
Identification of Potential COX-2 Inhibitors for the Treatment of Inflammatory Diseases Using Molecular Modeling Approaches

 ,
,  , , and
, , and 
Abstract
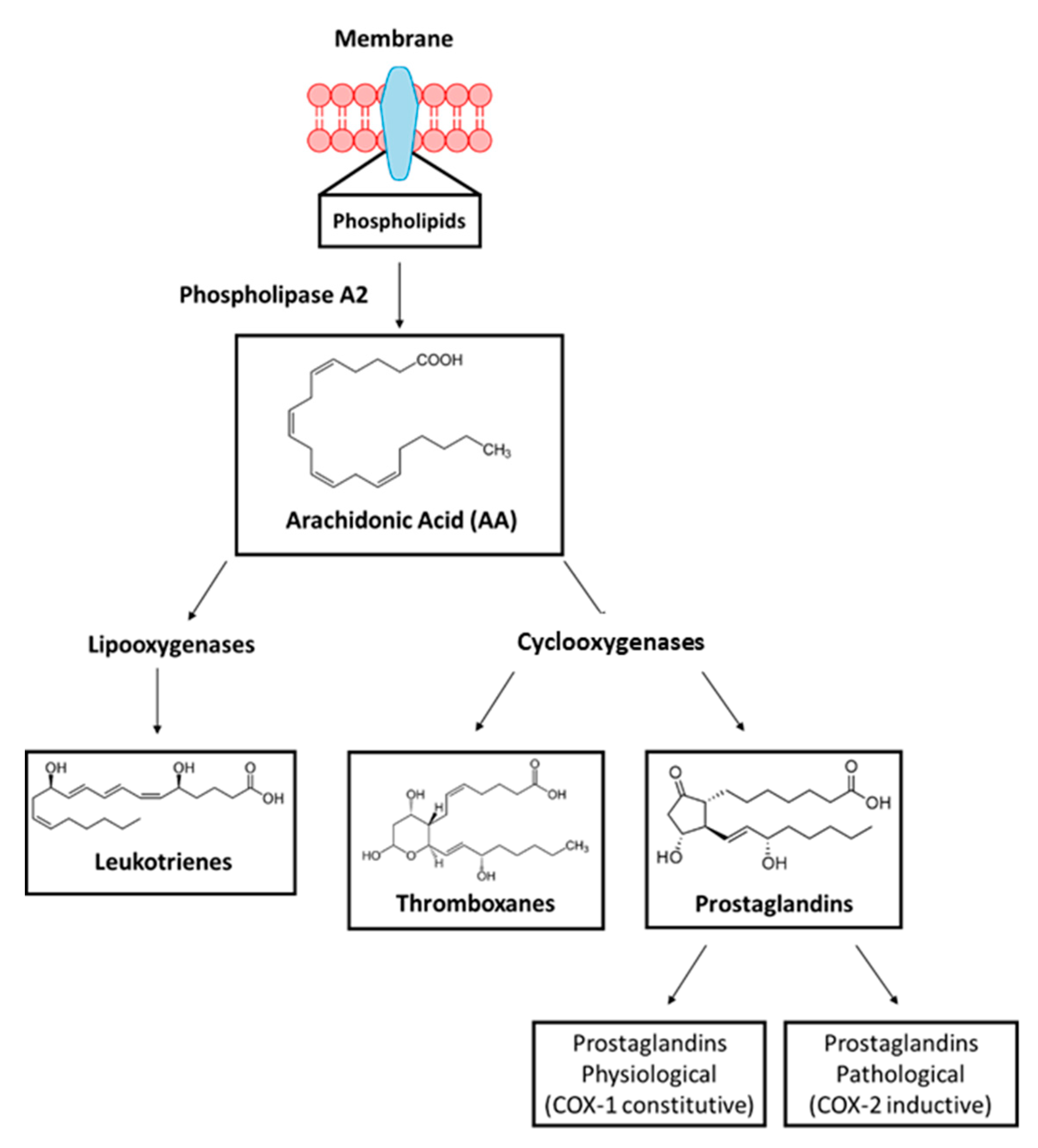
:1. Introduction
2. Results and Discussion
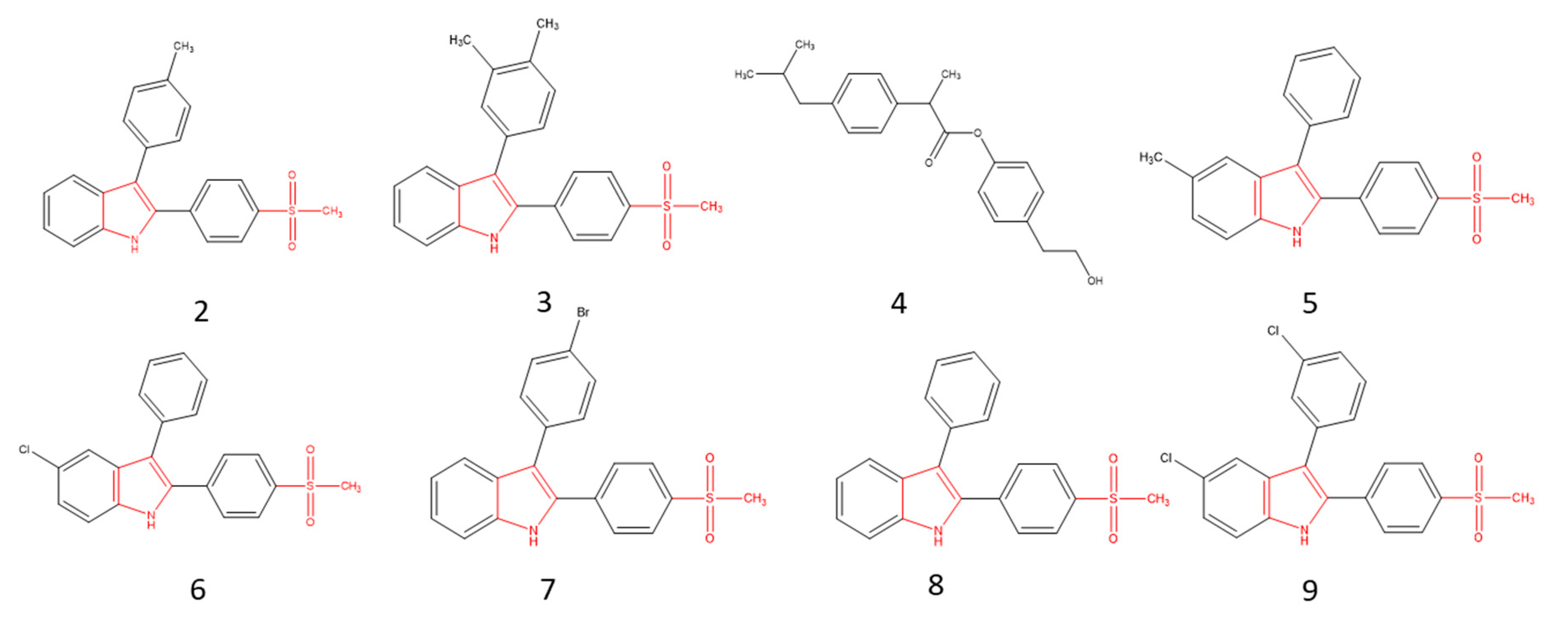
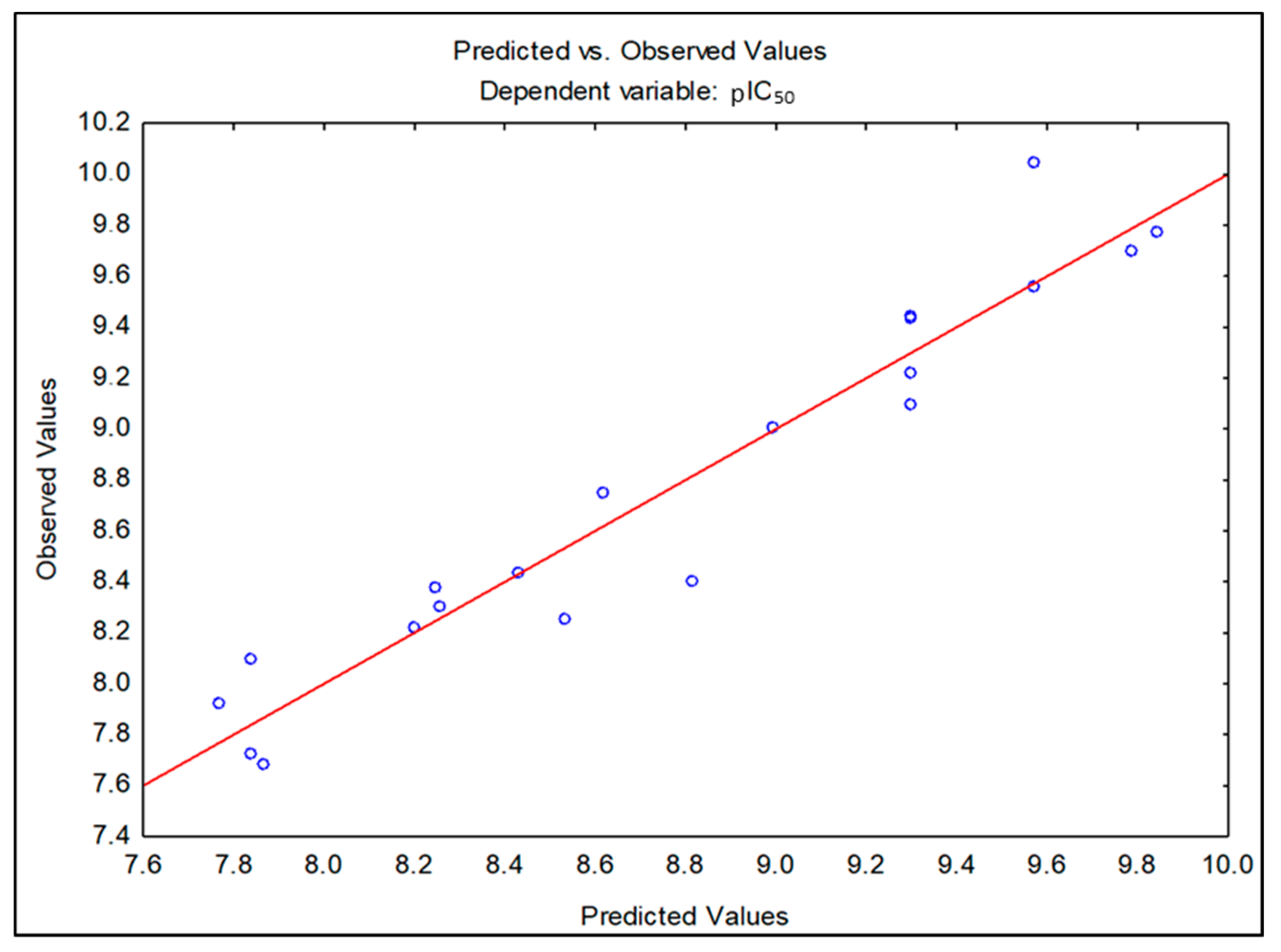
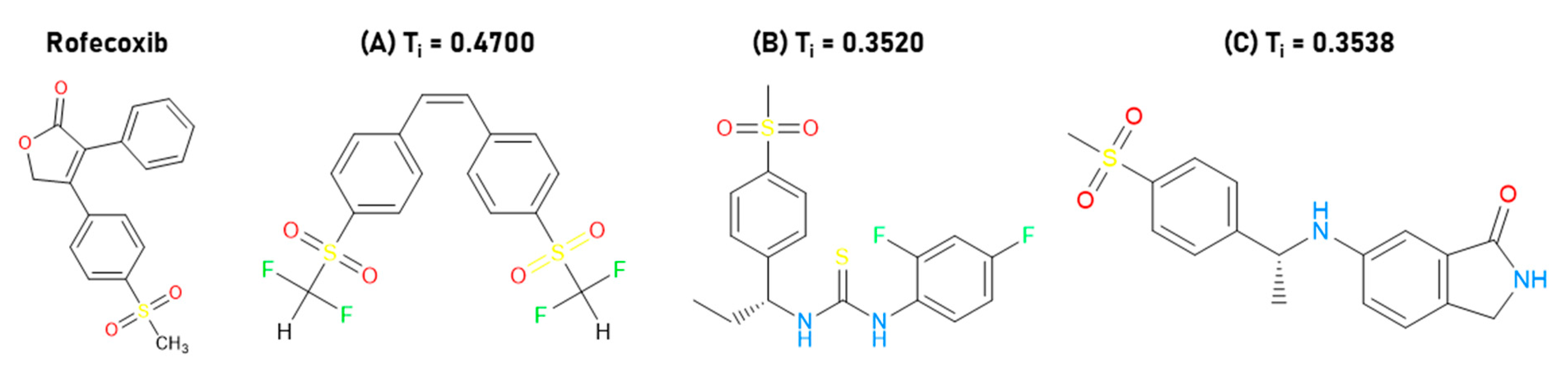
2.1. Molecular Optimization and QSAR Analysis
2.2. Virtual Screening and Analysis of Pharmacokinetic and Toxicological Properties
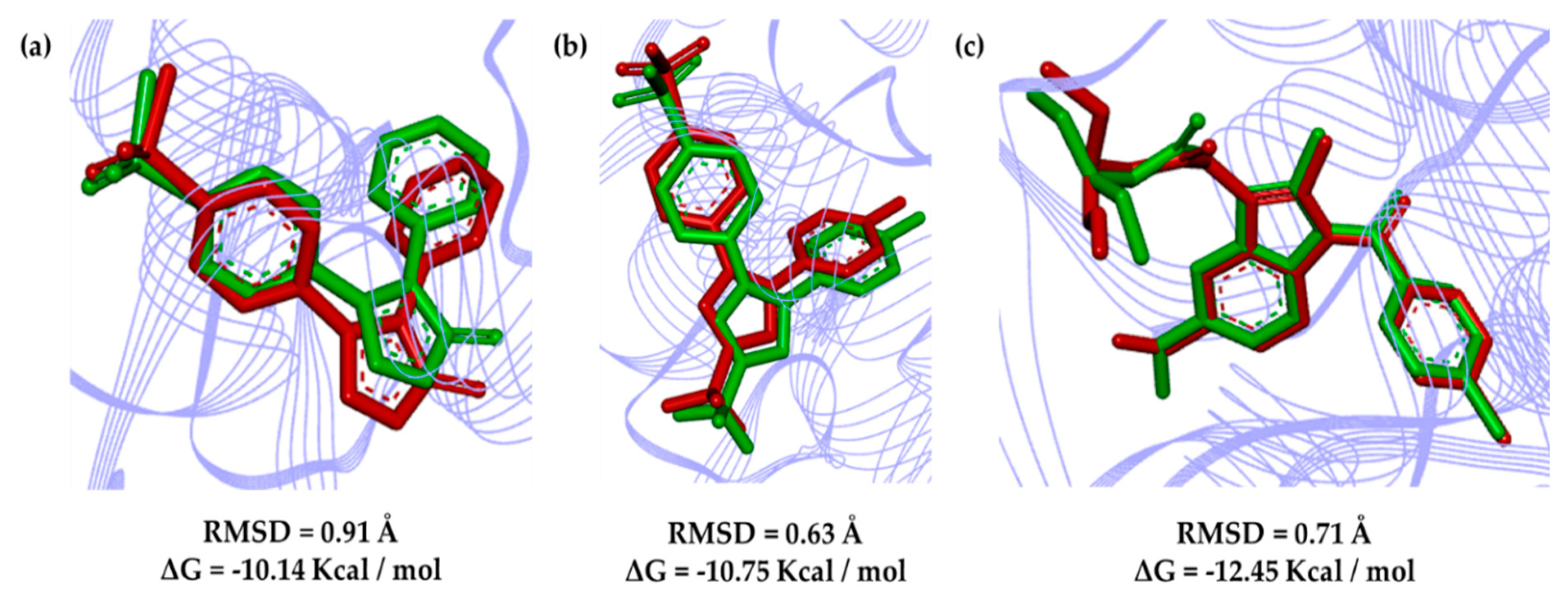
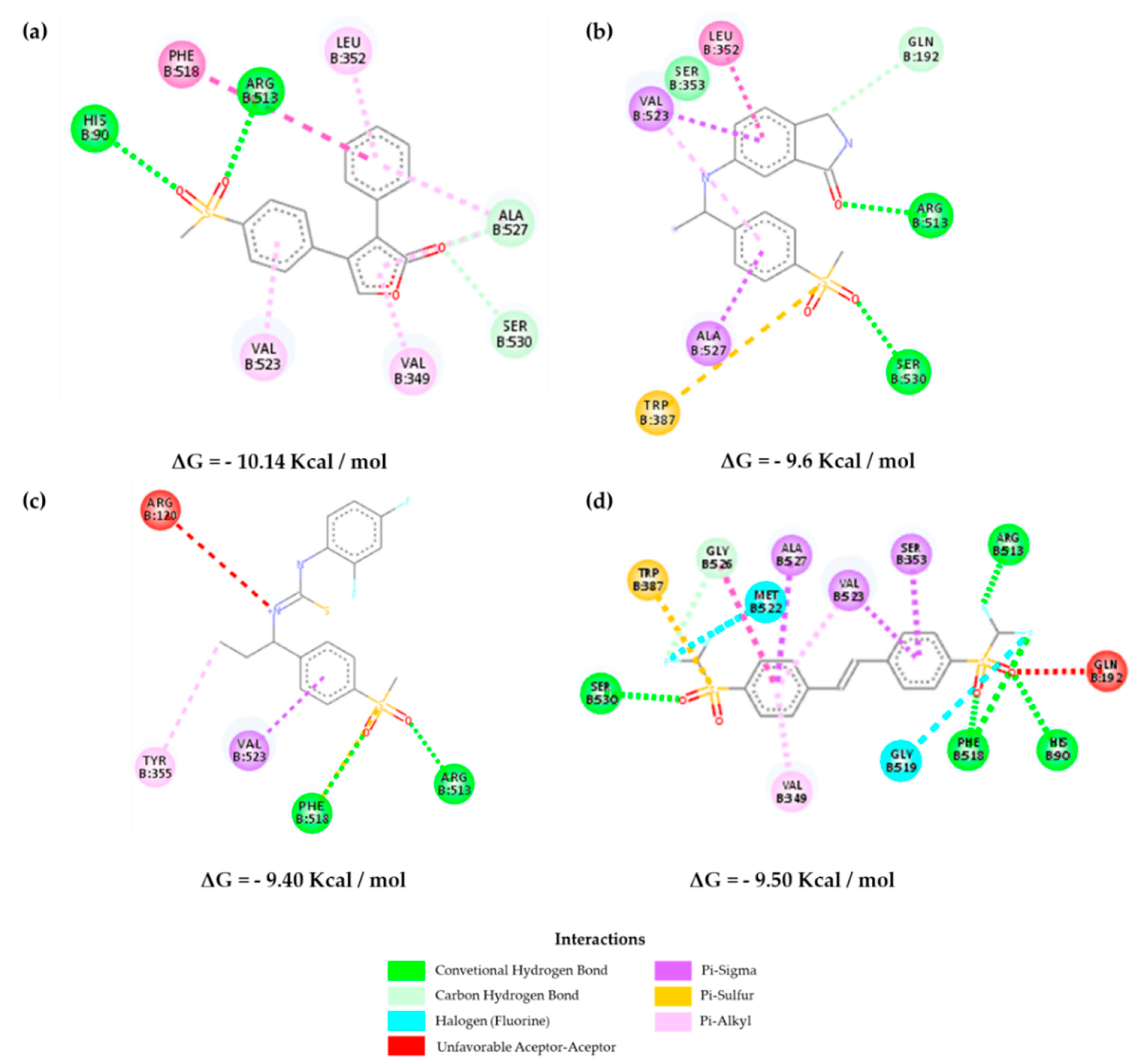
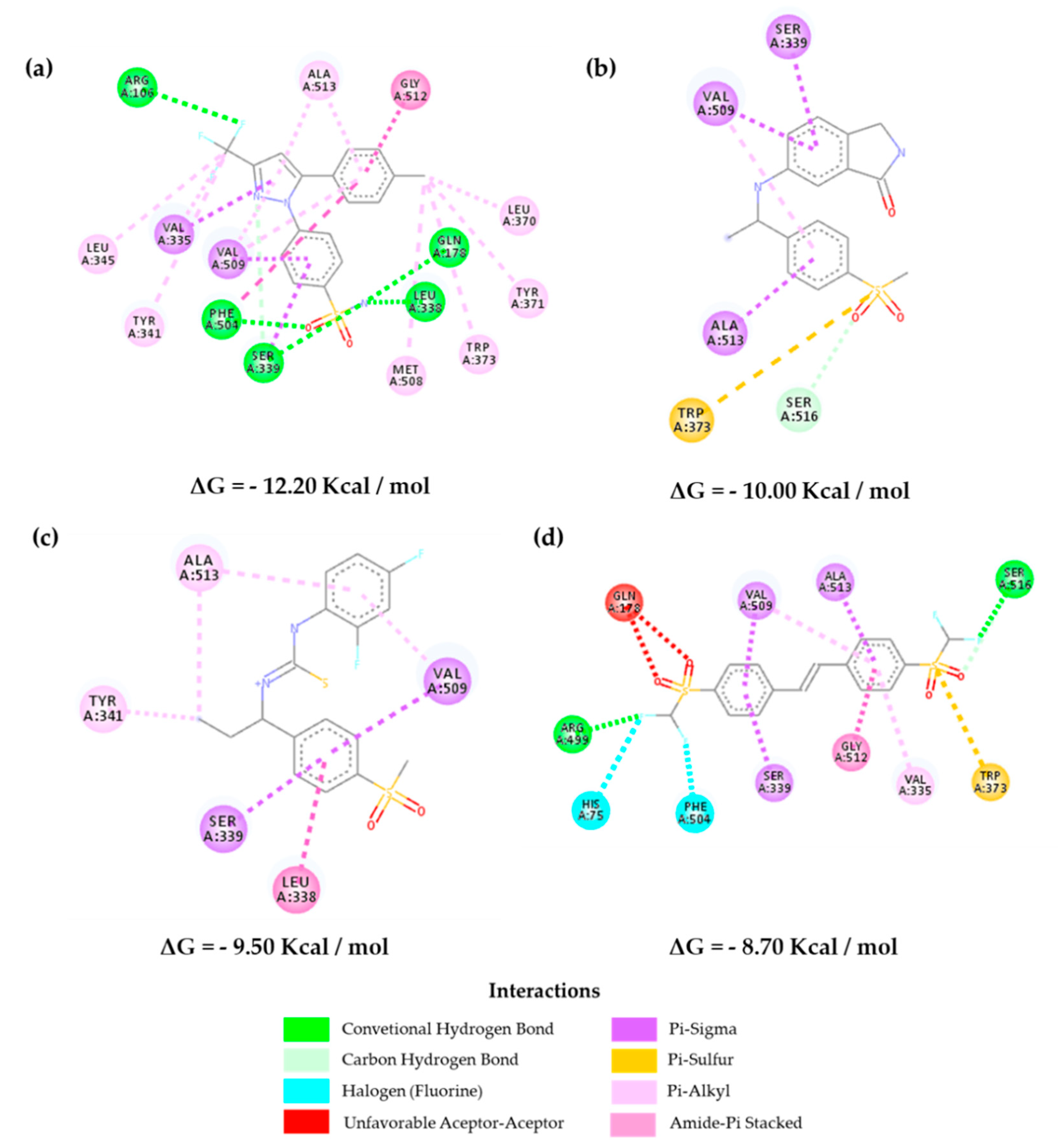
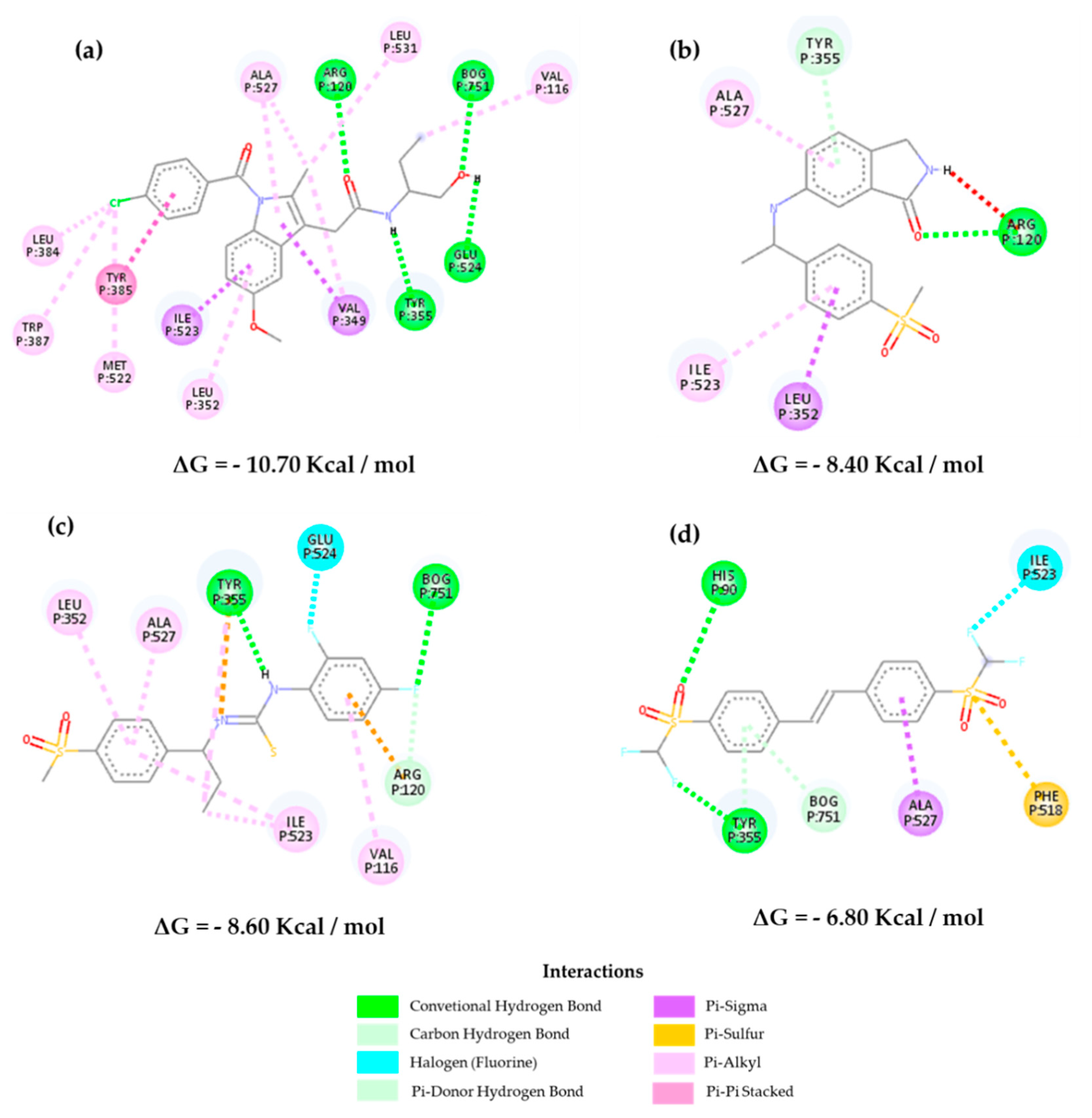
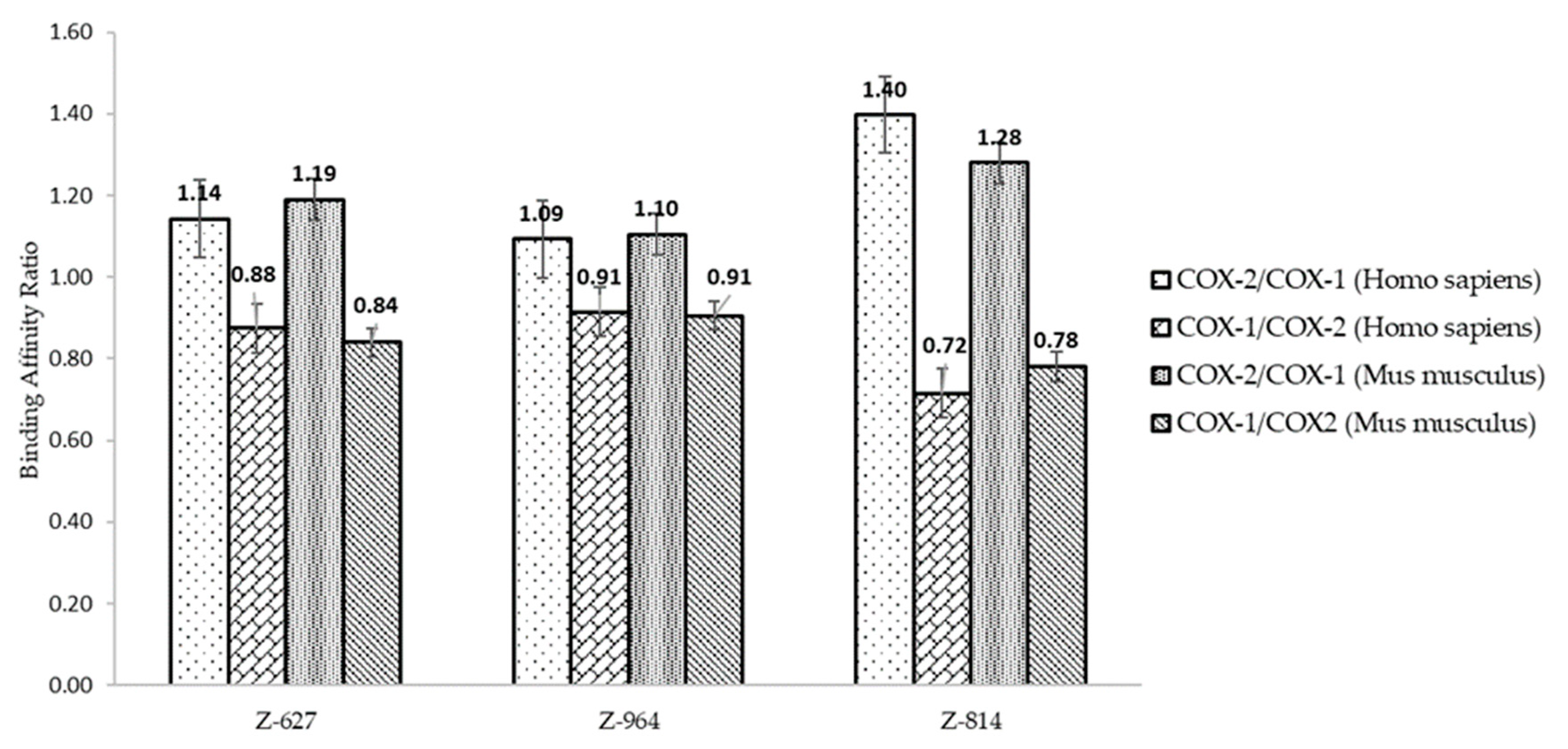
2.3. Molecular Docking
2.4. Structure–Activity Relationship of the Most Promising Molecules
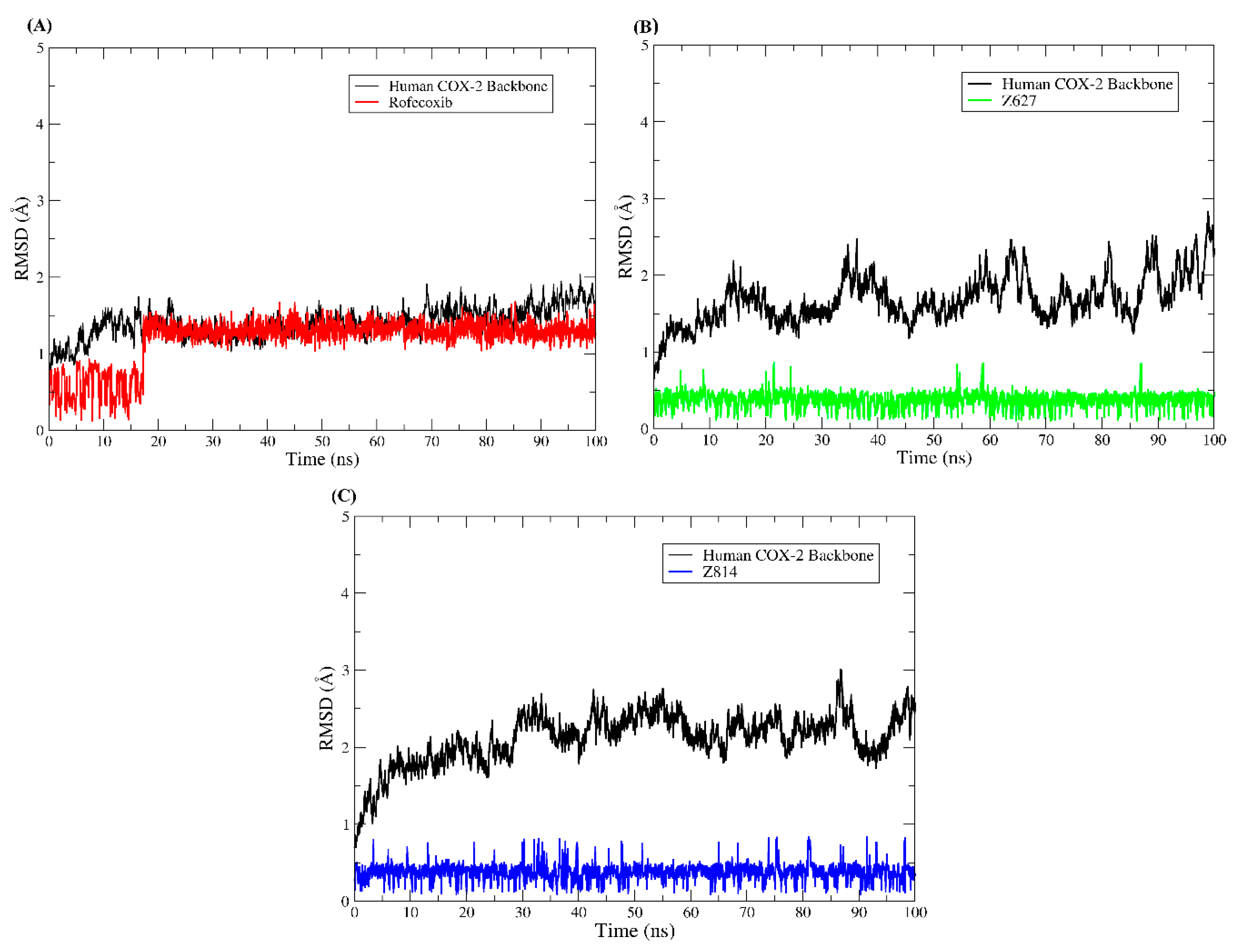
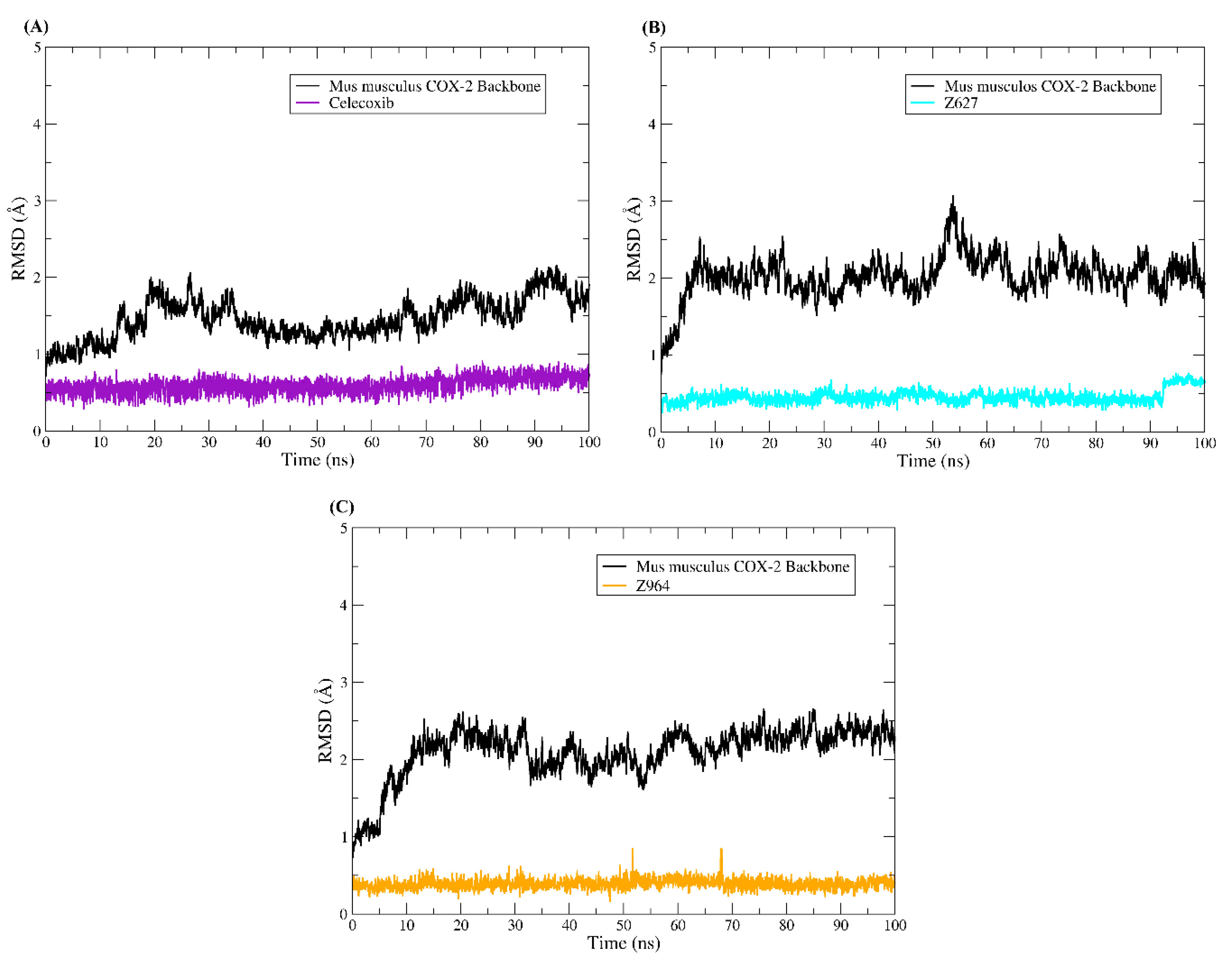
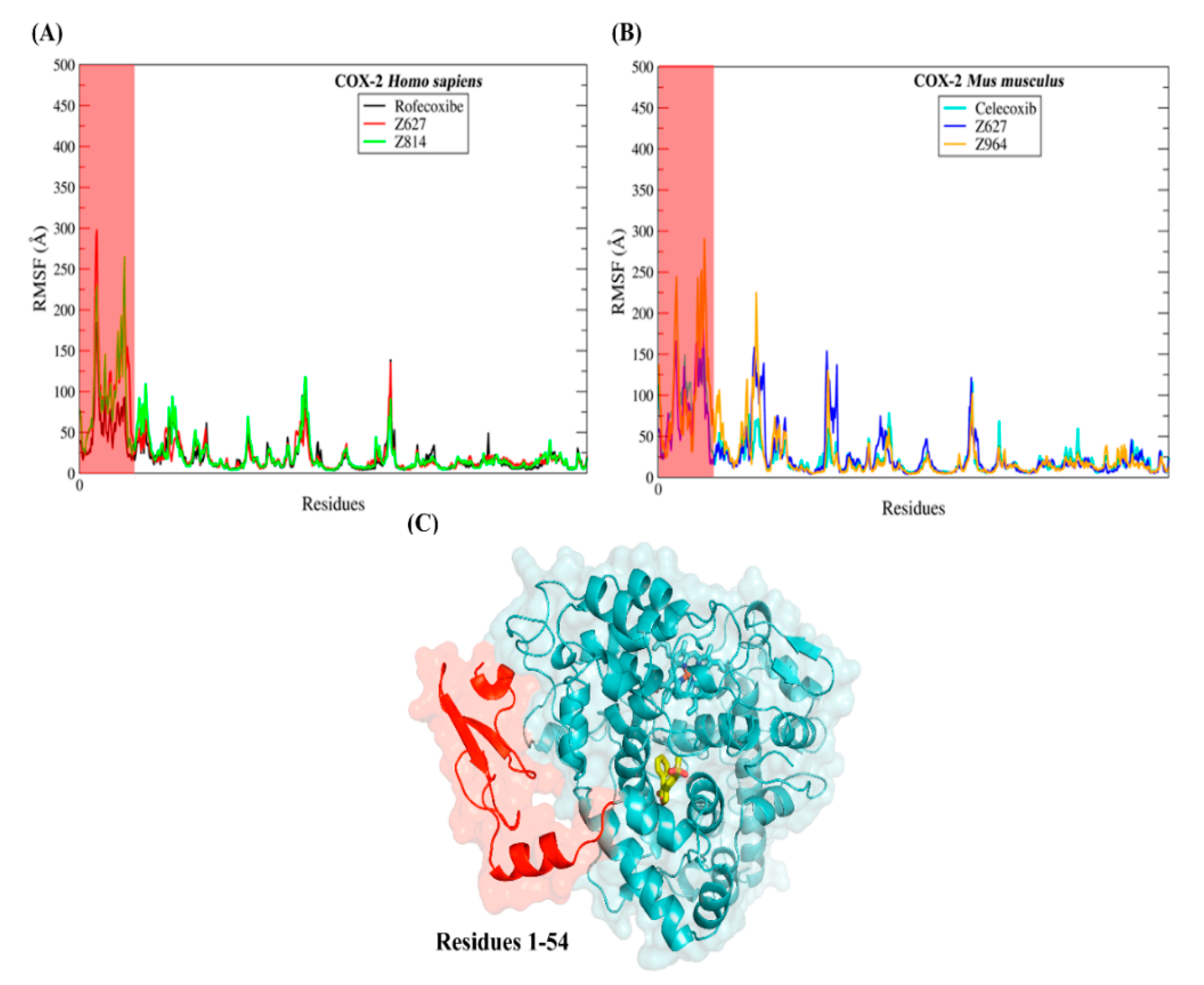
2.5. Molecular Dynamics Results and Affinity Energy
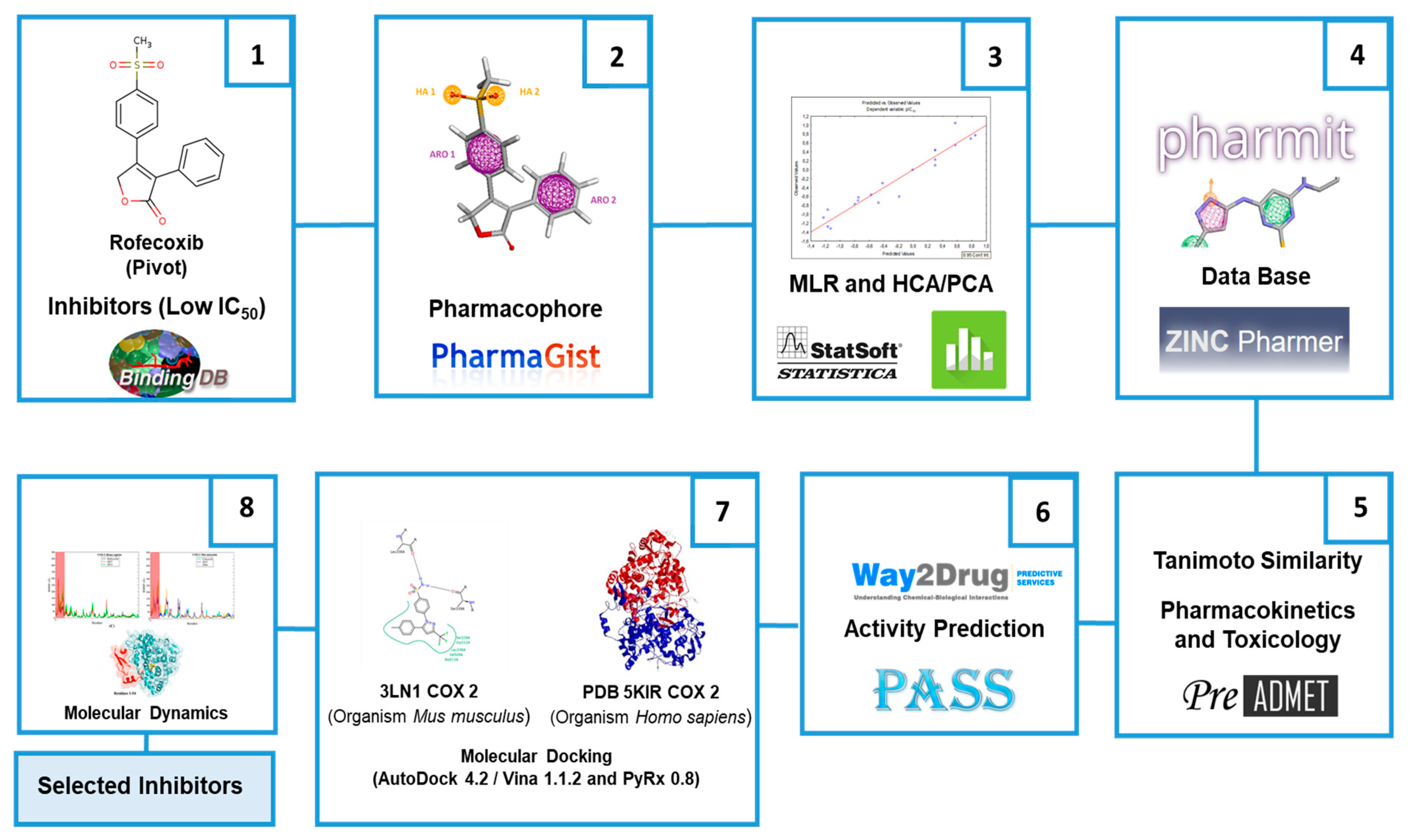
3. Materials and Methods
3.1. Selection of COX-2 Inhibitors
3.2. Optimization of Molecular Structures and Determination of Pharmacophore Characteristics
3.3. QSAR and PCA/HCA Studies
3.4. Virtual Screening and Selection of Inhibitor Compounds
3.5. Prediction of Toxicological and Pharmacokinetic Properties
3.6. Prediction of Biological Activity of Selected Inhibitors
3.7. Molecular Docking
Docking Study with AutoDock 4.2/Vina 1.1.2 via Graphical Interface PyRx (Version 0.8.30)
3.8. Molecular Dynamics Protocol
3.8.1. Affinity Energy Calculations
3.8.2. Per-Residue Free Energy Decomposition Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Kim, M.H.; Son, Y.-J.; Lee, S.Y.; Yang, W.S.; Yi, Y.-S.; Yoon, D.H.; Yang, Y.; Kim, S.H.; Lee, D.; Rhee, M.H.; et al. JAK2-targeted anti-inflammatory effect of a resveratrol derivative 2,4-dihydroxy-N-(4-hydroxyphenyl)benzamide. Biochem. Pharmacol. 2013, 86, 1747–1761. [Google Scholar] [CrossRef]
- Skinner, B.W.; Curtis, K.A.; Goodhart, A.L. Hypnotics and Sedatives. In Side Effects of Drugs Annual; Ray, S., Ed.; Elsevier: Indianapolis, IN, USA, 2018; Volume 40. [Google Scholar] [CrossRef]
