
Identification of New Inhibitors with Potential Antitumor Activity from Polypeptide Structures via Hierarchical Virtual Screening

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results and Discussion
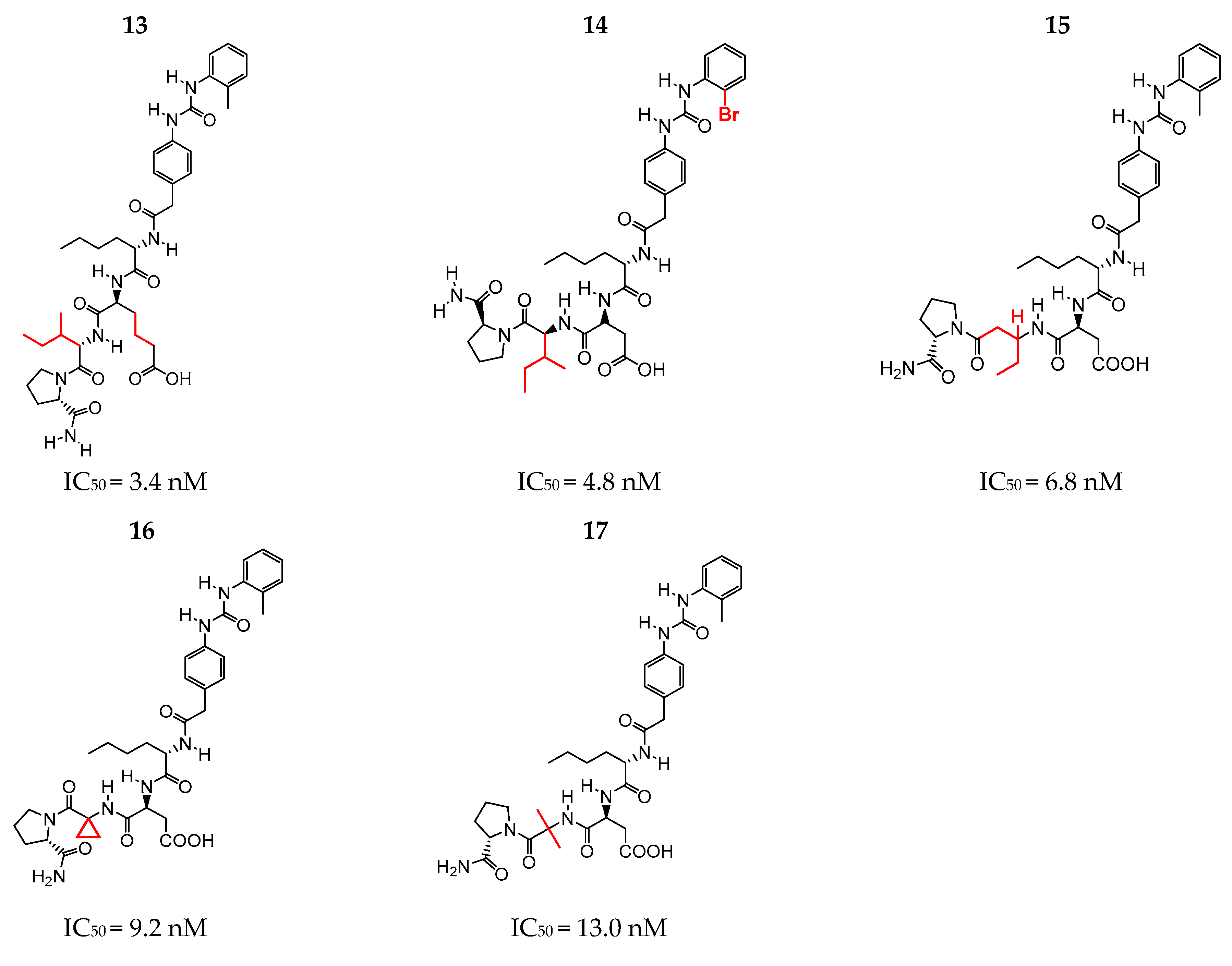
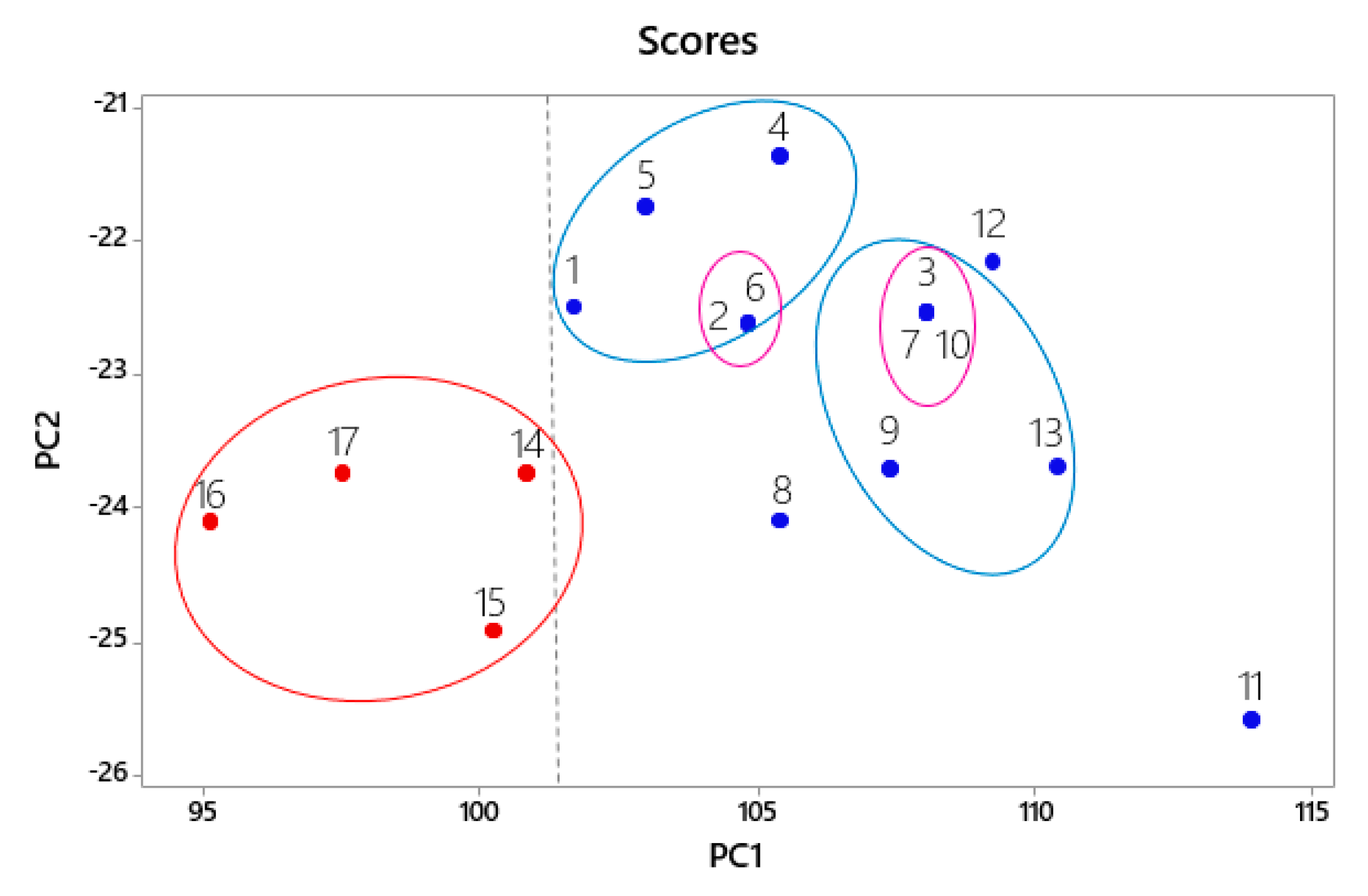
2.1. Selection of the Structures according to Inhibitory Activity (IC50)
2.2. Optimization of the Geometry of Selected Structures
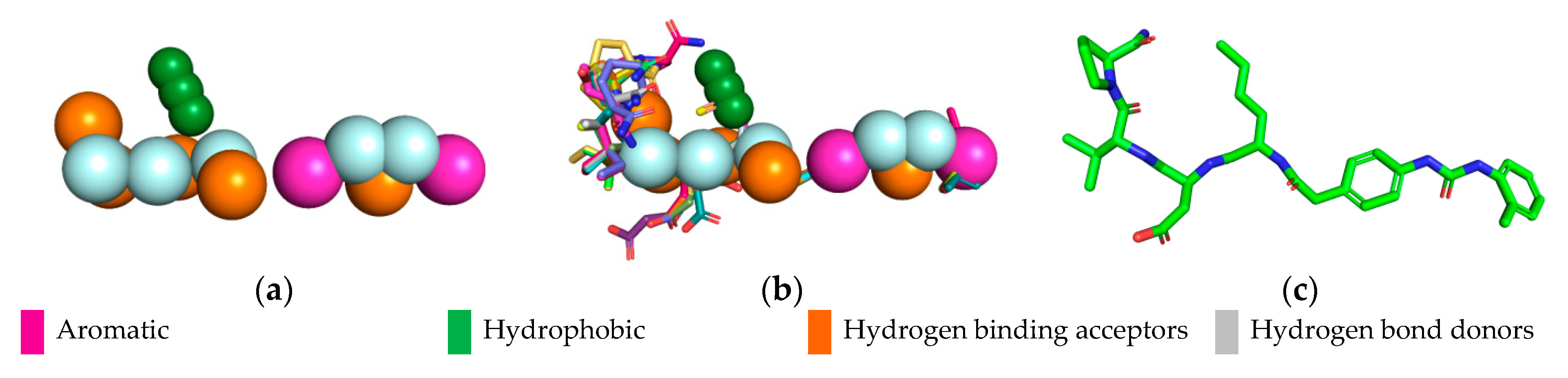
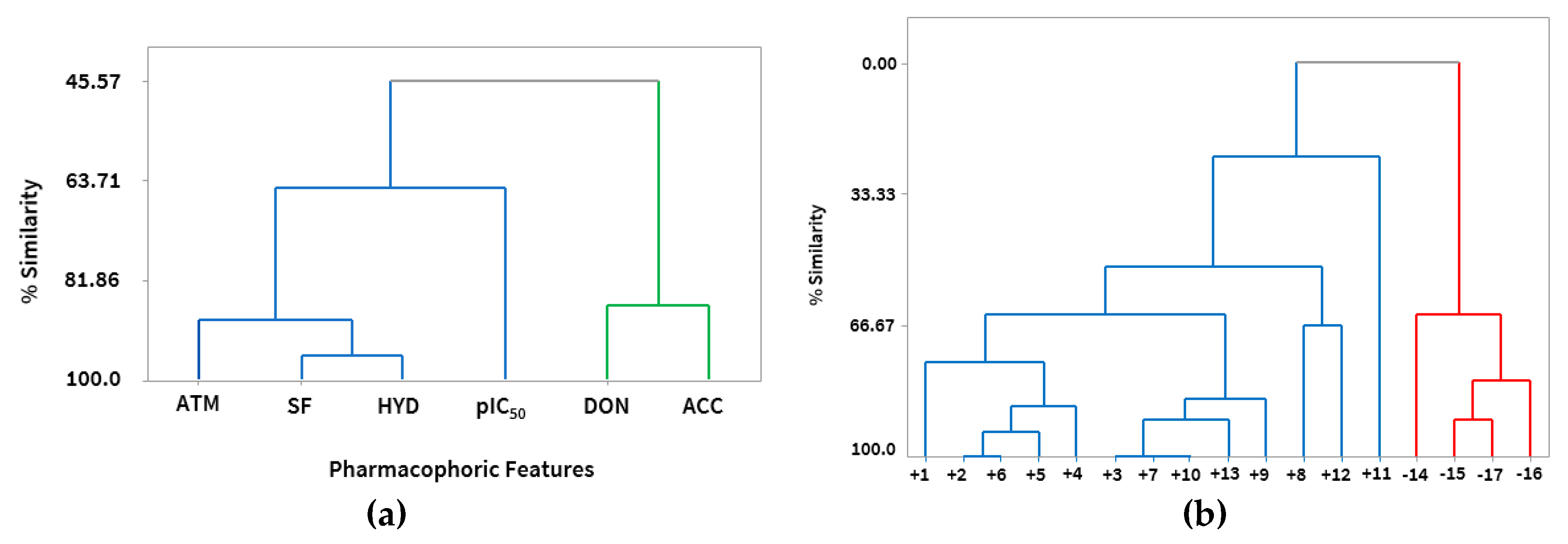
2.3. Pharmacophoric Model
2.3.1. Hypothesis Generation
2.3.2. Evaluation of the Pharmacophoric Hypothesis
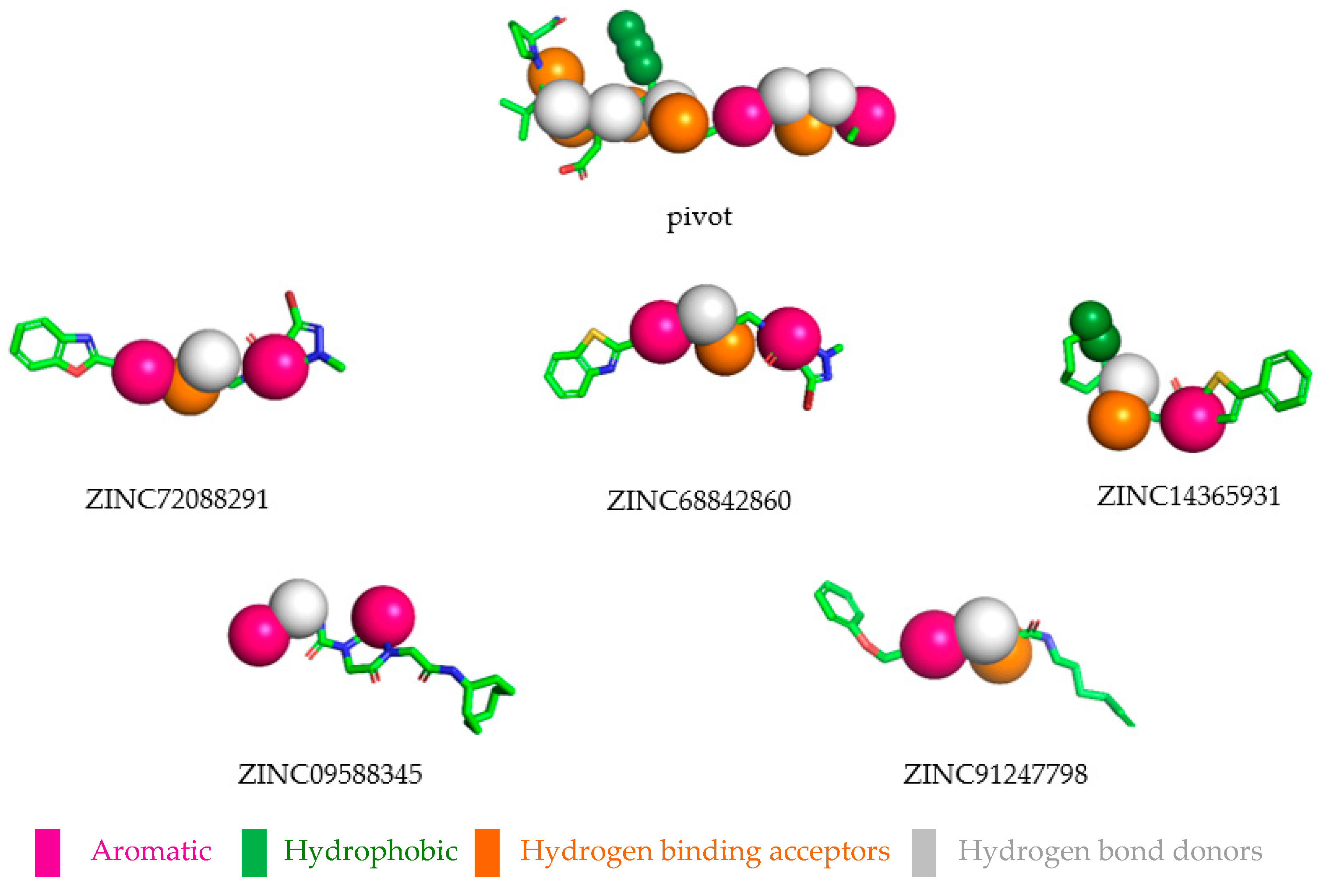
2.4. Pharmacophoric Hypothesis
2.5. Pharmacokinetic Properties Prediction
2.6. Prediction of Toxicological Properties
2.7. Predictions of Biological Activity
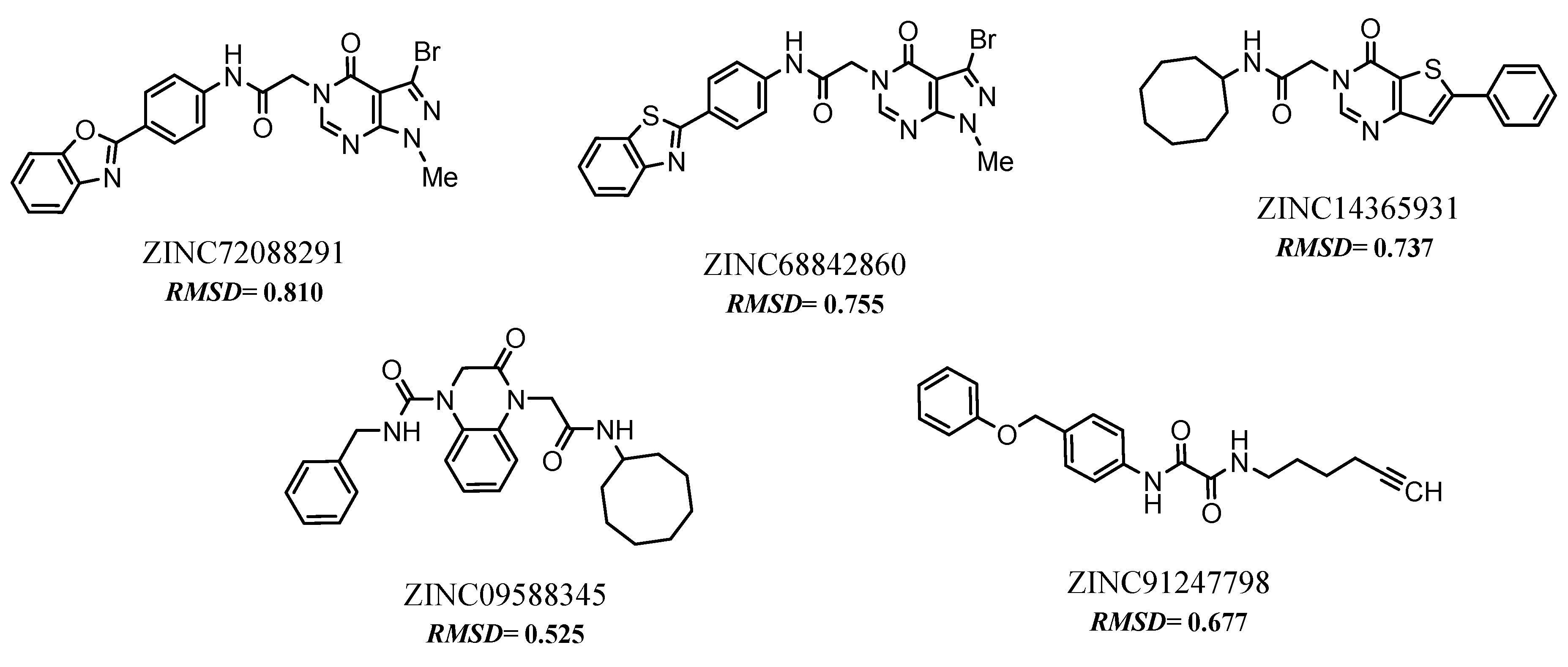
2.8. Selected Compounds via Pharmacophore-based Virtual Screening
3. Material and Methods
3.1. Selection of Structures
3.2. Generation of Pharmacophoric Hypotheses
3.2.1. Geometry Optimization
3.2.2. Generation of the Pharmacophoric Model
3.2.3. Evaluation of the Pharmacophoric Model
3.2.4. Pharmacophoric Hypotheses Refinement
3.3. Pharmacokinetic Predictions
3.4. Toxicological Predictions
3.5. Prediction of Biological Activity
