
Toward of Safer Phenylbutazone Derivatives by Exploration of Toxicity Mechanism

 , , and
, , and 
Abstract
:1. Introduction
2. Results and Discussion
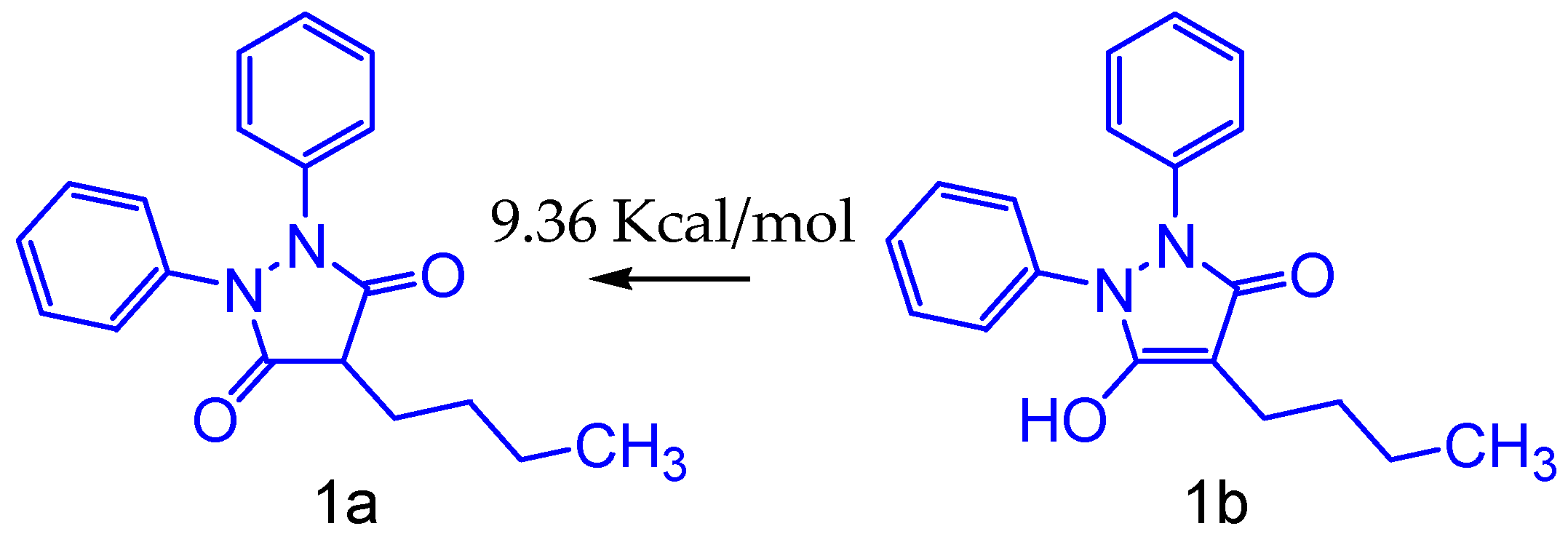
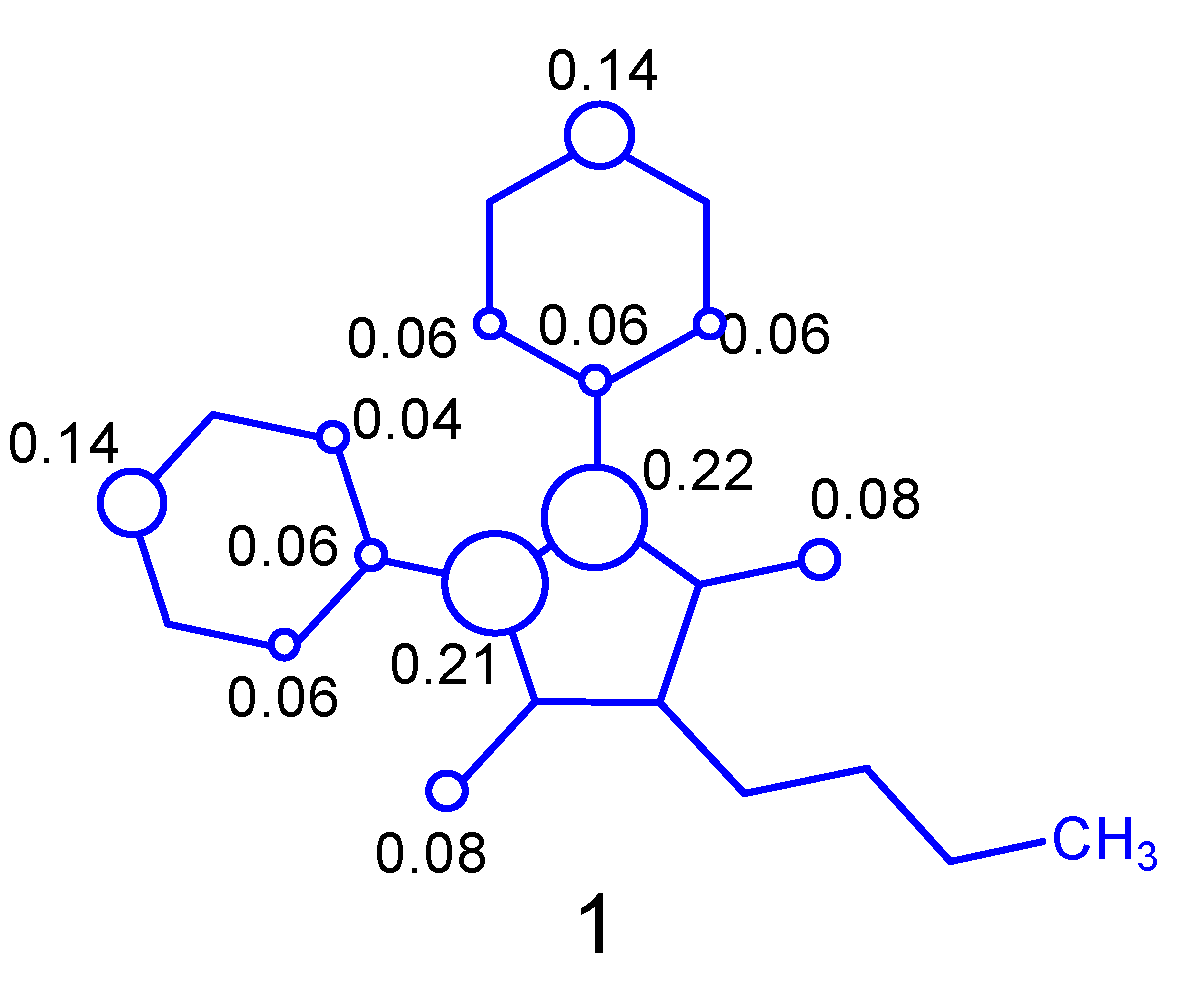
2.1. Tautomerism Study
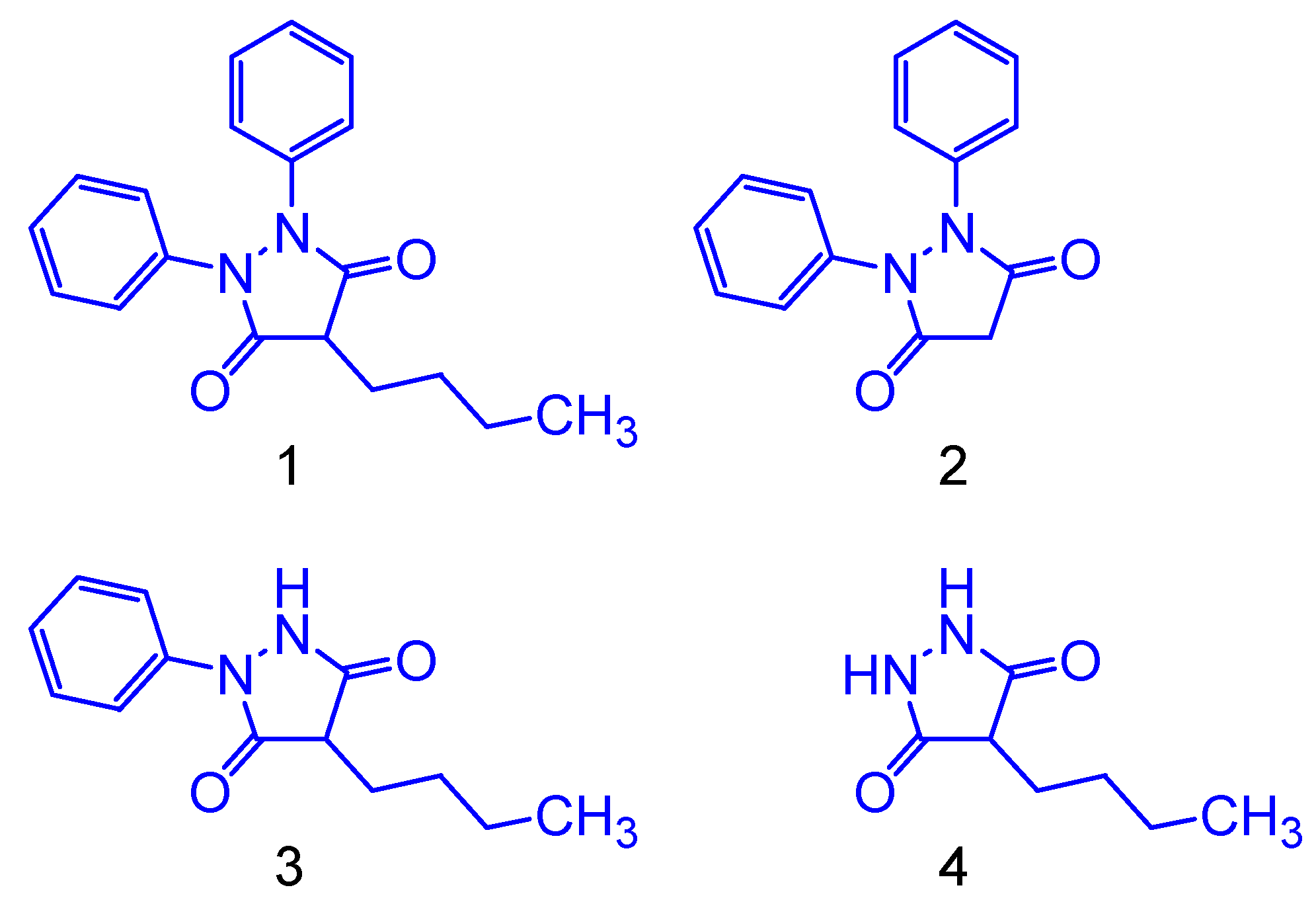
2.2. Molecular Simplification
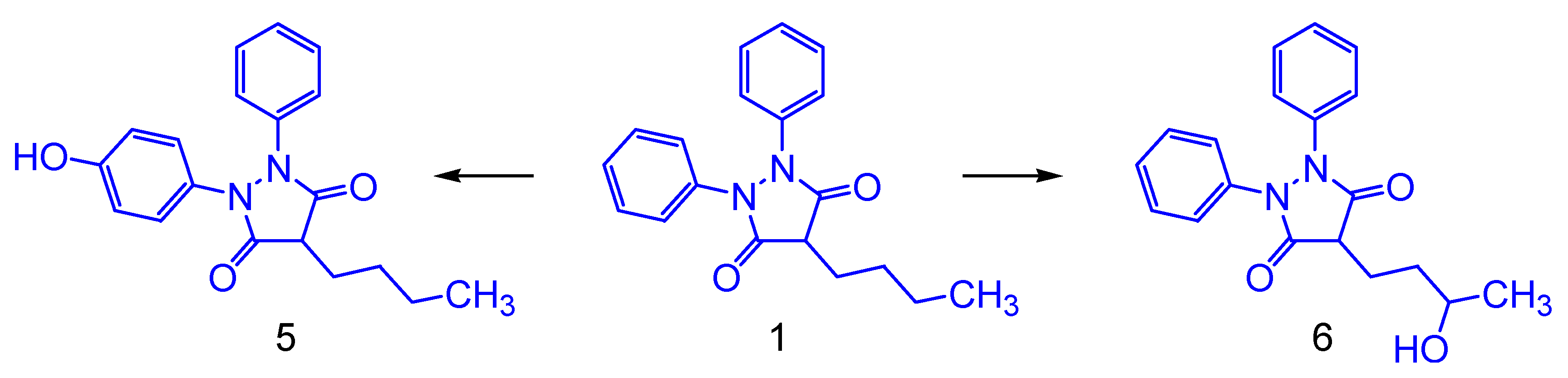
2.3. Theoretical Metabolism
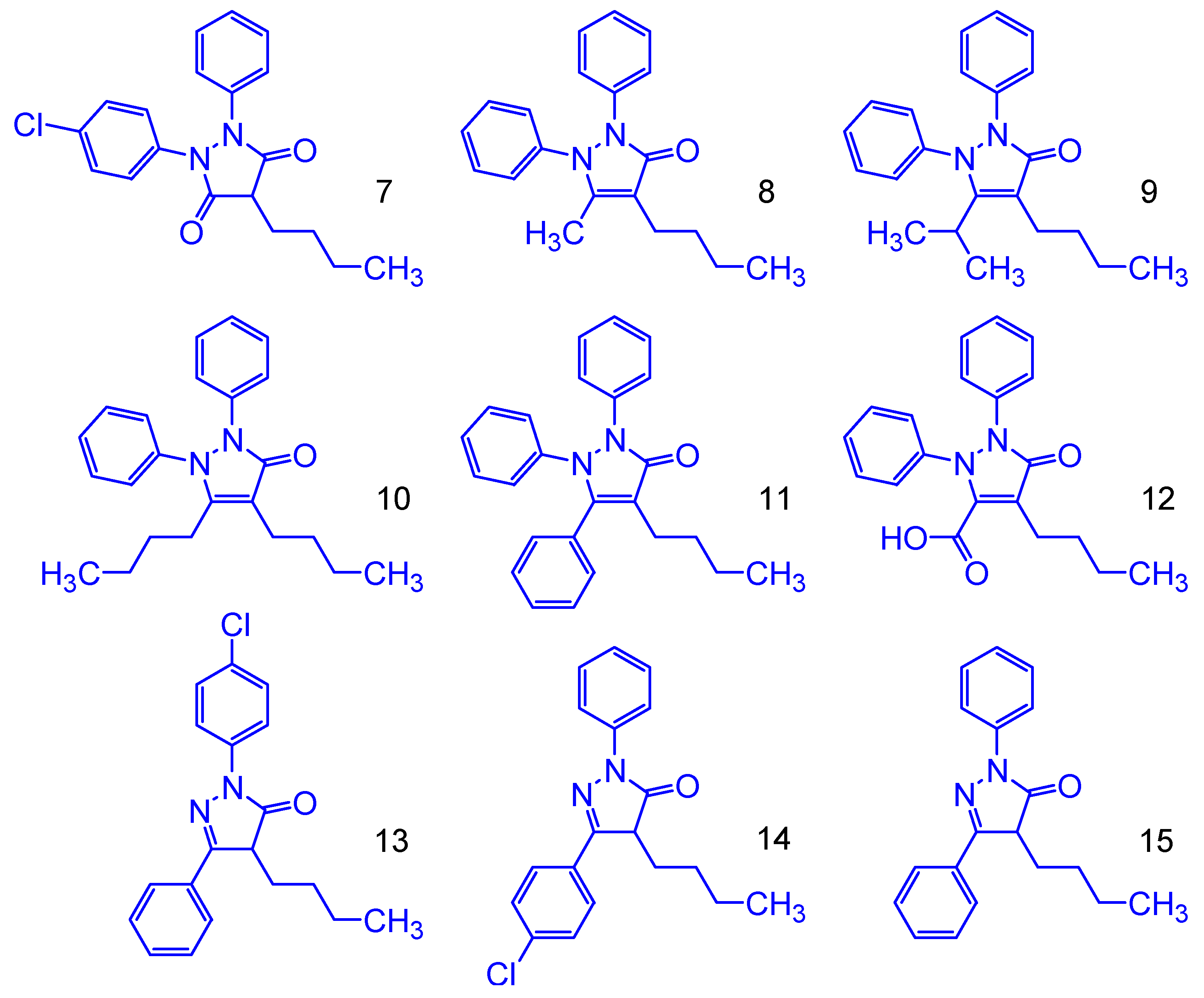
2.4. Proposed Phenylbutazone Derivatives
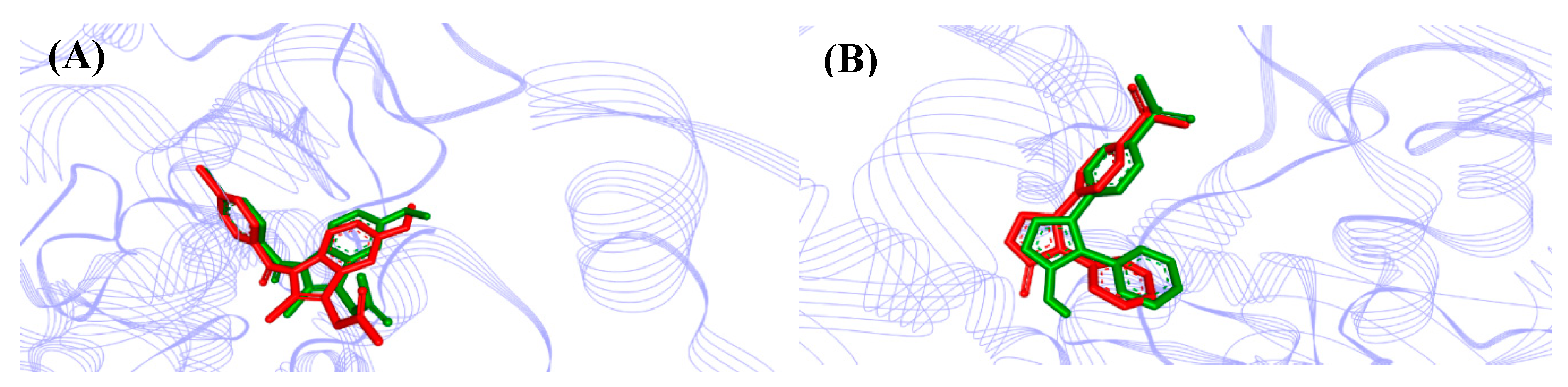
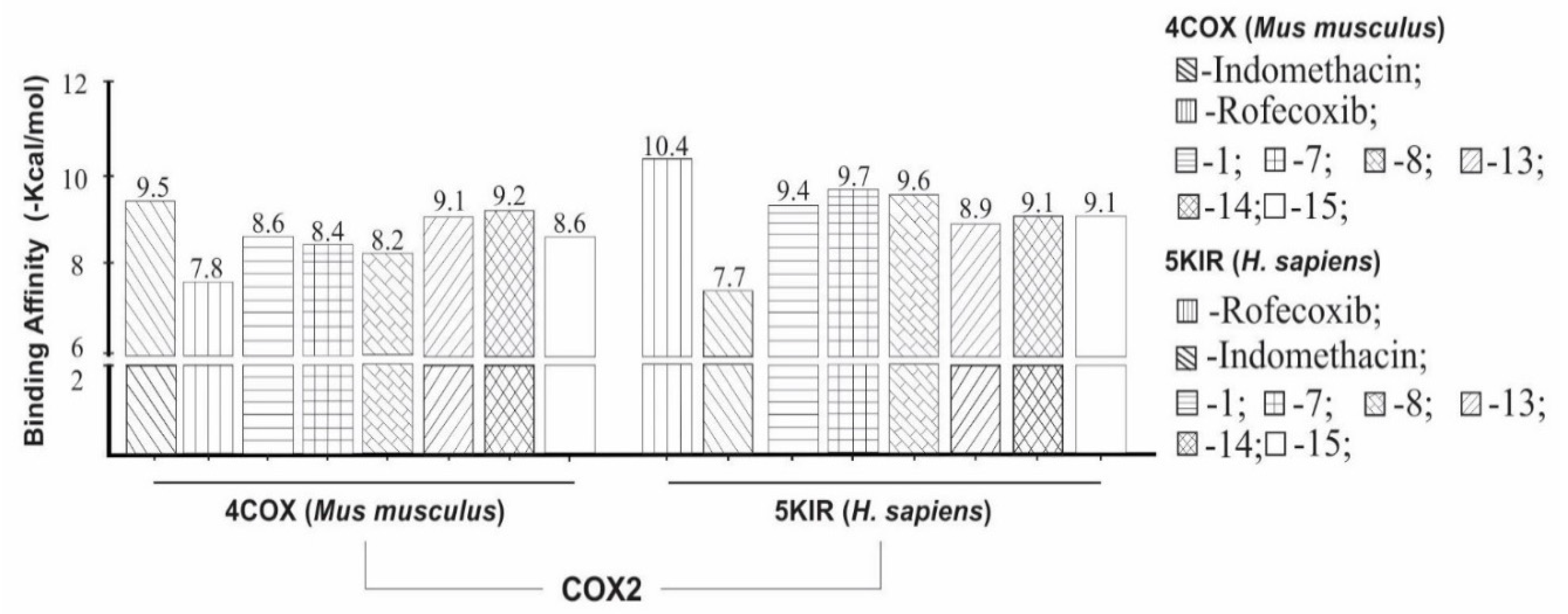
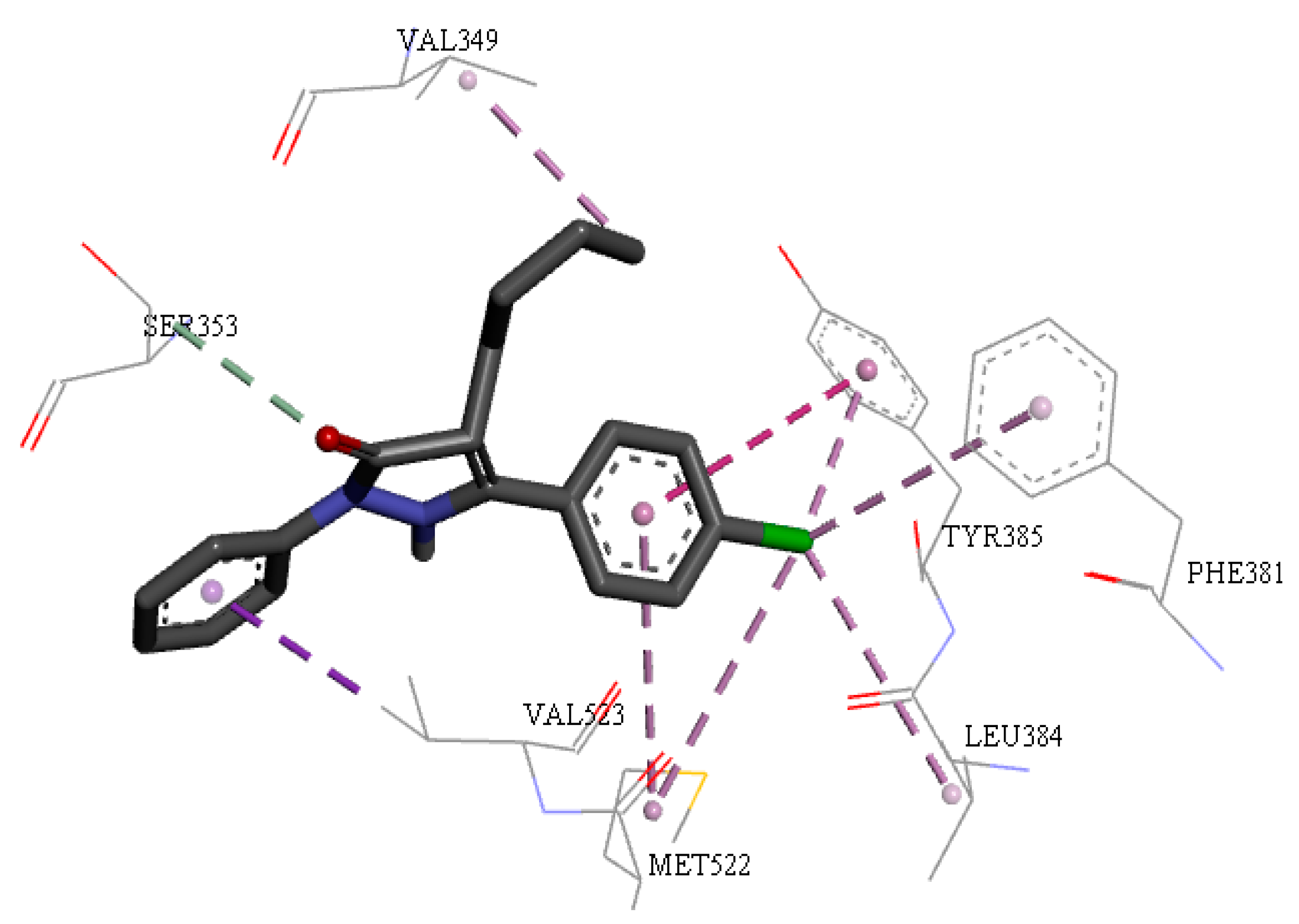
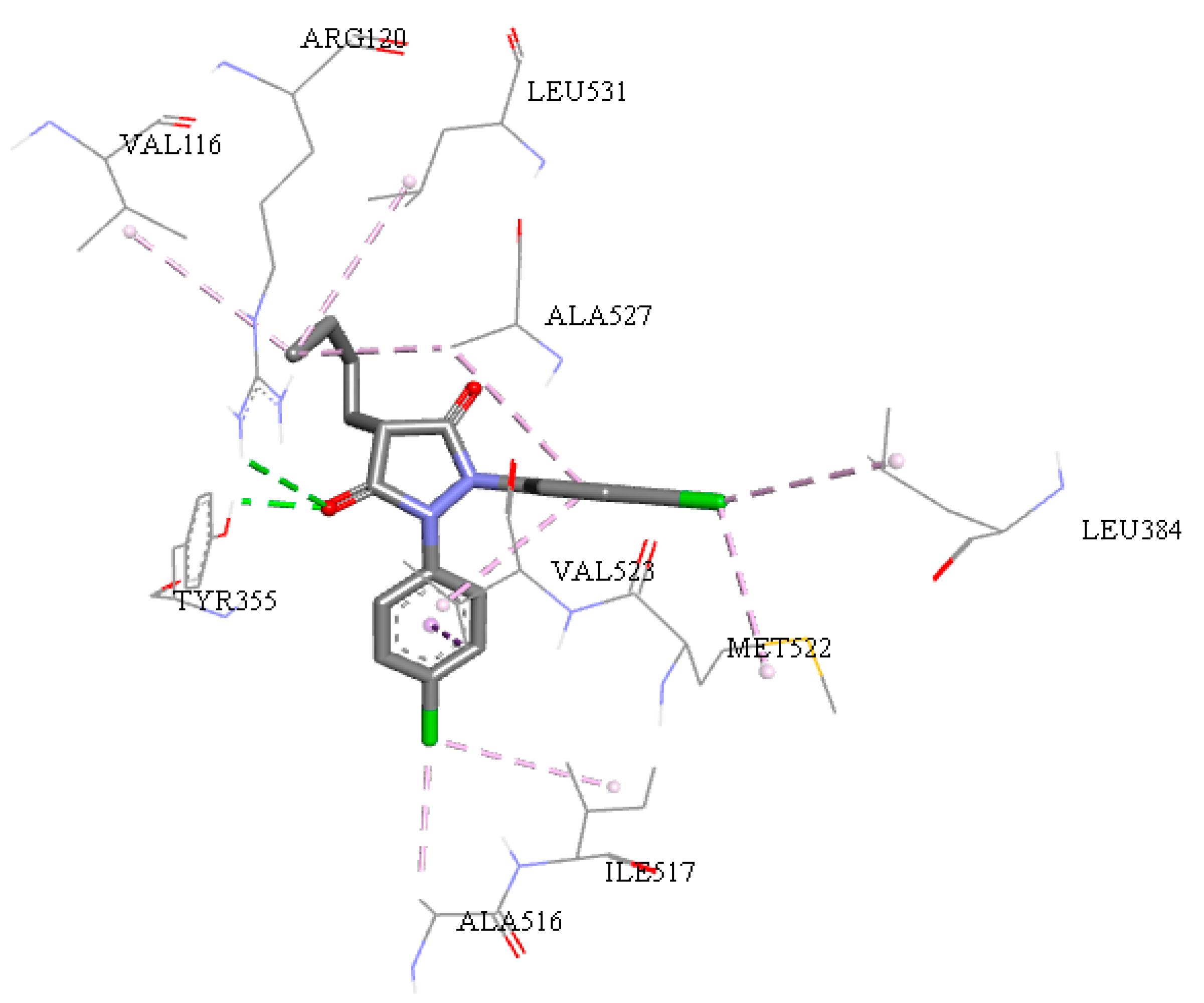
2.5. Molecular Docking Study
2.6. Toxicological Properties
3. Materials and Methods
3.1. Theoretical
3.2. Molecular Docking Simulations Study
3.3. Toxicological Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lees, P.; Toutain, P. Pharmacokinetics, pharmacodynamics, metabolism, toxicology and residues of phenylbutazone in humans and horses. Vet. J. 2013, 196, 294–303. [Google Scholar] [CrossRef] [PubMed]
- MacAllister, C.G.; Morgan, S.J.; Borne, A.T.; Pollet, R.A.J. Comparison of adverse effects of phenylbutazone, flunixin meglumine. and ketoprofen in horses. J. Am. Vet. Med. Assoc. 1993, 202, 71–77. [Google Scholar] [PubMed]
- Lees, P.; Higgins, A.J. Clinical pharmacology and therapeutic uses of non-steroidal anti-inflammatory drugs in the horse. Equine Vet. J. 1985, 17, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Mathews, K.A. Non-steroidal anti-inflammatory analgesics: A review of current practice. J. Vet. Emerg. Cris. Care 2002, 12, 89–97. [Google Scholar] [CrossRef]
- Fitzpatrick, J.L.; Nolan, A.M.; Lees, P.; May, S.A. Inflammation and pain. In Bovine Medicine Diseases Husandry Cattle, 2nd ed.; Andrews, A.H., Blowey, R.W., Boyd, H., Eddy, R.G., Eds.; Blackwell Science Ltd.: Oxford, UK, 2004; pp. 1050–1056. ISBN 0-632-05596-0. [Google Scholar]
- Collins, L.G.; Tyler, D.E. Phenylbutazone toxicosis in the horse: A clinical study. J. Am. Vet. Med. Assoc. 1984, 184, 699–703. [Google Scholar] [PubMed]