- Jain, S.; Tran, S.; El Gendy, M.A.; Kashfi, K.; Jurasz, P.; Velázquez-Martínez, C.A. Nitric Oxide Release Is Not Required to Decrease the Ulcerogenic Profile of Nonsteroidal Anti-inflammatory Drugs. J. Med. Chem. 2012, 55, 688–696. [Google Scholar] [CrossRef]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Carvalho, W.A.; Carvalho, R.D.S.; Rios-Santos, F. Analgésicos inibidores específicos da ciclooxigenase-2: Avanços terapêuticos. Braz. J. Anesthesiol. 2004, 54, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Araujo, L.F.; Soeiro, A.D.M.; Fernandes, J.D.L.; Júnior, C.V.S. [Cardiovascular events: A class effect by COX-2 inhibitors]. Arq. Bras. Cardiol. 2005, 85, 222–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryer, B.; Duboisø, A. The advent of highly selective inhibitors of cyclooxygenase—A review. Prostaglandins Other Lipid Mediat. 1998, 56, 341–361. [Google Scholar] [CrossRef]
- Crofford, L.J. COX-1 and COX-2 tissue expression: Implications and predictions. J. Rheumatol. Suppl. 1997, 49, 15–19. [Google Scholar]
- Birck, M.G.; Campos, L.J.; De Melo, E.B. Computacional Study Of 1 H -Imidazol-2-Yl-Pyrimidine-4,6-Diamines For Identification Of Potential Parent Compounds Of New Antimalarial Agents. Quím. Nova 2016, 39, 567–574. [Google Scholar] [CrossRef]
- Munhoz, V.H.O.; Alcantara, A.F.C.; Pilo-Veloso, D. Conformational analysis by theoretical calculations of distinctin, an antimicrobial peptide isolated from Phyllomedusa distincta. Quím. Nova 2008, 31, 822–827. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.B.R.; Vieira, J.B.; Lobato, C.C.; Hage-Melim, L.I.S.; Souto, R.N.P.; Lima, C.S.; Costa, E.V.M.; Brasil, D.S.B.; Macêdo, W.J.D.C.; Carvalho, J.C.T. A SAR and QSAR Study of New Artemisinin Compounds with Antimalarial Activity. Molecules 2013, 19, 367–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, J.V.; Serafim, R.B.; Da Silva, G.M.; Giuliatti, S.; Rosa, J.M.C.; Neto, M.F.A.; Leite, F.H.A.; Taft, C.A.; Silva, C.; Santos, C.B.R. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. J. Mol. Model. 2018, 24, 225. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, K.L.B.; Cruz, J.N.; Silva, L.B.; Ramos, R.S.; Neto, M.F.A.; Lobato, C.C.; Ota, S.S.B.; Leite, F.H.A.; Borges, R.S.; Silva, C.; et al. Identification of Novel Chemical Entities for Adenosine Receptor Type 2A Using Molecular Modeling Approaches. Molecules 2020, 25, 1245. [Google Scholar] [CrossRef] [Green Version]
