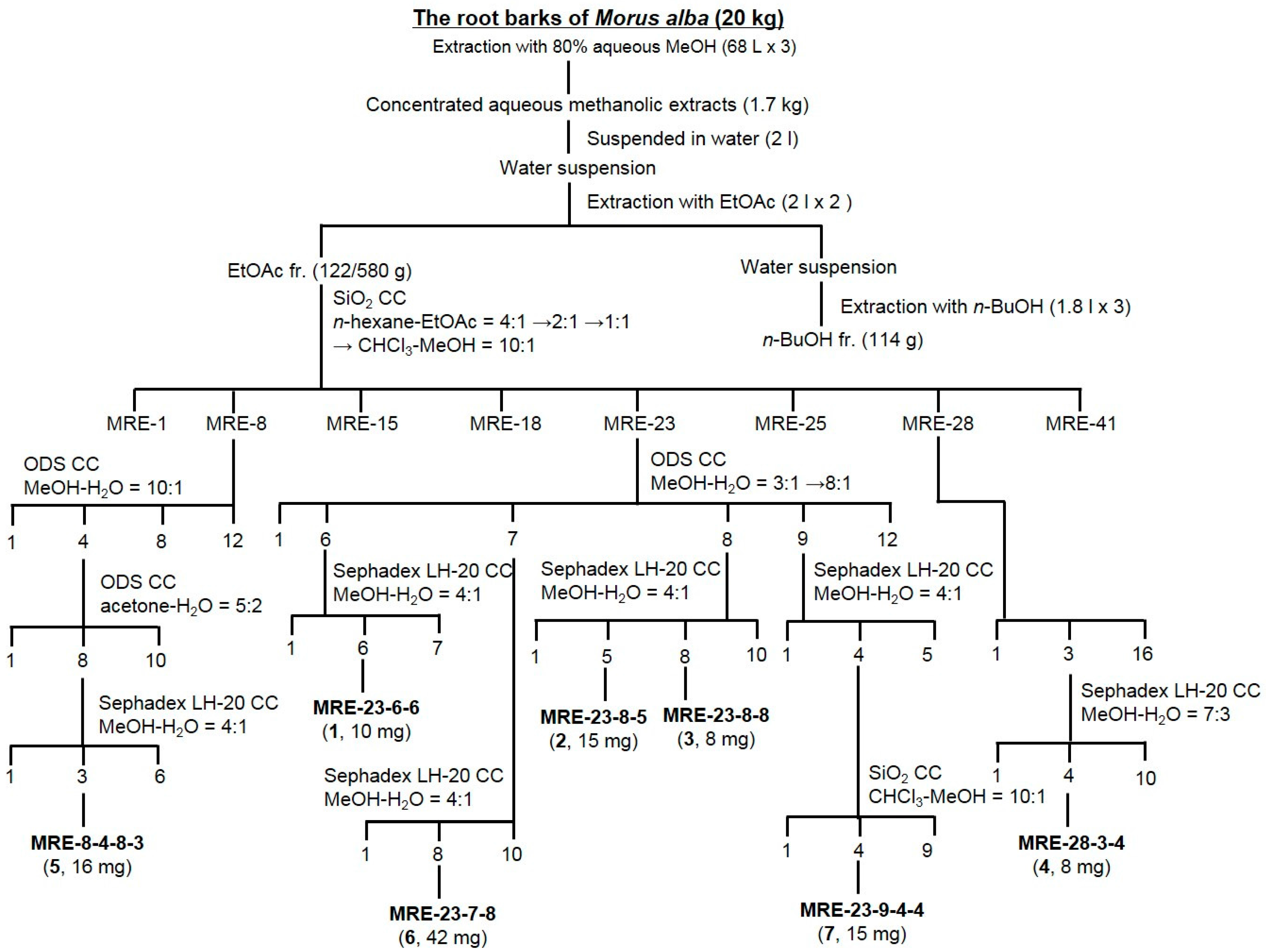
3.3. Extraction and Isolation
3.4. Spectroscopic Data
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barron, D.; Ibrahim, R.K. Isoprenylated flavonoids—A survey. Phytochemistry 1996, 43, 921–982. [Google Scholar] [CrossRef]
- Huang, X.; Zhu, D.; Lou, Y. A novel anticancer agent, icaritin, induced cell growth inhibition, G1 arrest and mitochondrial transmembrane potential drop in human prostate carcinoma PC-3 cells. Eur. J. Pharmacol. 2007, 564, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Lee, D.H.; Jung, Y.J.; Shin, S.Y.; Lee, Y.H. The natural flavone eupatorin induces cell cycle arrest at the G2/M phase and apoptosis in HeLa cells. Appl. Biol. Chem. 2016, 59, 193–199. [Google Scholar] [CrossRef]
- Han, A.R.; Kang, Y.J.; Windono, T.; Lee, S.K.; Seo, E.K. Prenylated flavonoids from the heartwood of Artocarpus communis with inhibitory activity on lipopolysaccharide-induced nitric oxide production. J. Nat. Prod. 2006, 69, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Maitrejean, M.; Comte, G.; Barron, D.; El Kirat, K.; Conseil, G.; Di Pietro, A. The flavanolignan silybin and its hemisynthetic derivatives, a novel series of potential modulators of P-glycoprotein. Bioorg. Med. Chem. Lett. 2000, 10, 157–160. [Google Scholar] [CrossRef]
- Jung, J.W.; Park, J.H.; Seo, K.H.; Oh, E.J.; Baek, N.I.; Ko, W.M.; Kim, Y.C.; Lee, D.Y.; Lee, D.S.; Lim, D.W. Isoprenylated flavonoids from the root bark of Morus alba and their hepatoprotective and neuroprotective activities. Arch. Pharm. Res. 2015, 38, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Elangovan, V. Nutritive contents of different varieties of mulberry leaves. J. Sci. Nat. 2011, 2, 254–258. [Google Scholar]
- Zhu, Y.P. Chinese Materia Medica; Harwood Academic Publisher: Amsterdam, The Netherlands, 1998; pp. 505–507. [Google Scholar]
- Hano, Y.; Suzuki, S.; Nomura, T.; Litakam, Y. Absolute configuration of natural diels-alder type adducts from the Morus root bark. Heterocycles 1988, 27, 2315–2325. [Google Scholar]
- Jiang, D.; He, Z.D.; Jiang, R.W.; Ye, W.C.; Xu, H.X.; But, P. Antiviral flavonoids from the root bark of Morus alba L. Phytochemistry 2003, 62, 1235–1238. [Google Scholar]
- Jung, J.W.; Park, J.H.; Jung, Y.J.; Lee, C.H.; Han, D.S.; Baek, N.I. Isolation and identification of triterpenoids from the Mulberry (Morus alba) root bark. J. Appl. Biol. Chem. 2014, 57, 295–299. [Google Scholar]
- Lee, H.J.; Lyu, D.H.; Nam, K.W.; Hong, S.S.; Kim, K.O.; Kim, K.H. Protection of prenylated flavonoids from Moricortex radicis (Moraceae) against nitric oxide-induced cell death in neuroblastoma SH-SY5Y cells. Arch. Pharm. Res. 2012, 35, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.J.; Qiu, F.; Chen, L.X.; Pan, Y.; Dou, D.Q. New stilbene, benzofuran, and coumarin glycosides from Morus alba. Helv. Chim. Acta 2009, 92, 579–587. [Google Scholar] [CrossRef]