- Triggs, E.J.; Whyatt, P.L.; Eckert, G. Bioavailability evaluation of an enteric-coated phenylbutazone formulation. and cross-over comparison with a sugar-coated product. Med. J. Aust. 1977, 2, 830–833. [Google Scholar] [PubMed]
- Silva, B.H.S.; Barros, T.G.; Barros, C.A.L.; Vieira, J.L.F.; Borges, R.S. An electronic study of biphenyl derivatives chlorinated. J. Comp. Theor. Nanosc. 2011, 8, 216–219. [Google Scholar] [CrossRef]
- Levy, G.N. Prostaglandin H synthases. nonsteroidal anti-inflammatory drugs and colon cancer. FASEB J. 1997, 11, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Kidd, B.L.; Urban, L.A. Mechanisms of inflammatory pain. Br. J. Anaesth. 2001, 87, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiroz, A.N.; Mendes, A.P.S.; Leal, M.R.S.; Chaves Neto, A.M.J.; Borges, R.S. Tautomerism and radical-scavenging activity of edaravone by DFT methods. J. Comp. Threoret. Nanosc. 2010, 7, 153–156. [Google Scholar] [CrossRef]
- Antonczak, S. Electronic description of four flavonoids revisited by DFT method. J. Mol. Struct. TheoChem. 2008, 856, 38–45. [Google Scholar] [CrossRef]
- Queiroz, A.N.; Gomes, B.A.Q.; Moraes, W.M.J.; Borges, R.S. A theoretical antioxidant pharmacophore for resveratrol. Eur. J. Med. Chem. 2009, 44, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.M. Toxic responses of the gill. In Target Organ Toxicity in Marine and Freshwater Teleosts, 2nd ed.; Schlenk, D., Benson, W.H., Eds.; CRC Press: London, UK, 2001; pp. 1–26. ISBN 0-415-24838-8. [Google Scholar]
- Borges, R.S.; Queiroz, A.N.; Mendes, A.P.S.; Araújo, S.C.; França, L.C.S.; Franco, E.C.S.; Gomes-Leal, W.; Silva, A.B.F. Density functional theory (DFT) study of edaravone derivatives as antioxidants. Int. J. Mol. Sci. 2012, 13, 7594–7606. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, E.S.; Santa-Brígida, S.A.; Queiroz, A.N.; Silva, J.R.; Silva, O.P.P.; Barros, C.A.L.; Borges, R.S. Edaravone toxicity can be related to redox properties of their oxidized derivatives. Chem. Data Collect. 