4. Conclusion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- National Institute of Cancer José Alencar Gomes da Silva. Coordination of Prevention and Surveillance. Estimate 2018: Incidence of cancer in Brazil/National Cancer Institute José Alencar Gomes da Silva. In Coordination of Prevention and Surveillance; INCA: Rio de Janeiro, Brazil, 2017. [Google Scholar]
- Moraes, E.S.; de Mello, M.S.; de Nogueira, F.; Otero, U.B.; Carvalho, F.N. Analysis of individuals with leukemia: Limitations of the cancer surveillance system. Sci. Collectiv. Health 2017, 22, 3321–3332. [Google Scholar]
- De Morais, E.F.; Lira, J.A.S.; Macedo, R.A.P.; Santos, K.S.; Elias, C.T.V.; Morais, M.L.S.A. Oral manifestations resulting from chemotherapy in children with acute lymphoblastic leukemia. Braz. J. Otorhinolaryngol. 2014, 80, 78–85. [Google Scholar] [CrossRef]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef]
- Soares, J.L.M.F.; Pasqualotto, A.C.; Rosa, D.D.; Leite, V.R.S. Métodos Diagnósticos: Consulta Rápida, 1st ed.; Artmed: Porto Alegre, Brazil, 2002. [Google Scholar]
- Da Silva, M.J.S.; Melo, E.C.P.; Osorio-de-Castro, C.G.S. Origin-destination flows in chemotherapy for breast cancer in Brazil: Implications for pharmaceutical services. Ciência Saúde Coletiva 2019, 24, 1153–1164. [Google Scholar] [CrossRef]
- Male, D.; Brostoff, J.; Roth, D.B.; Roitt, I. Immunology, 7th ed.; Mosby Elsevier: Philadelphia, PA, USA, 2006. [Google Scholar]
- Vincent, A.M.; Cawley, J.C.; Burthem, J. Integrin function in chronic lymphocytic leukemia. J. Blood 1996, 87, 4780–4788. [Google Scholar] [Green Version]
