EDR Peptide: Possible Mechanism of Gene Expression and Protein Synthesis Regulation Involved in the Pathogenesis of Alzheimer’s Disease


Abstract
:1. Introduction
2. EDR Peptide: Possible Molecular Aspects of the Regulation of Gene Expression and Protein Synthesis Involved in the Pathogenesis of Alzheimer’s Disease
2.1. MAPK/ERK Signaling Pathway: Role in the Pathogenesis of Alzheimer’s Disease, Regulation by the EDR Peptide
2.2. Antioxidant System Proteins SOD2, GPX1: Role in the Pathogenesis of Alzheimer’s Disease, Regulation by the EDR Peptide
2.3. Caspase-3 and p53 Protein: Role in the Pathogenesis of Alzheimer’s Disease, Regulation by the EDR Peptide
2.4. Transcription Factors PPARA, PPARG: Role in the Pathogenesis of Alzheimer’s Disease, Regulation by the EDR Peptide
3. Serotonin: Physiological Role in the Pathogenesis of Alzheimer’s Disease, Regulation by the EDR Peptide
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Villain, N.; Dubois, B. Alzheimer’s Disease Including Focal Presentations. Semin. Neurol. 2019, 39, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Fessel, W.J. Amyloid is essential but insufficient for Alzheimer causation: Addition of subcellular cofactors is required for dementia. Int. J. Geriatr. Psychiatry 2018, 33, e14–e21. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tan, L.; Yu, J.-T.; Tan, L. Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.; Lü, Y.; Zhang, M. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, I.A.; Dolotov, O.V.; Zolotarev, Y.A.; Andreeva, L.A.; Myasoedov, N.F.; Leacher, L. Effects of behaviorally active ACTH (4-10) analogue—Semax on rat basal forebrain cholinergic neurons. Restor. Neurol. Neurosci. 2008, 26, 35–43. [Google Scholar] [PubMed]
- Fedin, A. The efficacy of cortexin and memantinol (memantine) in the treatment of cognitive impairment in patients with chronic cerebral ischemia. Zhurnal Nevrol. i psikhiatrii im. S.S. Korsakova 2018, 118, 30–36. [Google Scholar] [CrossRef]
- Aliferova, V.M.; Dadasheva, M.N.; Doronin, B.M.; Kovalenko, A.V.; Lokshtanova, T.M.; MIu, M. Clinical efficacy and pharmacoeconomic characteristics of the neuroprotection with low doses of cortexin in the treatment of acute ischemic stroke. Zhurnal Nevrol. i psikhiatrii im. S.S. Korsakova 2014, 114, 41–46. [Google Scholar]
- Meloni, B.P.; Milani, D.; Cross, J.L.; Clark, V.W.; Edwards, A.B.; Anderton, R.S. Assessment of the neuroprotective effects of arginine-rich protamine peptides, poly-arginine peptides (r12-cyclic, r22) and arginine-tryptophan-containing peptides following in vitro excitotoxicity and/or permanent middle cerebral artery occlusion in rats. Neuromolecular Med. 2017, 19, 271–285. [Google Scholar] [CrossRef]
- Chiu, L.S.; Anderton, R.; Knuckey, N.W.; Meloni, B.P. The neuroprotective potential of arginine-rich peptides for the acute treatment of traumatic brain injury. Expert Rev. Neurother. 2016, 16, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Meloni, B.P.; Milani, D.; Edwards, A.B.; Anderton, R.; Doig, R.L.O.; Fitzgerald, M.; Palmer, T.N.; Knuckey, N.W. Neuroprotective peptides fused to arginine-rich cell penetrating peptides: Neuroprotective mechanism likely mediated by peptide endocytic properties. Pharmacol. Ther. 2015, 153, 36–54. [Google Scholar] [CrossRef] [Green Version]
- Umnov, R.S.; Linkova, N.S.; Khavinson, V.K. Neuroprotective effects of peptides bioregulators in people of various age. Adv. Gerontol. 2013, 26, 671–678. [Google Scholar] [PubMed]
- Mendzheritskiĭ, A.M.; Karantysh, G.V.; Ryzhak, G.A.; Dem’ianenko, S.V. Regulation of content of cytokines in blood serum and of caspase-3 activity in brains of old rats in model of sharp hypoxic hypoxia with Cortexin and Pinealon. Adv. Gerontol. 2014, 27, 94–97. [Google Scholar] [PubMed]
- Khavinson, V.K.; Grigoriev, E.I.; Malinin, V.V.; Ryzhak, G.A. Tripeptide Having a Stimulating Effect on the Regeneration of Neurons Regeneration and Pharmaceutical Composition It. Israel Patent 194346, 2013. [Google Scholar]