2017, 7–8, 51–57. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Gowtham, U.; Jayakanthan, M.; Sundar, D. Molecular docking studies of dithionitrobenzoic acid and its related compounds to protein disulfide isomerase: Computational screening of inhibitors to HIV-1 entry. BMC Bioinform. 2008, 9, 1–10. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inform. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; Mcdonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Iyashiro, J.M.; Penning, T.D.; Seibert, K.; et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef]
- Orlando, B.J.; Malkowski, M.G. Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallogr. 2016, 72, 772–776. [Google Scholar] [CrossRef]
- Beretta, C.; Garavaglia, G.; Cavalli, M. COX-1 and COX-2 inhibition in horse blood by phenylbutazone. flunixin. carprofen and meloxicam: An in vitro analysis. Pharm. Res. 2005, 52, 302–306. [Google Scholar] [CrossRef]
- Costa, J.S.; Costa, K.S.L.; Cruzb, J.V.; Ramos, R.S.; Silva, L.B.; Brasil, D.S.B.; Tomich, C.S.; Rodrigues, C.B.; Macêdo, W.J.C. Virtual screening and statistical analysis in the design of new caffeine analogues molecules with potential epithelial anticancer activity. Curr. Pharm. Des. 2018, 24, 576–594. [Google Scholar] [CrossRef]
- Cruz, J.V.; Neto, M.F.A.; Silva, L.B.; Ramos, R.; Costa, J.; Brasil, D.S.B.; Lobato, C.C.; Costa, G.V.; Bittencourt, J.A.H.M.; Silva, C.H.T.P.; et al. Identification of novel protein kinase receptor type 2 inhibitors using pharmacophore and structure-based virtual screening. Molecules 2018, 23, 453. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.B.R.; Ramos, R.S.; Ortiza, B.L.S.; Silva, G.M.; Giuliatti, S.; Navarrete, J.L.A.; Carvalho, J.C.T. Oil from the fruits of Pterodon emarginatus Vog.: A traditional anti-inflammatory. Study combining in vivo and in silico. J. Ethnopharmacol. 