- Da Silva, M.E.R.; Mory, D.; Davini, E. Genetic and Autoimmune Markers of Diabetes Type 1 Melito: From Theory to Practice. Braz. Arch. Endocrinol. Metab. 2008, 52, 166–180. [Google Scholar]
- Liu, R.; Peng, L.; Han, H.; Lam, K.S. Structure-Activity Relationship Studies of a Series of Ligands for a4b1 Integrin on Jurkat T-Leukemia Cells; Wiley InterScience: New York, NY, USA, 2006; pp. 595–604. [Google Scholar]
- Goldberg, A.C.; Rizzo, L.V. MHC Structure and Function—Antigen Presentation. Part 2. Einstein 2015, 13, 157–162. [Google Scholar] [CrossRef]
- Lin, K.; Ateeq, H.S.; Hsiung, S.H.; Ching, L.T.; Zimmerman, C.N.; Castro, A.; Lee, W.C.; Hammond, C.E.; Kalkunte, S.; Chen, L.L.; et al. Synthesis and Assay of retro-α4β Integrin-targeting Motifs. J. Med. Chem. 1999, 42, 920–934. [Google Scholar] [CrossRef]
- Carpenter, R.D.; Andrei, M.; Aina, O.H.; Lau, E.Y.; Lightstone, F.C.; Liu, R.; Kurth, M.J. Selectively Targeting T- and B-Cell Lymphomas: A Benzothiazole Antagonist of α4β1Integrin. J. Med. Chem. 2009, 52, 14–19. [Google Scholar] [CrossRef]
- Kutlushina, A.; Khakimova, A.; Madzhidov, T.; Polishchuk, P. Ligand-Based Pharmacophore Modeling Using Novel 3D Pharmacophore Signatures. Molecules 2018, 23, 3094. [Google Scholar] [CrossRef]
- Inbar, Y.; Schneidman-Duhovny, D.; Dror, O.; Nussinov, R.; Wolfson, H.J. Deterministic Pharmacophore Detection via Multiple Flexible Alignment of Drug-Like Molecules. Proc. RECOMB 2007, 3692, 423–434. [Google Scholar]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, 223–228. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; BeltránDebón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef]