- E Clark, D. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J. Pharm. Sci. 1999, 88, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Malkowski, M.G. Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallogr. Sect. F Struct. Boil. Commun. 2016, 72, 772–776. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Sengupta, C.; Roy, K. Exploring QSAR with E-state index: Selectivity requirements for COX-2 versus COX-1 binding of terphenyl methyl sulfones and sulfonamides. Bioorgan. Med. Chem. Lett. 2004, 14, 4665–4670. [Google Scholar] [CrossRef]
- Leão, R.P.; Cruz, J.V.; Da Costa, G.V.; Cruz, J.N.; Ferreira, E.F.B.; Silva, R.C.; De Lima, L.R.; Borges, R.S.; Dos Santos, G.B.; Santos, C.B.R. Identification of New Rofecoxib-Based Cyclooxygenase-2 Inhibitors: A Bioinformatics Approach. Pharmaceuticals 2020, 13, 209. [Google Scholar] [CrossRef]
- Psimadas, D.; Georgoulias, P.; Valotassiou, V.; Loudos, G. Molecular Nanomedicine Towards Cancer: 111In-Labeled Nanoparticles. J. Pharm. Sci. 2012, 101, 2271–2280. [Google Scholar] [CrossRef]
- Yan, A.; Wang, Z.; Cai, Z. Prediction of Human Intestinal Absorption by GA Feature Selection and Support Vector Machine Regression. Int. J. Mol. Sci. 2008, 9, 1961–1976. [Google Scholar] [CrossRef]
- Wang, Q.; Rager, J.D.; Weinstein, K.; Kardos, P.S.; Dobson, G.L.; Li, J.; Hidalgo, I.J. Evaluation of the MDR-MDCK cell line as a permeability screen for the blood–brain barrier. Int. J. Pharm. 2005, 288, 349–359. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Le, J.; Abraham, M.H.; Hersey, A.; Eddershaw, P.J.; Luscombe, C.N.; Boutina, D.; Beck, G.; Sherborne, B.; Cooper, I.; et al. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure–activity relationship (QSAR) with the Abraham descriptors. J. Pharm. Sci. 2001, 90, 749–784. [Google Scholar] [CrossRef] [Green Version]
- Yazdanian, M.; Glynn, S.L.; Wright, J.L.; Hawi, A. Correlating partitioning and caco-2 cell permeability of structurally diverse small molecular weight compounds. Pharm. Res. 1998, 15, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, E.; Amaral, A.T.-D. BUSCA VIRTUAL DE COMPOSTOS BIOATIVOS: CONCEITOS E APLICAÇÕES. Quím. Nova 2018, 41, 662–677. [Google Scholar] [CrossRef]
- Ramos, R.S.; Costa, J.D.S.; Silva, R.C.; Da Costa, G.V.; Rodrigues, A.B.L.; Rabelo Érica, M.; Souto, R.N.P.; Taft, C.A.; Silva, C.; Campos, J.M.; et al. Identification of Potential Inhibitors from Pyriproxyfen with Insecticidal Activity by Virtual Screening. Pharmaceuticals 2019, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascarenhas, A.M.S.; De Almeida, R.B.M.; Neto, M.F.D.A.; Mendes, G.O.; Cruz, J.N.; Santos, C.B.R.; Botura, M.B.; Leite, F.H.A. Pharmacophore-based virtual screening and molecular docking to identify promising dual inhibitors of human acetylcholinesterase and butyrylcholinesterase. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Garavito, R.M.; Malkowski, M.G.; DeWitt, D.L. The structures of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat. 2002, 68, 129–152. [Google Scholar] [CrossRef]
- Magalhães, W.S.; Corrêa, C.M.; De Alencastro, R.B.; Nagem, T.J. Bases moleculares da ação anti-inflamatória dos ácidos oleanólico e ursólico sobre as isoformas da ciclo-oxigenase por docking e dinâmica molecular. Quím. Nova 2012, 35, 241–248. [Google Scholar] [CrossRef]
- Fox, P.W.; Hershberger, S.L.; Bouchard, T.J. Genetic and enviromental contributions to the acquisition of a motor skill. Nature 1996, 384, 356–358. [Google Scholar] [CrossRef]
- Limongelli, V.; Bonomi, M.; Marinelli, L.; Gervasio, F.L.; Cavalli, A.; Novellino, E.; Parrinello, M. Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc. Natl. Acad. Sci. 2010, 107, 5411–5416. [Google Scholar] [CrossRef] [Green Version]
- Inhibitor, S. Program Studi Biomedik, Fakultas Kedokteran Universitas Brawijaya, Malang, Indonesia Dosen Program Studi Pendidikan Biologi STKIP Pembangunan Indonesia Makassar 6. 2018. Available online: http://ojs.stkippi.ac.id/index.php/jip/article/view/128 (accessed on 19 August 2019).
- Pham, V.C.; Shin, J.-S.; Choi, M.-J.; Kim, T.-W.; Lee, K.-J.; Kim, K.-J.; Huh, G.; Kim, J.-A.; Choo, D.J.; Lee, K.-T.; et al. Biological Evaluation and Molecular Docking Study of 3-(4-Sulfamoylphenyl)-4-phenyl-1H-pyrrole-2,5-dione as COX-2 Inhibitor. Bull. Korean Chem. Soc. 2012, 33, 721–724. [Google Scholar] [CrossRef] [Green Version]
- Harman, C.A.; Turman, M.V.; Kozak, K.R.; Marnett, L.J.; Smith, W.L.; Garavito, R.M. Structural Basis of Enantioselective Inhibition of Cyclooxygenase-1 by S- -Substituted Indomethacin Ethanolamides. J. Boil. Chem. 2007, 282, 28096–28105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gierse, J.K.; Koboldt, C.M.; Walker, M.C.; Seibert, K.; Isakson, P.C. Kinetic basis for selective inhibition of cyclo-oxygenases. Biochem. J. 1999, 339, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.C.; Kurumbail, R.G.; Kiefer, J.R.; Moreland, K.T.; Koboldt, C.M.; Isakson, P.C.; Seibert, K.; Gierse, J.K. A three-step kinetic mechanism for selective inhibition of cyclo-oxygenase-2 by diarylheterocyclic inhibitors. Biochem. J. 2001, 357, 709–718. [Google Scholar] [CrossRef]