2018, 222, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Teles Fujishima, M.A.; Silva, N.S.R.; Ramos, R.S.; Batista Ferreira, E.F.; Santos, K.L.B.; Silva, C.H.T.P.; Silva, J.O.; Campos Rosa, J.M.; Santos, C.B.R. An antioxidant potential, quantum-chemical and molecular docking study of the major chemical constituents present in the leaves of Curatella americana Linn. Pharmaceuticals 2018, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.V.; Serafim, R.B.; Silva, G.M.; Giuliatti, S.; Rosa, J.M.C.; Araújo Neto, M.F.; Leite, F.H.A.; Taft, C.A.; Silva, C.H.T.P.; Santos, C.B.R. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening and molecular dynamics. J. Mol. Model. 2018, 24, 225. [Google Scholar] [CrossRef] [PubMed]
- Oxenoid, K.; Dong, Y.S.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B.; et al. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, J.; Park, D.; Fu, Q.; Chen, J.; Ha, H.J.; Ghantous, F.; Herrmann, T.; Chang, W.; Liu, Z.; Frey, G.; et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science 2016, 353, 172–175. [Google Scholar] [CrossRef] [Green Version]
- OuYang, B.; Xie, S.; Berardi, M.J.; Zhao, X.M.; Dev, J.; Yu, W.; Sun, B.; Chou, J.J. Unusual architecture of the p7 channel from hepatitis C virus. Nature 2013, 498, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Fu, T.M.; Cruz, A.C.; Sengupta, P.; Thomas, S.K.; Wang, S.; Siegel, R.M.; Wu, H.; Chou, J.J. Structural basis and functional role of intramembrane trimerization of the Fas/CD95 death receptor. Mol. Cell 2016, 61, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Li, X.B.; Wang, S.Q.; Xu, W.R.; Wang, R.L. Novel Inhibitor Design for hemagglutinin against H1N1 influenza virus by core hopping method. PLoS ONE 2011, 6, e28111. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, S.Q.; Xu, W.R.; Wang, R.L. Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS ONE 2012, 7, e38546. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Review: Structural bioinformatics and its impact to biomedical science. Curr. Med. Chem. 2004, 11, 2105–2134. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, H.J. Hepatotoxicity: The Adverse Effects of Drugs and Other Chemicals on the Liver, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1999; p. 789. [Google Scholar]