- Yang, K.; Nong, K.; Gu, Q.; Dong, J.; Wang, J. Discovery of N -hydroxy-3-alkoxybenzamides as direct acid sphingomyelinase inhibitors using a ligand-based pharmacophore model. Eur. J. Med. Chem. 2018, 151, 389–400. [Google Scholar] [CrossRef]
- Yannakakis, M.-P.; Simal, C.; Tzoupis, H.; Rodi, M.; Dargahi, N.; Prakash, M.; Mouzaki, A.; Platts, J.A.; Apostolopoulos, V.; Tselios, T.V. Design and Synthesis of Non-Peptide Mimetics Mapping the Immunodominant Myelin Basic Protein (MBP83–96) Epitope to Function as T-Cell Receptor Antagonists. Int. J. Mol. Sci. 2017, 18, 1215. [Google Scholar] [CrossRef]
- Lee, Y.H.; Yi, G.-S. Prediction of Novel Anoctamin1 (ANO1) Inhibitors Using 3D-QSAR Pharmacophore Modeling and Molecular Docking. Int. J. Mol. Sci. 2018, 19, 3204. [Google Scholar] [CrossRef]
- Aykul, S.; Martinez-Hackert, E. Determination of half-maximal inhibitory concentration using biosensor-based protein interaction analysis. Anal. Biochem. 2016, 508, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Montanari, C.A. Quimica Medicinal: Metodos E Fundamentos Em Planejamento De Fármacos; Edusp: São Paulo, Brazil, 2011. [Google Scholar]
- ChemPlus, Modular Extensions to HyperChem, Release 6.02, Molecular Modeling for Windows; Hyper Inc.: Gainesville, FL, USA, 2000.