- Hayashi, S.; Ueno, N.; Murase, A.; Nakagawa, Y.; Takada, J. Novel acid-type cyclooxygenase-2 inhibitors: Design, synthesis, and structure–activity relationship for anti-inflammatory drug. Eur. J. Med. Chem. 2012, 50, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Nakata, E.; Morita, A.; Mizuno, K.; Yamamura, K.; Kato, A.; Ohashi, K. Discovery of {1-[4-(2-{hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl}-1H-benzimidazol-1-yl)piperidin-1-yl]cyclooctyl}methanol, systemically potent novel non-peptide agonist of nociceptin/orphanin FQ receptor as analgesic for the treatment of neuropathic pain: Design, synthesis, and structure–activity relationships. Bioorgan. Med. Chem. 2010, 18, 7675–7699. [Google Scholar] [CrossRef]
- Hayashi, S.; Hirao, A.; Imai, A.; Nakamura, H.; Murata, Y.; Ohashi, K.; Nakata, E. Novel Non-Peptide Nociceptin/Orphanin FQ Receptor Agonist, 1-[1-(1-Methylcyclooctyl)-4-piperidinyl]-2-[(3R)-3-piperidinyl]-1H-benzimidazole: Design, Synthesis, and Structure−Activity Relationship of Oral Receptor Occupancy in the Brain for Orally Potent Antianxiety Drug(1, 2). J. Med. Chem. 2009, 52, 610–625. [Google Scholar] [CrossRef]
- Silva, S.G.; Da Costa, R.A.; De Oliveira, M.S.; Cruz, J.N.; Figueiredo, P.L.B.; Brasil, D.D.S.B.; Nascimento, L.D.; Neto, A.D.J.C.; Junior, R.D.C.; Andrade, E.D.A. Chemical profile of Lippia thymoides, evaluation of the acetylcholinesterase inhibitory activity of its essential oil, and molecular docking and molecular dynamics simulations. PLoS ONE 2019, 14, e0213393. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, M.S.; Cruz, J.N.; Silva, S.G.; Da Costa, W.A.; De Sousa, S.H.B.; Bezerra, F.W.F.; Teixeira, E.; Da Silva, N.J.N.; Andrade, E.D.A.; Neto, A.M.D.J.C.; et al. Phytochemical profile, antioxidant activity, inhibition of acetylcholinesterase and interaction mechanism of the major components of the Piper divaricatum essential oil obtained by supercritical CO2. J. Supercrit. Fluids 2019, 145, 74–84. [Google Scholar] [CrossRef]
- Costa, E.; Silva, R.; Espejo-Román, J.; Neto, M.D.A.; Cruz, J.; Leite, F.; Silva, C.; Pinheiro, J.; Macêdo, W.; Santos, C. Chemometric methods in antimalarial drug design from 1,2,4,5-tetraoxanes analogues. SAR QSAR Environ. Res. 2020, 1–19. [Google Scholar] [CrossRef]
- Studio Discovery, Version 4.0; Accelrys Software Inc.: San Diego, CA, USA, 2009.