- McMurtry, R.J.; Mitchell, J.R. Renal and hepatic necrosis after metabolic activation of 2-substituted furans and thiophenes, including furosemide and cephaloridine. Toxicol. Appl. Pharmacol. 1977, 42, 285–300. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Potter, W.Z.; Hinson, J.A.; Jollow, D.J. Hepatic necrosis caused by furosemide. Nature 1974, 251, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Wiley, R.A.; Traiger, G.J.; Baraban, S.; Gammal, L.M. Toxicity-distribution relationships among 3-alkylfurans in mouse liver and kidney. Toxicol. Appl. Pharmacol. 1984, 74, 1–9. [Google Scholar] [CrossRef]
- Dalvie, D.K.; Kalgutkar, A.S.; Khojasteh-Bakht, S.C.; Obach, R.S.; O’Donnell, J.P. Biotransformation reactions of five-membered aromatic heterocyclic rings. Chem. Res. Toxicol. 2002, 15, 269–299. [Google Scholar] [CrossRef]
- Alvarez-Diez, T.M.; Zheng, J. Mechanism-based inactivation of cytochrome P450 3A4 by 4-ipomeanol. Chem. Res. Toxicol. 2004, 17, 150–157. [Google Scholar] [CrossRef]
- Gordon, W.P.; Forte, A.J.; McMurtry, R.J.; Gal, J.; Nelson, S.D. Hepatotoxicity and pulmonary toxicity of pennyroyal oil and its constituent terpenes in the mouse. Toxicol. Appl. Pharmacol. 1982, 65, 413–424. [Google Scholar] [CrossRef]
- Peterson, L.A.; Naruko, K.C.; Predecki, D.P. A reactive metabolite of furan, cis-2-butene-1,4-dial, is mutagenic in the Ames assay. Chem. Res. Toxicol. 2000, 13, 531–534. [Google Scholar] [CrossRef]
- Hamadeh, H.K.; Jayadev, S.; Gaillard, E.T.; Huang, Q.; Stoll, R.; Blanchard, K.; Chou, J.; Tucker, C.J.; Collins, J.; Maronpot, R.; et al. Integration of clinical and gene expression endpoints to explore furan-mediated hepatotoxicity. Mutat. Res. 2004, 549, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.D. Structure toxicity relationships—How useful are they in predicting toxicities of new drugs? Adv. Exp. Med. Biol. 2001, 500, 33–43. [Google Scholar] [PubMed]