- Costa, J.S.; Costa, K.S.L.; Cruz, J.V.; Ramos, R.S.; Silva, L.B.; Brasil, D.S.B.; Silva, C.H.T.P.; Santos, C.B.R.; Macêdo, W.J.C. Virtual Screening and Statistical Analysis in the Design of New Caffeine Analogues Molecules with Potential Epithelial Anticancer Activity. Curr. Pharm. Des. 2018, 24, 576–594. [Google Scholar] [CrossRef]
- Macêdo, W.J.C.; Braga, F.S.; Santos, C.F.; Costa, J.D.S.; de Melo, G.S.; de Mello, M.N.; dos Santos, C.B.R. Antimalarial Artemisinins Derivatives Study: Molecular Modeling and Multivariate Analysis (PCA, HCA, KNN, SIMCA and SDA). Journal of Computational and Theoretical Nanoscience 2015, 12, 3443–3458. [Google Scholar] [CrossRef]
- Santos, K.L.B. Drugs design with Anti-inflammatory Activity in the Receptor Adenosine Type A2A. Master’s Thesis, Institute of Health Sciences, Federal University of Pará, Belém, Brazil, 2017. [Google Scholar]
- Barreiro, E.J.; Kümmerle, A.E.; Fraga, C.A.M. The Methylation Effect in Medicinal Chemistry. Chem. Rev. 2011, 111, 5215–5246. [Google Scholar] [CrossRef]
- Koes, D.R.; Carlos, J.C. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2014, 40, W409–W414. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 23, 3–26. [Google Scholar] [CrossRef]
- Artursson, P.; Karlsson, J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 1991, 175, 880–885. [Google Scholar] [CrossRef]
- Horster, M.F.; Stopp, M. Transport and metabolic functions in cultured renal tubule cells. Kidney Int. 1986, 29, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Kujawski, J.; Popielarska, H.; Myka, A.; Drabinska, B.; Bernard, M. The Log P parameter as a molecular descriptor in the computer-aided drug design—An overview. Comput. Methods Sci. Technol. 2012, 18, 81–88. [Google Scholar] [CrossRef]
- Ames, B.N.; Gurney, E.G.; Miller, J.A.; Bartsch, H. Carcinogens as frameshift mutagens: Metabolites and derivatives of 2-acetylaminofluorene and other aromatic amine carcinogens. Proc. Natl. Acad. Sci. USA 1972, 69, 3128–3132. [Google Scholar] [CrossRef]
- Almeida, J.R. In silico planning, assay and optimization of new prototypes inhibiting the enzyme acetylcholinesterase. Ph.D. Thesis, Faculty of Pharmaceutical Sciences of Ribeirão Preto—University of São Paulo, Ribeirão Preto, Brazil, 2014. [Google Scholar]
- Cruz, J.V.; Neto, M.F.A.; Silva, L.B.; Ramos, R.D.S.; Costa, J.D.S.; Brasil, D.S.B.; Lobato, C.C.; Da Costa, G.V.; Bittencourt, J.A.H.M.; Da Silva, C.H.T.P.; et al. Identification of Novel Protein Kinase Receptor Type 2 Inhibitors Using Pharmacophore and Structure-Based Virtual Screening. Molecules 2018, 23, 453. [Google Scholar] [CrossRef]
- Lhasa Limited. Derek for Windows. Version 10.0.2. User Guide; University of Leeds: Leeds, UK, 2007. [Google Scholar]
- Poroikov, V.V.; Filimonov, D.A.; Ihlenfeldt, W.-D.; Gloriozova, T.A.; Lagunin, A.A.; Borodina, Y.V.; Stepanchikova, A.V.; Nicklaus, M.C. PASS Biological Activity Spectrum Predictions in the Enhanced Open NCI Database Browser. J. Chem. Inform. Comput. Sci. 2003, 43, 228–236. [Google Scholar] [CrossRef]
- De Almeida, V.L.; Leitão, A.; del Carmen Barrett Reina, L.; Montanari, C.A.; Donnici, C.L.; Lopes, M.T.P. Cancer and specific non-specific cyto-cellular and cyclo-cellular antineoplastic agents that interact with DNA: An introduction. New Chem. 2005, 28, 118–129. [Google Scholar] [CrossRef]
- Version, A.D.S. Accelrys Discovery Studio. 4.0; Biovia Inc.: San Diego, CA, USA, 2017. [Google Scholar]
- Pereira, A.L.E.; dos Santos, G.B.; Franco, M.S.F.; Federico, L.B.; da Silva, C.H.T.P.; Santos, C.B.R. Molecular modeling and statistical analysis in the design of derivatives of human dipeptidyl peptidase IV. J. Biomol. Struct. Dyn. 2018, 36, 318–334. [Google Scholar] [CrossRef]
- Vieira, J.B.; Braga, F.S.; Lobato, C.C.; Santos, C.F.; Costa, J.S.; Bittencourt, J.A.H.M.; Brasil, D.S.B.; Silva, J.O.; Hage-Melim, L.I.S.; Macêdo, W.J.C.; et al. A QSAR, Pharmacokinetic and Toxicological Study of New Artemisinin Compounds with Anticancer Activity. Molecules 2014, 19, 10670–10697. [Google Scholar] [CrossRef] [Green Version]
- Cruz, J.V.; Serafim, R.B.; da Silva, G.M.; Giuliatti, S.; Rosa, J.M.C.; Araújo Neto, M.F.; Leite, F.H.A.; Taft, C.A.; da Silva, C.H.T.P.; Santos, C.B.R. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. J. Mol. Model. 2018, 24, 225. [Google Scholar] [CrossRef]
- Santos, C.B.R.; Vieira, J.B.; Lobato, C.C.; Hage-Melim, L.I.S.; Souto, R.N.P.; Lima, C.S.; Costa, E.V.M.; Brasil, D.S.B.; Macêdo, W.J.C.; Carvalho, J.C.T. A SAR and QSAR Study of New Artemisinin Compounds with Antimalarial Activity. Molecules 2014, 19, 367–399. [Google Scholar] [CrossRef]
- Pirouette 3.01; Infometrix, Inc.: Woodinville, WA, USA, 2001.