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [Green Version]
- Biava, M.; Porretta, G.C.; Poce, G.; De Logu, A.; Saddi, M.; Meleddu, R.; Manetti, F.; De Rossi, E.; Botta, M. 1,5-Diphenylpyrrole Derivatives as Antimycobacterial Agents. Probing the Influence on Antimycobacterial Activity of Lipophilic Substituents at the Phenyl Rings. J. Med. Chem. 2008, 51, 3644–3648. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Gupta, R.A.; Soni, L.K.; Kaskhedikar, S.G. Exploration of physicochemical properties and molecular modelling studies of 2-sulfonyl-phenyl-3-phenyl-indole analogs as cyclooxygenase-2 inhibitors. Eur. J. Med. Chem. 2008, 43, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Li, J.J.; Garland, D.J.; Chamberlain, T.S.; Reinhard, E.J.; Manning, R.E.; Seibert, K.; Koboldt, C.M.; Gregory, S.A.; Anderson, G.D.; et al. Diarylspiro[2.4]heptenes as Orally Active, Highly Selective Cyclooxygenase-2 Inhibitors: Synthesis and Structure−Activity Relationships. J. Med. Chem. 1996, 39, 253–266. [Google Scholar] [CrossRef]
- Navidpour, L.; Amini, M.; Shafaroodi, H.; Abdi, K.; Ghahremani, M.H.; Dehpour, A.R.; Shafiee, A. Design and synthesis of new water-soluble tetrazolide derivatives of celecoxib and rofecoxib as selective cyclooxygenase-2 (COX-2) inhibitors. Bioorgan. Med. Chem. Lett. 2006, 16, 4483–4487. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, S.S.; Jorgensen, W.L. Estimation of binding affinities for celecoxib analogues with COX-2 via Monte Carlo-extended linear response. Bioorgan. Med. Chem. Lett. 2002, 12, 267–270. [Google Scholar] [CrossRef]
- Habeeb, A.G.; Rao, P.N.P.; Knaus, E.E. Design and synthesis of 4,5-diphenyl-4-isoxazolines: Novel inhibitors of cyclooxygenase-2 with analgesic and antiinflammatory activity. J. Med. Chem. 2001, 44, 2921–2927. [Google Scholar] [CrossRef]
- Stumpfe, D.; Bajorath, J. Exploring Activity Cliffs in Medicinal Chemistry. J. Med. Chem. 2012, 55, 2932–2942. [Google Scholar] [CrossRef]
- Swarbrick, M.E.; Beswick, P.J.; Gleave, R.J.; Green, R.H.; Bingham, S.; Bountra, C.; Carter, M.C.; Chambers, L.J.; Chessell, I.P.; Clayton, N.M.; et al. Identification of [4-[4-(methylsulfonyl)phenyl]-6-(trifluoromethyl)-2-pyrimidinyl] amines and ethers as potent and selective cyclooxygenase-2 inhibitors. Bioorgan. Med. Chem. Lett. 2009, 19, 4504–4508. [Google Scholar] [CrossRef]
- Wang, J.L.; Limburg, D.; Graneto, M.J.; Springer, J.; Hamper, J.R.B.; Liao, S.; Pawlitz, J.L.; Kurumbail, R.G.; Maziasz, T.; Talley, J.J.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorgan. Med. Chem. Lett. 2010, 20, 7159–7163. [Google Scholar] [CrossRef]
- Xing, L.; Hamper, B.C.; Fletcher, T.R.; Wendling, J.M.; Carter, J.; Gierse, J.K.; Liao, S. Structure-based parallel medicinal chemistry approach to improve metabolic stability of benzopyran COX-2 inhibitors. Bioorgan. Med. Chem. Lett. 2011, 21, 993–996. [Google Scholar] [CrossRef]
- Li, J.J.; Anderson, G.D.; Burton, E.G.; Cogburn, J.N.; Collins, J.T.; Garland, D.J.; Gregory, S.A.; Huang, H.-C.; Isakson, P.C. 1,2-Diarylcyclopentenes as Selective Cyclooxygenase-2 Inhibitors and Orally Active Anti-inflammatory Agents. J. Med. Chem. 1995, 38, 4570–4578. [Google Scholar] [CrossRef] [PubMed]
- Orjales, A.; Mosquera, R.; López, B.; Olivera, R.; Labeaga, L.; Nunez, M.T. Novel 2-(4-methylsulfonylphenyl)pyrimidine derivatives as highly potent and specific COX-2 inhibitors. Bioorgan. Med. Chem. 2008, 16, 2183–2199. [Google Scholar] [CrossRef] [PubMed]
- Blobaum, A.L.; Marnett, L.J. Molecular Determinants for the Selective Inhibition of Cyclooxygenase-2 by Lumiracoxib. J. Boil. Chem. 2007, 282, 16379–16390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, J.D.S.; Costa, K.D.S.L.; Cruz, J.V.; Ramos, R.D.S.; Silva, L.B.; Brasil, D.D.S.B.; Silva, C.; Santos, C.B.R.; Macedo, W.J.D.C. Virtual Screening and Statistical Analysis in the Design of New Caffeine Analogues Molecules with Potential Epithelial Anticancer Activity. Curr. Pharm. Des. 2018, 24, 576–594. [Google Scholar] [CrossRef]
- Cruz, J.V.; Neto, M.F.A.; Silva, L.B.; Ramos, R.S.; Costa, J.D.S.; Brasil, D.S.B.; Lobato, C.C.; Da Costa, G.V.; Bittencourt, J.A.H.M.; Silva, C.; et al. Identification of Novel Protein Kinase Receptor Type 2 Inhibitors Using Pharmacophore and Structure-Based Virtual Screening. Molecules 2018, 23, 453. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, E.F.B.; Silva, L.B.; Da Costa, G.V.; Costa, J.D.S.; Fujishima, M.A.T.; Leão, R.P.; Ferreira, A.L.S.; Federico, L.B.; Silva, C.; Campos, J.M.; et al. Identification of New Inhibitors with Potential Antitumor Activity from Polypeptide Structures via Hierarchical Virtual Screening. Molecules 2019, 24, 2943. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.B.R.; Braga, F.S.; Santos, C.F.; Costa, J.D.S.; De Melo, G.S.; De Mello, M.N.; Sousa, D.S.; Carvalho, J.C.T.; Socorro, D.D.; Brasil, B.; et al. Antimalarial Artemisinins Derivatives Study: Molecular Modeling and Multivariate Analysis (PCA, HCA, KNN, SIMCA and SDA). J. Comput. Theor. Nanosci. 2015, 12, 3443–3458. [Google Scholar] [CrossRef]
- Ferreira, J.; Chaves, G.A.; Marino, B.L.B.; Sousa, K.P.A.; Souza, L.R.; Brito, M.F.B.; Teixeira, H.R.C.; Da Silva, C.H.T.P.; Santos, C.B.R.; Hage-Melim, L.I.S. Cannabinoid Type 1 Receptor (CB1) Ligands with Therapeutic Potential for Withdrawal Syndrome in Chemical Dependents ofCannabis sativa. Chem. Med. Chem. 2017, 12, 1408–1416. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cieplak, P.; Kollman, P.A. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Pinto, V.D.S.; Araújo, J.S.C.; Silva, R.C.; Da Costa, G.V.; Cruz, J.N.; Neto, M.F.A.; Campos, J.M.; Santos, C.B.R.; Leite, F.H.A.; Junior, M.C.S. In Silico Study to Identify New Antituberculosis Molecules from Natural Sources by Hierarchical Virtual Screening and Molecular Dynamics Simulations. Pharmaceuticals 2019, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.A.; Cruz, J.N.; Nascimento, F.C.A.; Silva, S.G.; Silva, S.O.; Martelli, M.C.; Carvalho, S.M.L.; Santos, C.B.R.; Neto, A.M.J.C.; Brasil, D.S.B. Studies of NMR, molecular docking, and molecular dynamics simulation of new promising inhibitors of cruzaine from the parasite Trypanosoma cruzi. Med. Chem. Res. 2018, 28, 246–259. [Google Scholar] [CrossRef]
- Alves, F.S.; Rego, J.D.A.R.D.; Da Costa, M.L.; Da Silva, L.F.L.; Da Costa, R.A.; Cruz, J.N.; Brasil, D.D.S.B.; Brazil, D.D.S.B. Spectroscopic methods and in silico analyses using density functional theory to characterize and identify piperine alkaloid crystals isolated from pepper (Piper Nigrum L.). J. Biomol. Struct. Dyn. 2019, 38, 2792–2799. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, J.N.; De Oliveira, M.S.; Silva, S.G.; Filho, A.P.D.S.S.; Pereira, D.S.; E Lima, A.H.L.; Andrade, E.H.D.A. Insight into the Interaction Mechanism of Nicotine, NNK, and NNN with Cytochrome P450 2A13 Based on Molecular Dynamics Simulation. J. Chem. Inf. Model. 2019, 60, 766–776. [Google Scholar] [CrossRef]
- Santos, C.B.R.; Dos Santos, K.L.B.; Cruz, J.N.; Leite, F.H.A.; Borges, R.S.; Taft, C.A.; Campos, J.M.; Silva, C. Molecular modeling approaches of selective adenosine receptor type 2A agonists as potential anti-inflammatory drugs. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C.L. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, J.A.; Catarello, D.P.; Skeel, R.D.; Wozniak, J.M. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef]
- Cruz, J.N.; Da Costa, K.S.; De Carvalho, T.A.A.; De Alencar, N.A.N. Measuring the structural impact of mutations on cytochrome P450 21A2, the major steroid 21-hydroxylase related to congenital adrenal hyperplasia. J. Biomol. Struct. Dyn. 2019, 38, 1425–1434. [Google Scholar] [CrossRef]