- Benjamin, S.B.; Ishak, K.G.; Zimmerman, H.J.; Grushka, A. Phenylbutazone liver injury: A clinical-pathologic survey of 23 cases and review of the literature. Hepatology 1981, 1, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Gaisford, W. Fatality after oxyphenbutazone in Still’s disease. Br. Med. J. 1962, 2, 1517. [Google Scholar] [CrossRef] [PubMed]
- Kunze, K.D.; Porst, H.; Tschopel, L. Morphology and pathogenesis of liver injury produced by dihydralazine, propranolol and ketophenylbutazone. Zentralbl. Allg. Pathol. Pathol. 1985, 130, 509–518. (In German) [Google Scholar]
- Wiggins, J.; Scott, D.L. Hepatic injury following feprazone therapy. Rheumatol. Rehabil. 1981, 20, 44–45. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.G. Jaundice following sulfinpyrazone administration. Dtsch. Med. Wochenschr. 1981, 106, 1117. (In German) [Google Scholar]
- Kari, F.; Bucher, J.; Haseman, J.; Eustis, S.; Huff, J. Long-term exposure to the anti-inflammatory agent phenylbutazone induces kidney tumors in rats and liver tumors in mice. Jpn. J. Cancer Res. 1995, 86, 252–263. [Google Scholar] [CrossRef]
- Kayser, D.; Schlede, E. Chemikalien und Kontaktallergie—Eine bewertende Zusammenstellung; Verlag Urban & Vogel: Munique, Germany, 2001. (In German) [Google Scholar]
- Wahlberg, J.E.; Boman, A. Guinea pig maximization test. Curr. Problems Dermatol. 1985, 14, 59–106. [Google Scholar]
- Rycroft, R.J.G.; Wilkinson, J.D. Irritants and sensitisers. In Textbook of Dermatology, 5th ed.; Champion, R.H., Burton, J.L., Ebling, F.J.G., Eds.; Blackwell Science Ltd.: Oxford, UK, 1991; Volume 1, pp. 717–754. [Google Scholar]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Horton, W.; Peerannawar, S.; Török, B.; Török, M. Theoretical and experimental analysis of the antioxidant features of substituted phenol and aniline model compounds. Struct. Chem. 2018. [Google Scholar] [CrossRef]
- Becke, A.D.J. Density-funtional thermochemistry. III. The role of exact exchange. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1998, 37, 785–789. [Google Scholar] [CrossRef]