- Version, Q. Rapid ADME predictions of drug candidates, Version 3.4; Schrödinger: New York, NY, USA, 2011. [Google Scholar]
- Sanderson, D.M.; Earnshaw, C.G. Computer prediction of possible toxic action from chemical structure; the DEREK system. Hum. Exp. Toxicol. 1991, 10, 261–273. [Google Scholar] [CrossRef]
- Bittencourt, J.A.H.M.; Neto, M.F.A.; Lacerda, P.S.; Bittencourt, R.C.V.S.; Silva, R.C.; Lobato, C.C.; Silva, L.B.; Leite, F.H.A.; Zuliani, J.P.; Rosa, J.M.C.; et al. In Silico Evaluation of Ibuprofen and Two Benzoylpropionic Acid Derivatives with Potential Anti-Inflammatory Activity. Molecules 2019, 24, 1476. [Google Scholar] [CrossRef]
- Borges, R.S.; Palheta, I.C.; Ota, S.S.B.; Morais, R.B.; Barros, V.A.; Ramos, R.S.; Silva, R.C.; Costa, J.S.; Silva, C.H.T.P.; Campos, J.M.; et al. Toward of Safer Phenylbutazone Derivatives by Exploration of Toxicity Mechanism. Molecules 2019, 24, 143. [Google Scholar] [CrossRef]
- Ferreira, J.V.; Chaves, G.A.; Marino, B.L.B.; Sousa, K.P.A.; Souza, L.R.; Brito, M.F.B.; Texeira, H.R.C.; Silva, C.H.T.P.; Santos, C.B.R.; Hage-Melim, L.I.S. Cannabinoid type 1 receptor (CB1) bioligand with therapeutic potential for withdrawal syndrome in chemical dependents Cannabis sativa. ChemMedChem 2017, 12, 1408–1416. [Google Scholar] [CrossRef]
- Ramos, R.S.; Costa, J.S.; Silva, R.C.; da Costa, G.V.; Rodrigues, A.B.L.; Rabelo, É.M.; Souto, R.N.P.; Taft, C.A.; Silva, C.H.T.P.; Rosa, J.M.C.; et al. Identification of Potential Inhibitors from Pyriproxyfen with Insecticidal Activity by Virtual Screening. Pharmaceuticals 2019, 12, 20. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








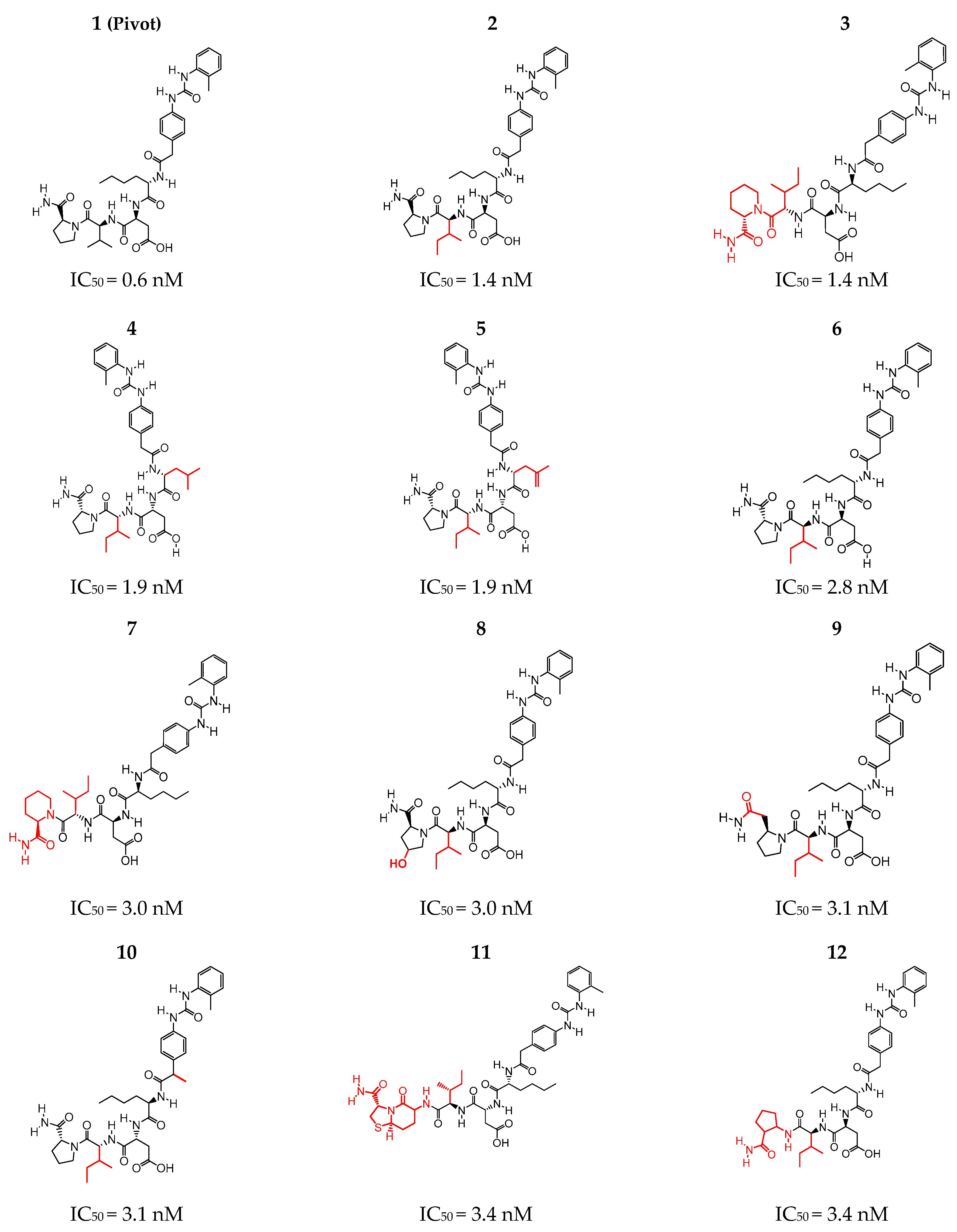{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | ATM | SF | HYD | DON | ACC | pIC50 | ARO |
|---|---|---|---|---|---|---|---|
| 1 | 99 | 28 | 11 | 6 | 8 | 9.2219 | 2 |
| 2 | 102 | 29 | 12 | 6 | 8 | 8.8539 | 2 |
| 3 | 105 | 30 | 13 | 6 | 8 | 8.8539 | 2 |
| 4 | 102 | 30 | 13 | 6 | 8 | 8.7212 | 2 |
| 5 | 100 | 29 | 12 | 6 | 8 | 8.5528 | 2 |
| 6 | 102 | 29 | 12 | 6 | 8 | 8.5376 | 2 |
| 7 | 105 | 30 | 13 | 6 | 8 | 8.5229 | 2 |
| 8 | 103 | 29 | 11 | 7 | 9 | 8.5229 | 2 |
| 9 | 105 | 29 | 12 | 6 | 8 | 8.5086 | 2 |
| 10 | 105 | 30 | 13 | 6 | 8 | 8.5086 | 2 |
| 11 | 111 | 32 | 12 | 7 | 10 | 8.4685 | 2 |
| 12 | 105 | 31 | 13 | 7 | 8 | 8.4685 | 2 |
| 13 | 108 | 30 | 13 | 6 | 8 | 8.4685 | 2 |
| 14 | 99 | 27 | 10 | 6 | 9 | 8.3188 | 2 |
| 15 | 99 | 26 | 9 | 6 | 8 | 8.1675 | 2 |
| 16 | 94 | 25 | 8 | 6 | 8 | 8.0362 | 2 |
| 17 | 96 | 26 | 9 | 6 | 8 | 7.8861 | 2 |
| SF | 0.912 | ||||||
| * | 0.000 | ||||||
| HYD | 0.791 | 0.913 | |||||
| * | 0.000 | 0.000 | |||||
| DON | 0445 | 0.468 | 0.138 | ||||
| * | 0.074 | 0.058 | 0.597 | ||||
| ACC | 0.380 | 0.278 | −0.076 | 0.649 | |||
| * | 0.133 | 0.279 | 0.771 | 0.005 | |||
| pIC50 | 0.340 | 0.508 | 0.604 | −0.031 | −0.089 | ||
| * | 0.182 | 0.037 | 0.010 | 0.906 | 0.735 |
| Parameters | Main Component | |||
| PC1 | PC2 | PC3 | ||
| Variance (%) | 93.3 | 0.05 | 0.014 | |
| Cumulative variance (%) | 93.3 | 98.3 | 99.8 | |
| Pharmacophoric Characteristics | Contribution | |||
| PC1 | PC2 | |||
| ATM | 0.882 | −0.395 | ||
| SF | 0.370 | 0.400 | ||
| HYD | 0.286 | 0.765 | ||
| DON | 0.036 | −0.123 | ||
| ACC | 0.050 | −0.290 | ||
| Pharmacophoric Characteristics | Coordinates | ||||
|---|---|---|---|---|---|
| x | y | z | Radius | ||
| Aromatic | Aro 1 | 12.73 | 2.75 | −0.03 | 1.1 |
| Aromatic | Aro 2 | 16.33 | 9.31 | −0.09 | 1.1 |
| Hydrophobic | Hyd 1 | 6.95 | −1.05 | −0.44 | 1.0 |
| Hydrophobic | Hyd 2 | 5.68 | −0.94 | −0.33 | 1.0 |
| Hydrophobic | Hyd 3 | 4.41 | −0.84 | −0.26 | 1.0 |
| Hydrogen binding acceptors | Acc 1 | 4.71 | −6.08 | −1.08 | 0.5 |
| Hydrogen binding acceptors | Acc 2 | 7.51 | −6.48 | −1.85 | 0.5 |