- Da Costa, K.S.; Galúcio, J.M.; Da Costa, C.H.S.; Santana, A.R.; Carvalho, V.D.S.; Nascimento, L.D.D.; E Lima, A.H.L.; Cruz, J.N.; Alves, C.N.; Lameira, J. Exploring the Potentiality of Natural Products from Essential Oils as Inhibitors of Odorant-Binding Proteins: A Structure- and Ligand-Based Virtual Screening Approach To Find Novel Mosquito Repellents. ACS Omega 2019, 4, 22475–22486. [Google Scholar] [CrossRef] [Green Version]
- Vale, V.V.; Cruz, J.N.; Viana, G.M.R.; Póvoa, M.M.; Brasil, D.D.S.B.; Dolabela, M.F. Naphthoquinones isolated from Eleutherine plicata herb: In Vitro antimalarial activity and molecular modeling to investigate their binding modes. Med. Chem. Res. 2020, 29, 487–494. [Google Scholar] [CrossRef]
- Ramos, R.S.; Macêdo, W.J.C.; Costa, J.S.; Silva, C.H.T.D.P.D.; Rosa, J.M.C.; Da Cruz, J.N.; De Oliveira, M.S.; Andrade, E.H.D.A.; E Silva, R.B.L.; Souto, R.N.P.; et al. Potential inhibitors of the enzyme acetylcholinesterase and juvenile hormone with insecticidal activity: Study of the binding mode via docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 1–23. [Google Scholar] [CrossRef]
- Cruz, J.N.; Costa, J.F.S.; Khayat, A.S.; Kuca, K.; Barros, C.A.L.; Neto, A.M.J.C. Molecular dynamics simulation and binding free energy studies of novel leads belonging to the benzofuran class inhibitors of Mycobacterium tuberculosis Polyketide Synthase 13. J. Biomol. Struct. Dyn. 2018, 37, 1616–1627. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds not available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Characteristics | |||||
|---|---|---|---|---|---|---|
| ATM a | ARO b | DONN c | ACC d | pIC50 | IC50 (nM) | |
| 1 * | 36 | 2 | 0 | 4 | 7.9208 | 12.00 |
| 2 | 45 | 4 | 1 | 2 | 10.0457 | 0.09 |
| 3 | 48 | 4 | 1 | 2 | 9.7695 | 0.17 |
| 4 | 50 | 2 | 1 | 3 | 9.6989 | 0.20 |
| 5 | 45 | 4 | 1 | 2 | 9.5528 | 0.28 |
| 6 | 42 | 4 | 1 | 2 | 9.4436 | 0.36 |
| 7 | 42 | 4 | 1 | 2 | 9.4317 | 0.37 |
| 8 | 42 | 4 | 1 | 2 | 9.2218 | 0.60 |
| 9 | 42 | 4 | 1 | 2 | 9.0969 | 0.80 |
| 10 | 42 | 3 | 1 | 3 | 9.0000 | 1.00 |
| 11 | 37 | 2 | 3 | 5 | 8.7447 | 1.80 |
| 12 | 44 | 3 | 1 | 5 | 8.4317 | 3.70 |
| 13 | 40 | 3 | 1 | 3 | 8.3979 | 4.00 |
| 14 | 42 | 3 | 0 | 4 | 8.3767 | 4.20 |
| 15 | 29 | 2 | 1 | 2 | 8.3000 | 5.00 |
| 16 | 41 | 3 | 1 | 4 | 8.2518 | 5.60 |
| 17 | 40 | 1 | 0 | 4 | 8.2218 | 6.00 |
| 18 | 36 | 1 | 0 | 4 | 8.0969 | 8.00 |
| 19 | 36 | 1 | 0 | 4 | 7.7212 | 19.00 |
| 20 | 33 | 2 | 0 | 3 | 7.6777 | 21.00 |
| ATM | 1.0000 | 0.5659 | 0.2027 | −0.2280 | 0.7651 | − |
| ARO | 1.0000 | 0.3559 | −0.6492 | 0.7358 | − | |
| DONN | 1.0000 | −0.0423 | 0.4743 | − | ||
| ACC | 1.0000 | −0.6399 | − | |||
| Mono Parametric | ||||||
| Eq. | Descriptors used | R | R2 | RA2 | SEE | F |
| 1 | ATM | 0.7651 | 0.5854 | 0.5623 | 0.4804 | 25.4183 |
| 2 | ARO | 0.7358 | 0.5414 | 0.5159 | 0.5053 | 21.2522 |
| 3 | ACC | 0.6399 | 0.4095 | 0.3767 | 0.5733 | 12.4871 |
| 4 | DONN | 0.4743 | 0.2249 | 0.1819 | 0.6569 | 5.2251 |
| Bi Parametric | ||||||
| Eq. | Descriptors used | R | R2 | RA2 | SEE | F |
| 1 | ATM + ACC | 0.9022 | 0.8140 | 0.7921 | 0.3311 | 37.2030 |
| 2 | ARO + ATM | 0.8487 | 0.7203 | 0.6874 | 0.4060 | 21.8906 |
| 3 | ATM + DONN | 0.8316 | 0.6916 | 0.6554 | 0.4263 | 19.0684 |
| 4 | ACC + DONN | 0.7809 | 0.6099 | 0.5640 | 0.4795 | 13.2914 |
| 5 | ARO + DONN | 0.7701 | 0.5930 | 0.5452 | 0.4898 | 12.3893 |
| 6 | ARO + ACC | 0.7661 | 0.5869 | 0.5383 | 0.4934 | 12.0789 |
| Tri Parametric | ||||||
| Eq. | Descriptors used | R | R2 | RA2 | SEE | F |
| 1 | ATM + ACC + DONN | 0.9599 | 0.9215 | 0.9068 | 0.2217 | 62.6376 |
| 2 | ARO + ATM + ACC | 0.9053 | 0.8197 | 0.7859 | 0.3360 | 24.2481 |
| 3 | ARO + ACC + DONN | 0.8207 | 0.6736 | 0.6125 | 0.4521 | 11.0113 |
| Tetra Parametric | ||||||
| Eq. | Descriptors used | R | R2 | RA2 | SEE | F |
| 1 | ATM + ACC + DONN + ARO | 0.9617 | 0.9250 | 0.9050 | 0.2238 | 46.2719 |
| Molecule | pIC50 | Mono | Bi | Tri | Tetra | ||||
|---|---|---|---|---|---|---|---|---|---|
| Eq. (1) | Δ1 | Eq. (2) | Δ2 | Eq. (3) | Δ3 | Eq. (4) | Δ4 | ||
| 1 * | 7.9208 | 8.2562 | −0.3354 | 8.0305 | −0.1097 | 7.7961 | 0.1247 | 7.7670 | 0.1538 |
| 2 | 10.0457 | 9.2561 | 0.7896 | 9.5495 | 0.4962 | 9.5773 | 0.4684 | 9.5693 | 0.4764 |
| 3 | 9.7695 | 9.5894 | 0.1801 | 9.8339 | −0.0644 | 9.832 | −0.0625 | 9.8414 | −0.0719 |
| 4 | 9.6989 | 9.8116 | −0.1127 | 9.6906 | 0.0083 | 9.6680 | 0.0309 | 9.7855 | −0.0866 |
| 5 | 9.5528 | 9.2561 | 0.2967 | 9.5495 | 0.0033 | 9.5773 | −0.0245 | 9.5693 | −0.0165 |
| 6 | 9.4436 | 8.9228 | 0.5208 | 9.2651 | 0.1785 | 9.3226 | 0.1210 | 9.2972 | 0.1464 |
| 7 | 9.4317 | 8.9228 | 0.5089 | 9.2651 | 0.1666 | 9.3226 | 0.1091 | 9.2972 | 0.1345 |
| 8 | 9.2218 | 8.9228 | 0.2990 | 9.2651 | −0.0433 | 9.3226 | −0.1008 | 9.2972 | −0.0754 |
| 9 | 9.0969 | 8.9228 | 0.1741 | 9.2651 | −0.1682 | 9.3226 | −0.2257 | 9.2972 | −0.2003 |
| 10 | 9.0000 | 8.9228 | 0.0772 | 8.9322 | 0.0678 | 8.9888 | 0.0112 | 8.9925 | 0.0075 |
| 11 | 8.7447 | 8.3673 | 0.3774 | 7.7924 | 0.9523 | 8.5957 | 0.1490 | 8.6154 | 0.1293 |
| 12 | 8.4317 | 9.1450 | −0.7133 | 8.4560 | −0.0243 | 8.4910 | −0.0593 | 8.4297 | 0.0020 |
| 13 | 8.3979 | 8.7006 | −0.3027 | 8.7426 | −0.3447 | 8.8190 | −0.4211 | 8.8111 | −0.4132 |
| 14 | 8.3767 | 8.9228 | −0.5461 | 8.5993 | −0.2226 | 8.3055 | 0.0712 | 8.2438 | 0.1329 |
| 15 | 8.3000 | 7.4785 | 0.8215 | 8.0327 | 0.2673 | 8.2189 | 0.0811 | 8.2529 | 0.0471 |
| 16 | 8.2518 | 8.8117 | −0.5599 | 8.5045 | −0.2527 | 8.5701 | −0.3183 | 8.5297 | −0.2779 |
| 17 | 8.2218 | 8.7006 | −0.4788 | 8.4097 | −0.1879 | 8.1357 | 0.0861 | 8.1972 | 0.0246 |
| 18 | 8.0969 | 8.2562 | −0.1593 | 8.0305 | 0.0664 | 7.7961 | 0.3008 | 7.8344 | 0.2625 |
| 19 | 7.7212 | 8.2562 | −0.5350 | 8.0305 | −0.3093 | 7.7961 | −0.0749 | 7.8344 | −0.1132 |
| 20 | 7.6777 | 7.9229 | −0.2452 | 8.0790 | −0.4013 | 7.8752 | −0.1975 | 7.8670 | −0.1893 |
| 21[a] | 9.3010 | 9.0339 | 0.2671 | 9.0270 | 0.2740 | 8.7242 | 0.5768 | 8.7066 | 0.5944 |
| 22[a] | 8.8508 | 8.9228 | −0.0720 | 9.2651 | −0.4143 | 9.3226 | −0.4718 | 9.2972 | −0.4464 |
| 23[a] | 8.8239 | 9.3672 | −0.5433 | 9.6443 | −0.8204 | 9.3127 | −0.4888 | 9.3508 | −0.5269 |
| 24[a] | 8.6990 | 8.8117 | −0.1127 | 8.8374 | −0.1384 | 9.2534 | −0.5544 | 9.2110 | −0.5120 |
| 25[a] | 8.4815 | 9.0339 | −0.5524 | 9.3599 | −0.8784 | 9.0580 | −0.5765 | 9.0787 | −0.5972 |
| Prediction Equations | |||||||||
| Mono | pIC50 = 4.2566 + (0.1111 × ATM) | ||||||||
| Bi | pIC50 = 5.9493 + (0.0948 ×ATM) + (−0.3329 × ACC) | ||||||||
| Tri | pIC50 = 6.0749 + (0.0849 × ATM) + (−0.3338 × ACC) + (0.3495 × DONN) | ||||||||
| Tetra | pIC50 = 6.1250 + (0.0907 × ATM) + (−0.3721 × ACC) + (0.3766 × DONN) + (−0.0674 × ARO) | ||||||||
| Molecule | pIC50 | Mono | Bi | Tri | Tetra | ||||
|---|---|---|---|---|---|---|---|---|---|
| Eq. (1) | Δ1 | Eq. (2) | Δ2 | Eq. (3) | Δ3 | Eq. (4) | Δ4 | ||
| 26 | 9.7447 | 9.3672 | 0.3775 | 9.3114 | 0.4333 | 9.6779 | 0.0668 | 9.6645 | 0.0802 |
| 27 | 9.2676 | 8.8117 | 0.4559 | 8.8374 | 0.4302 | 9.2534 | 0.0142 | 9.2110 | 0.0566 |
| 28 | 8.8508 | 8.9228 | −0.0720 | 9.2651 | −0.4143 | 9.3226 | −0.4718 | 9.2972 | −0.4464 |
| 29 | 8.6198 | 9.7005 | −1.0807 | 8.2642 | 0.3556 | 8.2479 | 0.3719 | 8.1390 | 0.4808 |
| 30 | 8.2757 | 8.3673 | −0.0916 | 8.1253 | 0.1504 | 8.2305 | 0.0452 | 8.1669 | 0.1088 |
| 31 | 8.1549 | 7.8118 | 0.3431 | 8.3171 | −0.1622 | 8.4736 | −0.3187 | 8.5250 | −0.3701 |
| Celecoxib | 9.2839 | 8.7006 | 0.5833 | 8.4097 | 0.8742 | 8.4852 | 0.7987 | 8.4390 | 0.8449 |
 | Pharmacophore Characteristics | Coordinates | |||
| x | y | z | Radius | ||
| Hydrogen Acceptor (HA1) | 15.30 | −10.69 | −1.79 | 0.50 | |
| Hydrogen Acceptor (HA2) | 17.52 | −11.00 | −2.88 | 0.50 | |
| Aromatic (ARO1) | 17.28 | −7.30 | −1.54 | 1.10 | |
| Aromatic (ARO2) | 21.57 | −4.64 | −1.16 | 1.10 | |
| Zinc | Tanimoto Index (Ti) | ||
|---|---|---|---|
| >0.25 | >0.30 | >0.35 | |
| Top Hits | 1451 | 491 | 58 |
| Molecule | Toxicity | ||||
|---|---|---|---|---|---|
| LD50 (mg/kg) [a] | Toxicity Class [a] | Ames_test | Carcino-Mouse | Carcino-Rat | |
| Rofecoxib | 4500 | V | Mutagen | Negative | Negative |