- Santos, C.B.R.; Lobato, C.C.; Braga, F.S.; Costa, J.S.; Favacho, H.A.S.; Carvalho, J.C.T.; Macedo, W.J.C.; Brasil, D.S.B.; Silva, C.H.T.P.; Hage-Melim, L.I.S. Rational design of antimalarial drugs using molecular modeling and statistical analysis. Curr. Pharm. Desig. 2015, 21, 4112–4127. [Google Scholar] [CrossRef]
- Discovery Studio Modeling Environment, version 5.0; Dassault Systemes BIOVIA: San Diego, CA, USA, 2015.
- Macêdo, W.J.C.; Braga, F.S.; Santos, C.F.; Costa, J.S.; Melo, G.S.; Mello, M.N.; Sousa, D.S.; Carvalho, J.C.T.; Brasil, D.S.B.; Santos, C.B.R. Antimalarial artemisinins derivatives study: Molecular modeling and multivariate analysis: PCA. HCA. KNN. SIMCA and SDA. J. Comp. Threoret. Nanosc. 2015, 12, 3443–3458. [Google Scholar] [CrossRef]
- Federico, L.; Santos, C.B.R.; Lobato, C.; Gomes, J.; Rosa, J.M.C.; Silva, C.H.T.P. Ligand- and structure-based drug design of novel calcium channel blockers. J. Comp. Threoret. Nanosc. 2017, 14, 3489–3502. [Google Scholar] [CrossRef]
- Padilha, E.C.; Serafim, R.B.; Sarmiento, D.Y.R.; Santos, C.F.; Santos, C.B.R.; Silva, C.H.T.P. New PPARα/γ/δ optimal activator rationally designed by computational methods. Braz. Chem. Soc. 2016, 27, 1636–1647. [Google Scholar] [CrossRef]
- Pereira, A.L.E.; Santos, G.B.; Franco, M.S.F.; Federico, L.B.; Silva, C.H.T.P.; Santos, C.B.R. Molecular modeling and statistical analysis in the design of derivatives of human dipeptidyl peptidase IV. J. Biomol. Struct. Dyn. 2018, 36, 318–334. [Google Scholar] [CrossRef]
- Costa, J.S.; Ramos, R.S.; Costa, K.S.L.; Brasil, D.S.B.; Silva, C.H.T.P.; Ferreira, E.F.B.; Borges, R.S.; Campos, J.M.; Macêdo, W.J.C.; Santos, C.B.R. An in silico study of the antioxidant ability for two caffeine analogs using molecular docking and quantum chemical methods. Molecules 2018, 23, 2801. [Google Scholar] [CrossRef]
- Derek for Windows, version 10.0.2; User Guide. Lhasa Limited; Department of Chemistry, University of Leeds: Leeds, UK, 2007.