| Hydrogen binding acceptors | Acc 3 | 8.96 | −3.08 | −2.15 | 0.5 |
| Hydrogen binding acceptors | Acc 4 | 11.29 | −1.10 | 1.10 | 0.5 |
| Hydrogen binding acceptors | Acc 5 | 15.07 | 5.78 | −0.36 | 0.5 |
| Hydrogen bond donors | Don 1 | 7.09 | −6.68 | 0.42 | 0.5 |
| Hydrogen bond donors | Don 2 | 8.72 | −4.06 | −0.09 | 0.5 |
| Hydrogen bond donors | Don 3 | 10.06 | −0.85 | −0.83 | 0.5 |
| Hydrogen bond donors | Don 4 | 13.0 | 5.5 | 0.59 | 0.5 |
| Hydrogen bond donors | Don 5 | 14.23 | 7.61 | 0.75 | 0.5 |
| Hypothesis 1 | ||||||
 | Pharmacophoric Properties | Coordinates | Number of Compounds Obtained (ZINCpharmer) | |||
| x | y | z | R | |||
| Aro 1 | 12.73 | 2.75 | −0.03 | 1.1 | 942 structures | |
| Aro 2 | 16.33 | 9.31 | −0.09 | 1.1 | ||
| Hyd 1 | 5.68 | −0.94 | −0.33 | 1.0 | ||
| Hyd 2 | 6.95 | −1.05 | −0.44 | 1.0 | ||
| Hyd 3 | 4.41 | −0.84 | −0.26 | 1.0 | ||
| Hypothesis 2 | ||||||
 | Aro 1 | 12.73 | 2.75 | −0.03 | 1.1 | 141 structures |
| Aro 2 | 16.33 | 9.31 | −0.09 | 1.1 | ||
| Hyd 1 | 5.68 | −0.94 | −0.33 | 1.0 | ||
| Hyd 2 | 6.95 | −1.05 | −0.44 | 1.0 | ||
| Hyd 3 | 4.41 | −0.84 | −0.26 | 1.0 | ||
| Don 3 | 10.06 | −0.85 | −0.83 | 0.5 | ||
| Hypothesis 3 | ||||||
 | Aro 1 | 12.73 | 2.75 | −0.03 | 1.1 | 9 structures |
| Aro 2 | 16.33 | 9.31 | −0.09 | 1.1 | ||
| Hyd 1 | 5.68 | −0.94 | −0.33 | 1.0 | ||
| Hyd 2 | 6.95 | −1.05 | −0.44 | 1.0 | ||
| Hyd 3 | 4.41 | −0.84 | −0.26 | 1.0 | ||
| Don 5 | 14.23 | 7.61 | 0.75 | 0.5 | ||
| Hypothesis 4 | ||||||
 | Aro 1 | 12.73 | 2.75 | −0.03 | 1.1 | 386 structures |
| Aro 2 | 16.33 | 9.31 | −0.09 | 1.1 | ||
| Hyd 1 | 5.68 | −0.94 | −0.33 | 1.0 | ||
| Hyd 2 | 6.95 | −1.05 | −0.44 | 1.0 | ||
| Hyd 3 | 4.41 | −0.84 | −0.26 | 1.0 | ||
| Acc 5 | 15.07 | 5.78 | −0.36 | 0.5 | ||
| Compounds | RMSD | #star | ROF * | %HOA * | QPPCaco | QPPMDCK | QPlogPo/w | CNS * | QPlogBB |
|---|---|---|---|---|---|---|---|---|---|
| Hypothesis 1 | |||||||||
| pivot | - | 10 | 3 | 0.0 | 0.614 | 1.864 | 33.047 | −2 | −4.115 |
| ZINC78538125 | 0.756 | 0 | 0 | 100 | 1711.2 | 884,159 | 4.383 | −1 | −0.788 |
| ZINC91247798 ** | 0.677 | 0 | 0 | 100 | 1104.2 | 550,677 | 4.845 | −2 | −1.148 |
| ZINC91247798 ** | 0.718 | 0 | 0 | 100 | 1104.3 | 550,732 | 4.845 | −2 | −1.148 |
| ZINC32143540 | 0.756 | 0 | 0 | 100 | 1819.3 | 944,695 | 4.621 | −1 | −0.811 |
| ZINC78538137 | 0.792 | 0 | 0 | 100 | 1182.2 | 592,819 | 4.225 | −2 | −1.121 |
| ZINC02134226 | 0.749 | 0 | 0 | 100 | 1153.4 | 577,244 | 4.758 | −2 | −1.042 |
| ZINC14365931 | 0.737 | 0 | 0 | 100 | 1003.7 | 1146.8 | 3.682 | 0 | −0.456 |
| ZINC01902746 | 0.749 | 0 | 0 | 100 | 1153.5 | 1039.7 | 4.995 | −1 | −0.936 |
| ZINC19716136 | 0.760 | 0 | 0 | 100 | 717,939 | 528,751 | 3.335 | −2 | −1.005 |
| ZINC09588345 ** | 0.677 | 0 | 0 | 96.2 | 567,435 | 567,156 | 3.418 | −1 | −0.792 |
| ZINC09588345 ** | 0.525 | 0 | 0 | 96.6 | 592,582 | 572,285 | 3.430 | −1 | −0.789 |
| ZINC64971623 | 0.528 | 0 | 0 | 100 | 559,685 | 557,242 | 3.629 | −1 | −0.943 |
| ZINC72088291 ** | 0.818 | 0 | 0 | 92.6 | 505,985 | 594,490 | 2.959 | −1 | −0.921 |
| ZINC72088291 ** | 0.810 | 0 | 0 | 92.8 | 514,975 | 605,917 | 2.958 | −1 | −0.900 |
| ZINC72088291 ** | 0.841 | 0 | 0 | 92.8 | 514,997 | 605,942 | 2.958 | −1 | −0.900 |
| ZINC23592367 | 0.967 | 0 | 0 | 100 | 655,789 | 564,084 | 4.505 | −2 | −1.275 |
| ZINC68842860 ** | 0.803 | 0 | 0 | 96.5 | 568,385 | 1055.6 | 3.467 | −1 | −0.775 |
| ZINC68842860 ** | 0.785 | 0 | 0 | 96.5 | 568,468 | 1055.8 | 3.467 | −1 | −0.774 |
| ZINC68842860 ** | 0.755 | 0 | 0 | 96.2 | 555,629 | 1030.0 | 3.450 | −1 | −0.797 |
| ZINC68842860 ** | 0.827 | 0 | 0 | 100 | 656,889 | 1234.4 | 3.501 | −1 | −0.711 |
| Hypothesis 2 | |||||||||
| ZINC91247798 ** | 0.704 | 0 | 0 | 100 | 1104.2 | 550,682 | 4.845 | −2 | −1.148 |
| ZINC91247798 ** | 0.730 | 0 | 0 | 100 | 1104.3 | 550,758 | 4.845 | −2 | −1.148 |