- Dat, N.T.; Binh, P.T.X.; Quynh, L.T.P.; Minh, C.V.; Huong, H.T.; Lee, J.J. Cytotoxic prenylated flavonoids from Morus alba. Fitoterapia 2010, 81, 1224–1227. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.A.; Ma, Y.B.; Zhang, X.M.; Yao, S.Y.; Xue, D.Q.; Zhang, R.P.; Chen, J.J. Mulberrofuran G and isomulberrofuran G from Morus alba L.: Anti-hepatitis B virus activity and mass spectrometric fragmentation. J. Agric. Food Chem. 2012, 60, 8197–8202. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.; Harmalkar, D.S.; Xuezhen, X.; Jang, K.; Lee, K. Bioactive benzofuran derivatives: Moracins A–Z in medicinal chemistry. Eur. J. Med. Chem. 2015, 90, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.P.; Tan, H.Y.; Wang, M.F. Tyrosinase inhibition constituents from the roots of Morus australis. Fitoterapia 2012, 83, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.W.; Kim, Y.T.; Park, J.H.; Baek, N.I.; Han, D. Antidepressant-like effects of the ethyl acetate soluble fraction of the root bark of Morus alba on the immobility behavior of rats in the forced swim test. Molecules 2014, 19, 7981–7989. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-W.; Park, J.-H.; Seo, K.-H.; Oh, E.-J.; Lee, D.-Y.; Lim, D.-W.; Han, D.; Song, M.-C.; Baek, N.-I. New hydroxy fatty acid from the root bark of Morus alba L. J. Korean Soc. Appl. Biol. Chem. 2015, 58, 541–543. [Google Scholar] [CrossRef]
- Fukai, T.; Hano, Y.; Hirakura, K.; Nomura, T.; Uzawa, J. Structures of a novel 2-arylbenzofuran derivative and two flavones derivatives from the cultivated mulberry tree (Morus lhou Koidz.). Chem. Pharm. Bull. 1985, 33, 4288–4295. [Google Scholar] [CrossRef]
- Hano, Y.; Nomura, T. Constituents of the Chinese crude drug “Sang-Bai-Pi” (Morus root barks). IV. Structure of Four new flavanoids, sanggenons H, I, J, and K. Heterocycles 1983, 20, 1071–1076. [Google Scholar]
- Jeong, S.H.; Ryu, Y.B.; Curtis-Long, M.J.; Ryu, H.W.; Baek, Y.S.; Kang, J.E.; Lee, W.S.; Park, K.H. Tyrosinase inhibitory polyphenols from roots of Morus lhou. J. Agric. Food Chem. 2009, 57, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Tanaka, T.; Matsuura, N.; Linuma, M.; Cheih, C. Two flavanones from Euchresta horsfieldii. Phytochemistry 1990, 29, 2738–2740. [Google Scholar] [CrossRef]
- Slade, D.; Ferreira, D.; Marais, P.J. Circular dichroism, a powerful tool for the assessment of absolute configuration of flavonoids. Phytochemistry 2005, 66, 2177–2215. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1–7 are available from the authors.