- Rindings, J.E.; Barratt, M.D.; Cary, R. Computer prediction of possible toxic action from chemical structure, an update of the DEREK system. Toxicology 1996, 106, 267–279. [Google Scholar] [CrossRef]
- Barcellos, M.P.; Santos, C.B.R.; Federico, L.B.; Almeida, P.F.; Silva, C.H.T.P.; Taft, C.A. Pharmacophore and structure-based drug design, molecular dynamics and admet/tox studies to design novel potential pad4 inhibitors. J. Biomol. Struct. Dyn. 2018, 1–33. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Derivatives | HOMO (eV) | LUMO (eV) | GAPL-H (eV) | IP (kcal/mol) |
|---|---|---|---|---|
| 1 | −6.24 | −1.25 | 4.98 | 177.46 |
| 2 | −6.31 | −1.31 | 5.01 | 180.32 |
| 3 | −6.35 | −1.30 | 5.05 | 185.11 |
| 4 | −7.15 | −1.21 | 5.94 | 215.45 |
| 5 | −6.04 | −1.20 | 4.83 | 166.43 |
| 6 | −6.20 | −1.22 | 4.99 | 170.12 |
| Derivatives | HOMO (eV) | LUMO (eV) | GAPL-H (eV) | IP (kcal/mol) | ΔEiso (kcal/mol) | Log P | Vol (Å3) |
|---|---|---|---|---|---|---|---|
| 1 | −6.24 | −1.25 | 4.98 | 177.46 | 0 | 4.22 | 941.89 |
| 7 | −6.44 | −1.55 | 4.88 | 174.44 | −3.01 | 5.26 | 1027.15 |
| 8 | −5.83 | −0.94 | 4.89 | 161.07 | −16.38 | 3.94 | 979.66 |
| 9 | −5.83 | −0.99 | 4.83 | 159.79 | −17.66 | 4.81 | 1051.55 |
| 10 | −5.80 | −0.97 | 4.83 | 159.54 | −17.91 | 5.20 | 1119.93 |
| 11 | −5.91 | −1.46 | 4.44 | 159.69 | −17.76 | 5.37 | 1030.56 |
| 12 | −6.11 | −2.21 | 3.90 | 168.09 | −9.37 | 3.39 | 1010.77 |
| 13 | −6.02 | −2.10 | 3.91 | 167.21 | −10.25 | 5.37 | 960.47 |
| 14 | −6.02 | −2.02 | 4.00 | 167.15 | −10.30 | 5.37 | 963.55 |
| 15 | −5.93 | −1.90 | 4.02 | 166.03 | −11.42 | 4.85 | 920.66 |
| Structures | Toxicity Prediction Alert (Lhasa Prediction) | Toxicophoric Group | Toxicity Alert | Toxicity Prediction (Custom Prediction) |
|---|---|---|---|---|
| Indometacin | - | - | - | Nothing to declare |
| Refecoxib | Hepatotoxicity in human, mouse and rat | Furan | PLAUSIBLE | Nothing to declare |
| Phenylbutazone (1) | Hepatotoxicity in human and mouse | 1,2-Diphenyl-3,5-pyrazolidinedione derivative | CERTAIN | Nothing to declare |
| Hepatotoxicity in rat | 1,2-Diphenyl-3,5-pyrazolidinedione derivative | PROBABLE | ||
| Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | ||
| 7 | Hepatotoxicity in human, mouse and rat | 1,2-Diphenyl-3,5-pyrazolidinedione derivative | PLAUSIBLE | Nothing to declare |
| Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | ||
| 8 | Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | Nothing to declare |
| 13 | Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | Nothing to declare |
| 14 | Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | Nothing to declare |
| 15 | Skin sensitization in human, mouse and rat | Hydrazine or precursor | PLAUSIBLE | Nothing to declare |
| Receptor | Ligand | Ligand Coordinates of the Grid Center | Grid Size (Points) |
|---|---|---|---|
| COX-2 (PDB code: 4COX) Mus musculus | Indomethacin | X = 11.5685 Y = 12.3903 Z = 7.2430 | 26 x 30 y 28 z |
| COX-2 (PDB code: 5KIR) Homo sapiens | Rofecoxib | X = 26.6293 Y = 40.1834 Z = 3.8484 | 26 x 30 y 28 z |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borges, R.S.; Palheta, I.C.; Ota, S.S.B.; Morais, R.B.; Barros, V.A.; Ramos, R.S.; Silva, R.C.; Costa, J.d.S.; Silva, C.H.T.P.; Campos, J.M.; et al. Toward of Safer Phenylbutazone Derivatives by Exploration of Toxicity Mechanism. Molecules 2019, 24, 143. https://doi.org/10.3390/molecules24010143
Borges RS, Palheta IC, Ota SSB, Morais RB, Barros VA, Ramos RS, Silva RC, Costa JdS, Silva CHTP, Campos JM, et al. Toward of Safer Phenylbutazone Derivatives by Exploration of Toxicity Mechanism. Molecules. 2019; 24(1):143. https://doi.org/10.3390/molecules24010143
Chicago/Turabian StyleBorges, Rosivaldo S., Ivanete C. Palheta, Sirlene S. B. Ota, Roberto B. Morais, Valéria A. Barros, Ryan S. Ramos, Rai C. Silva, Josivan da S. Costa, Carlos H. T. P. Silva, Joaquín M. Campos, and et al. 2019. "Toward of Safer Phenylbutazone Derivatives by Exploration of Toxicity Mechanism" Molecules 24, no. 1: 143. https://doi.org/10.3390/molecules24010143