| Hypothesis 3 | |||||||||
| ZINC64971623 | 0.635 | 0 | 0 | 100 | 559,718 | 557,260 | 3.629 | −1 | −0.943 |
| Hypothesis 4 | |||||||||
| ZINC68842860 | 0.772 | 0 | 0 | 96.2 | 555,605 | 1030.0 | 3.450 | −1 | −0.797 |
| Compounds Code | Toxicity Prediction Alert (Lhasa Prediction) | Toxicophoric Group | Toxicity Alert |
|---|---|---|---|
| ZINC78538125 | Methaemoglobinaemia | Aniline or precursor | Plausible |
| Carcinogenicity | - | Plausible | |
| Peroxisome proliferation | Alkylaryl or bisaryl, carboxylic acid or precursor | Plausible | |
| ZINC91247798 * | - | - | No alerts |
| ZINC32143540 | Carcinogenicity | - | Plausible |
| Peroxisome proliferation | Alkylaryl or bisaryl carboxylic acid or precursor | Plausible | |
| ZINC78538137 | Carcinogenicity | - | Plausible |
| Peroxisome proliferation | beta-O/S-Substituted carboxylic acid or precursor | Plausible | |
| ZINC02134226 | Photoallergenicity | Coumarin | Plausible |
| Skin sensitization | Resorcinol or precursor | Plausible | |
| ZINC14365931 | - | - | No alerts |
| ZINC01902746 | Photoallergenicity | Coumarin | Plausible |
| Skin sensitization | Resorcinol or precursor | Plausible | |
| ZINC19716136 | Hepatotoxicity | p-Aminophenol or derivative | Plausible |
| ZINC09588345 * | - | - | No alerts |
| ZINC64971623 | Hepatotoxicity | p-Aminophenol or derivative | Plausible |
| ZINC72088291 * | - | - | No alerts |
| ZINC23592367 | Chromosome damage in vitro | Xanthine | Plausible |
| Ocular toxicity | Phosphodiesterase 6 inhibitor and purine base or analogue | Plausible | |
| Teratogenicity | Xanthine | Plausible | |
| ZINC68842860 * | - | - | No alerts |
| Compound | Biological Activity | Pa | Pi |
|---|---|---|---|
| pivot | Inhibitor of the cell adhesion molecule | 0.629 | 0.008 |
| Integrin alpha4 antagonist | 0.617 | 0.002 | |
| Cell adhesion inhibitor | 0.558 | 0.003 | |
| Antineoplastic (Non-Hodgkin’s Lymphoma) | 0.442 | 0.071 | |
| Immunomodulator | 0.337 | 0.052 | |
| Antineoplastic (multiple myelomas) | 0.236 | 0.184 | |
| Integrin alpha4 beta1 antagonist | 0.068 | 0.007 | |
| ZINC72088291 | Antineoplastic | 0.618 | 0.041 |
| Immunomodulator | 0.337 | 0.012 | |
| ZINC68842860 | Antineoplastic | 0.638 | 0.037 |
| Immunomodulator | 0.371 | 0.037 | |
| ZINC14365931 | Antineoplastic | 0.327 | 0.042 |
| ZINC09588345 | Antimetastatic | 0.336 | 0.067 |
| Antineoplastic (multiple myelomas) | 0.321 | 0.062 | |
| ZINC91247728 | Antineoplastic (Lymphocytic Leukemia) | 0.447 | 0.007 |
| Antineoplastic (Non-Hodgkin’s Lymphoma) | 0.388 | 0.119 | |
| Antileukemic | 0.320 | 0.039 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, E.F.B.; Silva, L.B.; Costa, G.V.; Costa, J.S.; Fujishima, M.A.T.; Leão, R.P.; Ferreira, A.L.S.; Federico, L.B.; Silva, C.H.T.P.; Rosa, J.M.C.; et al. Identification of New Inhibitors with Potential Antitumor Activity from Polypeptide Structures via Hierarchical Virtual Screening. Molecules 2019, 24, 2943. https://doi.org/10.3390/molecules24162943
Ferreira EFB, Silva LB, Costa GV, Costa JS, Fujishima MAT, Leão RP, Ferreira ALS, Federico LB, Silva CHTP, Rosa JMC, et al. Identification of New Inhibitors with Potential Antitumor Activity from Polypeptide Structures via Hierarchical Virtual Screening. Molecules. 2019; 24(16):2943. https://doi.org/10.3390/molecules24162943
Chicago/Turabian StyleFerreira, Elenilze F. B., Luciane B. Silva, Glauber V. Costa, Josivan S. Costa, Mayara A. T. Fujishima, Rozires P. Leão, André L. S. Ferreira, Leonardo B. Federico, Carlos H. T. P. Silva, Joaquín M. C. Rosa, and et al. 2019. "Identification of New Inhibitors with Potential Antitumor Activity from Polypeptide Structures via Hierarchical Virtual Screening" Molecules 24, no. 16: 2943. https://doi.org/10.3390/molecules24162943