| Celecoxib | 1400 | IV | Mutagen | Negative | Negative |
| Z-814 | 2000 | IV | Non-mutagen | Negative | Negative |
| Z-627 | 61 | III | Non-mutagen | Negative | Negative |
| Z-964 | 5000 | V | Non-mutagen | Negative | Negative |
| Inhibitor | Distribution | |
|---|---|---|
| Cbrain/CBlood [a] | PPB (%) [b] | |
| Rofecoxib | 0.0137 | 94.8036 |
| Celecoxib | 0.0272 | 91.0772 |
| Z-814 | 0.9553 | 95.5607 |
| Z-627 | 0.0344 | 85.3783 |
| Z-964 | 0.4328 | 100.0000 |
| Inhibitor | Absorption | |||
|---|---|---|---|---|
| PCaco-2 (nm/sec) [a] | HIA (%) [b] | PMDCK (nm/sec) [c] | P-gp inhibition [d] | |
| Rofecoxib | 2.7294 | 98.2248 | 11.2740 | Non |
| Celecoxib | 0.4994 | 96.6870 | 45.0486 | Inhibitor |
| Z-814 | 12.4185 | 98.6322 | 28.3061 | Inhibitor |
| Z-627 | 0.6460 | 94.4692 | 1.4352 | Non |
| Z-964 | 42.9100 | 95.5974 | 0.0517 | Inhibitor |
| Inhibitor | Pa [a] | Pi [b] | Biological Activity | Pa [a] | Pi [b] | Adverse Effects |
|---|---|---|---|---|---|---|
| Rofecoxib | 0.951 | 0.004 | Antiarthritic | 0.831 | 0.006 | Extrapyramidal effect |
| 0.855 | 0.004 | Non-Steroidal Anti-Inflammatory | 0.640 | 0.006 | Ulcer, peptic | |
| 0.828 | 0.005 | Anti-Inflammatory | 0.531 | 0.040 | Ulceration | |
| 0.726 | 0.002 | Cyclooxygenase-2 Inhibitor | 0.385 | 0.024 | Carcinogenic, group 1 | |
| Celecoxib | 0.940 | 0.001 | Cyclooxygenase Inhibitor | 0.671 | 0.016 | Pseudoporphyria |
| 0.936 | 0.001 | Cyclooxygenase-2 Inhibitor | 0.569 | 0.013 | Ulcer, peptic | |
| 0.809 | 0.007 | Antiarthritic | 0.498 | 0.024 | Ulceration | |
| 0.663 | 0.021 | Anti-Inflammatory | 0.385 | 0.134 | Acidosis | |
| Z-627 | 0.985 | 0.003 | Antiarthritic | 0.489 | 0.088 | Extrapyramidal effect |
| 0.983 | 0.003 | Antiosteoporotic | 0.352 | 0.102 | Visual acuity impairment | |
| 0.852 | 0.005 | Anti-Inflammatory | 0.349 | 0.130 | Fasciculation | |
| 0.414 | 0.004 | Cyclooxygenase Inhibitor | 0.291 | 0.187 | Interstitial nephritis | |
| 0.192 | 0.016 | Cyclooxygenase-2 Inhibitor | 0.239 | 0.189 | Ulcer, peptic | |
| Z-814 | 0.821 | 0.006 | Antiarthritic | 0.761 | 0.015 | Extrapyramidal effect |
| 0.569 | 0.024 | Analgesic | 0.747 | 0.037 | Neutrophilic dermatoses | |
| 0.527 | 0.049 | Anti-Inflammatory | 0.627 | 0.051 | Hypercholesterolemic | |
| 0.109 | 0.077 | Cyclooxygenase Inhibitor | 0.539 | 0.032 | Adrenal contex hypoplasia | |
| 0.084 | 0.065 | Cyclooxygenase-2 Inhibitor | 0.441 | 0.041 | Ulcer | |
| Z-964 | 0.689 | 0.007 | Analgesic, Non-Opioid | 0.428 | 0.106 | Extrapyramidal effect |
| 0.688 | 0.018 | Antiarthritic | 0.383 | 0.083 | Visual acuity impairment | |
| 0.642 | 0.015 | Analgesic | 0.347 | 0.198 | Nephrotic syndrome | |
| 0.497 | 0.058 | Anti-Inflammatory | 0.313 | 0.166 | Interstitial nephritis | |
| 0.190 | 0.016 | Cyclooxygenase-2 Inhibitor | 0.309 | 0.207 | Nephritis |
| Inhibitor | Pa [a] | Pi [b] | Adverse Effect [c] | hERG [d] |
|---|---|---|---|---|
| Rofecoxib | 0.561 | 0.031 | Cardiac Failure | Medium Risk |
| 0.547 | 0.036 | Myocardial Infarction | ||
| 0.345 | 0.305 | Hepatotoxicity | ||
| Celecoxib | 0.528 | 0.043 | Cardiac Failure | Medium Risk |
| 0.494 | 0.044 | Myocardial Infarction | ||
| Z-814 | 0.557 | 0.034 | Myocardial Infarction | Low Risk |
| 0.464 | 0.074 | Cardiac Failure | ||
| Z-627 | 0.516 | 0.041 | Myocardial Infarction | Medium Risk |
| 0.479 | 0.066 | Cardiac Failure | ||
| 0.710 | 0.096 | Hepatotoxicity | ||
| Z-964 | 0.503 | 0.054 | Cardiac Failure | High Risk |
| 0.462 | 0.051 | Myocardial infarction |
| Inhibitor | Properties | |||||||
|---|---|---|---|---|---|---|---|---|
| Milog P [a] | TPSA [b] | MW [c] | nHA [d] | nHD [e] | Nv [f] | Nrotb [g] | Volume | |
| Rofecoxib | 0.71 | 60.45 | 314.36 | 4 | 0 | 0 | 3 | 264.79 |
| Celecoxib | 3.61 | 77.99 | 381.38 | 5 | 2 | 0 | 4 | 298.65 |
| Z-814 | 3.35 | 68.28 | 408.39 | 4 | 0 | 0 | 6 | 299.11 |
| Z-627 | 1.38 | 75.27 | 330.41 | 5 | 2 | 0 | 4 | 286.58 |
| Z-964 | 2.03 | 58.20 | 384.47 | 4 | 2 | 0 | 7 | 316.16 |
| Inhibitor | Molecular Properties | Parametric QSAR Models (pIC50 = −logIC50) | ||||||
|---|---|---|---|---|---|---|---|---|
| ATM [a] | ARO [b] | DONN [c] | ACC [d] | Mono | Bi | Tri | Tetra | |
| Rofecoxib | 36 | 2 | 0 | 4 | 8.2562 | 8.0305 | 7.7961 | 7.7670 |
| Celecoxib | 40 | 3 | 1 | 4 | 8.7006 | 8.4097 | 8.4852 | 8.4390 |
| Z-814 | 38 | 2 | 0 | 4 | 8.4784 | 8.2201 | 7.9659 | 7.9484 |
| Z-627 | 41 | 2 | 2 | 3 | 8.8117 | 8.8374 | 9.2534 | 9.3458 |
| Z-964 | 43 | 2 | 2 | 3 | 9.0339 | 9.0270 | 9.4232 | 9.5272 |
| Organism | Ligand | ΔGbind (Kcal/mol) |
|---|---|---|
| Homo sapiens | Rofecoxib | −48.15 |
| Z814 | −45.51 | |
| Z627 | −42.76 | |
| Mus musculus | Celecoxib | −47.78 |
| Z627 | −41.63 | |
| Z964 | −44.27 |
| Inhibitor | Molecule (PDB Code) | IC50 (nM) | pIC50 | References |
|---|---|---|---|---|
| 1 * | Rofecoxib (BDBM22369) | 12.000 | 7.9208 | [44] |
| 2 | BDBM50272105 | 0.090 | 10.0457 | [45] |
| 3 | BDBM50272097 | 0.170 | 9.7695 | [45] |
| 4 | BDBM50365267 | 0.200 | 9.6989 | [4] |
| 5 | BDBM50272130 | 0.280 | 9.5528 | [45] |
| 6 | BDBM50272111 | 0.360 | 9.4436 | [45] |
| 7 | BDBM50272124 | 0.370 | 9.4317 | [45] |
| 8 | BDBM50272106 | 0.600 | 9.2218 | [45] |
| 9 | BDBM50272092 | 0.800 | 9.0969 | [45] |
| 10 | BDBM50049011 | 1.000 | 9.0000 | [46] |
| 11 | BDBM50189988 | 1.800 | 8.7447 | [47] |
| 12 | BDBM50057618 | 3.700 | 8.4317 | [48] |
| 13 | BDBM50151689 | 4.000 | 8.3979 | [17] |
| 14 | BDBM50103310 | 4.200 | 8.3767 | [49] |
| 15 | BDBM13066 | 5.000 | 8.3000 | [50] |
| 16 | BDBM50297680 | 5.600 | 8.2518 | [51] |
| 17 | BDBM50026234 | 6.000 | 8.2218 | [52] |
| 18 | BDBM50332773 | 8.000 | 8.0969 | [52] |
| 19 | BDBM50332765 | 19.000 | 7.7212 | [52] |
| 20 | BDBM50336012 | 21.000 | 7.6777 | [53] |
| 21 a | BDBM50029613 | 0.500 | 9.3010 | [54] |
| 22 a | BDBM50272113 | 1.410 | 8.8508 | [45] |
| 23 a | BDBM50049030 | 1.500 | 8.8239 | [46] |
| 24 a | BDBM50272109 | 2.000 | 8.6990 | [45] |
| 25 a | BDBM50049013 | 3.300 | 8.4815 | [46] |
| 26 b | BDBM50272128 | 0.180 | 9.7447 | [45] |
| 27 b | BDBM50272090 | 0.540 | 9.2676 | [45] |
| 28 b | BDBM50272113 | 1.410 | 8.8508 | [45] |
| 29 b | BDBM50373566 | 2.400 | 8.6198 | [55] |
| 30 b | BDBM50057606 | 5.300 | 8.2757 | [48] |
| 31 b | BDBM50207446 | 7.000 | 8.1549 | [56] |
| Celecoxib b | BDBM11639 | 0.520 | 8.4390 | [45] |
| Enzyme | Resolution | Inhibitor | Coordinates of the Grid Center | Grid Size (Points) |
|---|---|---|---|---|
| COX-2 (PDB code: 5KIR) Homo sapiens | 2.400 Å | Rofecoxib | X = 23.214 Y = 41.353 Z = 3.517 | 36 x 26 y 28 z |
| COX-2 (PDB code: 3LN1) Mus musculus | 2.697 Å | Celecoxib | X = 30.486 Y = −22.364 Z = -15.725 | 36 x 26 y 24 z |
| COX-1 (PDB code: 2OYE) Ovis aries | 2.850 Å | Indomethacin (R) | X = 251.492 Y = 109.817 Z = −40.751 | 36 x 36 y 24 z |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araújo, P.H.F.; Ramos, R.S.; da Cruz, J.N.; Silva, S.G.; Ferreira, E.F.B.; de Lima, L.R.; Macêdo, W.J.C.; Espejo-Román, J.M.; Campos, J.M.; Santos, C.B.R. Identification of Potential COX-2 Inhibitors for the Treatment of Inflammatory Diseases Using Molecular Modeling Approaches. Molecules 2020, 25, 4183. https://doi.org/10.3390/molecules25184183
Araújo PHF, Ramos RS, da Cruz JN, Silva SG, Ferreira EFB, de Lima LR, Macêdo WJC, Espejo-Román JM, Campos JM, Santos CBR. Identification of Potential COX-2 Inhibitors for the Treatment of Inflammatory Diseases Using Molecular Modeling Approaches. Molecules. 2020; 25(18):4183. https://doi.org/10.3390/molecules25184183
Chicago/Turabian StyleAraújo, Pedro H. F., Ryan S. Ramos, Jorddy N. da Cruz, Sebastião G. Silva, Elenilze F. B. Ferreira, Lúcio R. de Lima, Williams J. C. Macêdo, José M. Espejo-Román, Joaquín M. Campos, and Cleydson B. R. Santos. 2020. "Identification of Potential COX-2 Inhibitors for the Treatment of Inflammatory Diseases Using Molecular Modeling Approaches" Molecules 25, no. 18: 4183. https://doi.org/10.3390/molecules25184183