{kind=link}
{kind=link}
| Compound 1 | Compound 2 | Compound 3 | ||||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 2 | 5.64 (dd, J = 12.8, 2.8 Hz) | 76.72 | 163.49 | 165.74 | ||
| 3 | 3.08 (dd, J = 17.2, 12.8 Hz) | 43.35 | 7.04 (s) | 108.59 | 6.86 (s) | 108.38 |
| 2.69 (dd, J = 17.2, 2.8 Hz) | ||||||
| 4 | 198.25 | 184.20 | 184.16 | |||
| 4a | 103.23 | 105.12 | 105.13 | |||
| 5 | 165.22 | 163.06 | 163.09 | |||
| 6 | 5.90 (d, J = 2.0 Hz) | 96.40 | 6.17 (s) | 99.92 | 6.17 (s) | 100.04 |
| 7 | 168.85 | 166.00 | 166.27 | |||
| 8 | 5.86 (d, J = 2.0 Hz) | 97.18 | 6.38 (s) | 94.86 | 6.39 (s) | 95.08 |
| 8a | 165.52 | 159.42 | 159.67 | |||
| 1′ | 119.62 | 111.53 | 112.62 | |||
| 2′ | 151.77 | 155.25 | 160.87 | |||
| 3′ | 118.74 | 110.84 | 118.10 | |||
| 4′ | 154.46 | 158.05 | 156.70 | |||
| 5′ | 123.40 | 6.49 (d, J = 8.4 Hz) | 109.41 | 6.50 (d, J = 8.4 Hz) | 109.20 | |
| 6′ | 7.03 (s) | 125.93 | 7.62 (d, J = 8.4 Hz) | 129.80 | 7.46 (d, J = 8.4 Hz) | 128.25 |
| 1′′ | 3.41 (d, J = 6.8 Hz) | 23.77 | 6.72 (d, J = 10.0 Hz) | 118.19 | 3.40 (d, J = 6.8 Hz) | 23.05 |
| 2′′ | 5.18 (t, J = 6.8 Hz) | 123.99 | 5.63 (d, J = 10.0 Hz) | 128.46 | 5.20 (t, J = 6.8 Hz) | 123.49 |
| 3′′ | 136.44 | 81.37 | 136.45 | |||
| 4′′ | 1.78 (s) | 16.36 | 1.47 (s) | 26.89 | 1.78 (s) | 16.36 |
| 5′′ | 1.98 (d, J = 6.8 Hz) | 40.90 | 1.79 (m) | 42.17 | 1.97 (t, J = 6.8 Hz) | 40.91 |
| 1.69 (m) | ||||||
| 1′′′ | 2.06 (dt, J = 6.8, 6.8 Hz) | 27.63 | 2.08 (m) | 23.98 | 2.05 (dt, J = 6.8, 6.8 Hz) | 27.67 |
| 2′′′ | 5.06 (t, J = 6.8 Hz ) | 125.37 | 5.08 (t, J = 6.8 Hz) | 125.04 | 5.05 (t, J = 6.8 Hz) | 125.38 |
| 3′′′ | 132.24 | 132.64 | 132.19 | |||
| 4′′′ | 1.62 (s) | 25.89 | 1.58 (s) | 25.79 | 1.59 (s) | 25.85 |
| 5′′′ | 1.56 (s) | 17.74 | 1.47 (s) | 17.60 | 1.54 (s) | 17.72 |
| 1′′′′ | 2.63 (m) | 25.97 | ||||
| 2′′′′ | 1.71 (m) | 45.05 | ||||
| 3′′′′ | 71.58 | |||||
| 4′′′′ | 1.24 (s) | 29.31 | ||||
| 5′′′′ | 1.24 (s) | 29.31 | ||||
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, J.-W.; Park, J.-H.; Lee, Y.-G.; Seo, K.-H.; Oh, E.-J.; Lee, D.-Y.; Lim, D.-W.; Han, D.; Baek, N.-I. Three New Isoprenylated Flavonoids from the Root Bark of Morus alba. Molecules 2016, 21, 1112. https://doi.org/10.3390/molecules21091112
Jung J-W, Park J-H, Lee Y-G, Seo K-H, Oh E-J, Lee D-Y, Lim D-W, Han D, Baek N-I. Three New Isoprenylated Flavonoids from the Root Bark of Morus alba. Molecules. 2016; 21(9):1112. https://doi.org/10.3390/molecules21091112
Chicago/Turabian StyleJung, Jae-Woo, Ji-Hae Park, Yeong-Geun Lee, Kyeong-Hwa Seo, Eun-Ji Oh, Dae-Young Lee, Dong-Wook Lim, Daeseok Han, and Nam-In Baek. 2016. "Three New Isoprenylated Flavonoids from the Root Bark of Morus alba" Molecules 21, no. 9: 1112. https://doi.org/10.3390/molecules21091112