- Khavinson, V.; Ribakova, Y.; Kulebiakin, K.; Vladychenskaya, E.; Kozina, L.; Arutjunyan, A.; Boldyrev, A. Pinealon Increases Cell Viability by Suppression of Free Radical Levels and Activating Proliferative Processes. Rejuvenation Res. 2011, 14, 535–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arutjunyan, A.; Kozina, L.; Stvolinskiy, S.; Bulygina, Y.; Mashkina, A.; Khavinson, V. Pinealon protects the rat offspring from prenatal hyperhomocysteinemia. Int. J. Clin. Exp. Med. 2012, 5, 179–185. [Google Scholar] [PubMed]
- Boros, B.D.; Bs, K.M.G.; Bs, E.G.G.; Curtis, K.A.; Bs, E.L.B.; Gearing, M.; Herskowitz, J.H. Dendritic spines provide cognitive resilience against Alzheimer’s disease. Ann. Neurol. 2017, 82, 602–614. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, M.A.; Stallings, N.R.; Malter, J.S. Alzheimer’s Disease, Dendritic Spines, and Calcineurin Inhibitors: A New Approach? ACS Chem. Neurosci. 2018, 9, 1233–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Liu, Q.; Wen, T. Dendritic cell factor 1 deletion leads to developmental defects in mushroom-shaped dendritic spines. NeuroReport 2019, 30, 1008–1015. [Google Scholar] [CrossRef]
- Kraskovskaya, N.A.; Kukanova, E.O.; Linkova, N.S.; Popugaeva, E.A.; Khavinson, V. Tripeptides Restore the Number of Neuronal Spines under Conditions of In Vitro Modeled Alzheimer’s Disease. Bull. Exp. Biol. Med. 2017, 163, 550–553. [Google Scholar] [CrossRef]
- Flores, K.; Yadav, S.S.; Katz, A.A.; Seger, R. The Nuclear Translocation of Mitogen-Activated Protein Kinases: Molecular Mechanisms and Use as Novel Therapeutic Target. Neuroendocrine 2019, 108, 121–131. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Flores, K.; Maik-Rachline, G.; Zehorai, E.; Kapri-Pardes, E.; Berti, D.A.; Hanoch, T.; Besser, M.J.; Seger, R. The nuclear translocation of ERK1/2 as an anticancer target. Nat. Commun. 2015, 6, 6685. [Google Scholar] [CrossRef]
- Moens, U.; Kostenko, S. Structure and function of MK5/PRAK: The loner among the mitogen-activated protein kinase-activated protein kinases. Biol. Chem. 2013, 394, 1115–1132. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Deng, Y.; Zhang, B.; Gong, C.-X. Deregulation of brain insulin signaling in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 282–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, L.; Balazs, R.; Thornton, P.L.; Cotman, C.W. -Amyloid Peptide at Sublethal Concentrations Downregulates Brain-Derived Neurotrophic Factor Functions in Cultured Cortical Neurons. J. Neurosci. 2004, 24, 6799–6809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-Z.; Liu, F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol. 2008, 85, 148–175. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Tabner, B.J.; El-Agnaf, O.M.A.; Turnbull, S.; German, M.J.; Paleologou, K.E.; Hayashi, Y.; Cooper, L.J.; Fullwood, N.J.; Allsop, D. Hydrogen Peroxide Is Generated during the Very Early Stages of Aggregation of the Amyloid Peptides Implicated in Alzheimer Disease and Familial British Dementia. J. Biol. Chem. 2005, 280, 35789–35792. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, A.; Prà, I.D.; Marconi, M.; Chakravarthy, B.; Whitfield, J.; Armato, U. Calcium-Sensing Receptor (CaSR) in Human Brains Pathophysiology: Roles in Late-Onset Alzheimers Disease (LOAD). Curr. Pharm. Biotechnol. 2009, 10, 317–326. [Google Scholar] [CrossRef]
- Puig, B.; Gómez-Isla, T.; Ribé, E.; Cuadrado, M.; Torrejón-Escribano, B.; Dalfó, E.; Ferrer, I. Expression of stress-activated kinases c-Jun N-terminal kinase (SAPK/JNK-P) and p38 kinase (p38-P), and tau hyperphosphorylation in neurites surrounding βA plaques in APP Tg2576 mice. Neuropathol. Appl. Neurobiol. 2004, 30, 491–502. [Google Scholar] [CrossRef]
- Shen, C.; Chen, Y.; Liu, H.; Zhang, K.; Zhang, T.; Lin, A.; Jing, N. Hydrogen peroxide promotes Aβ production through JNK-dependent activation of γ-secretase. J. Biol. Chem. 2008, 283, 17721–17730. [Google Scholar] [CrossRef]
- Colombo, A.; Bastone, A.; Ploia, C.; Sclip, A.; Salmona, M.; Forloni, G.; Borsello, T. JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol. Dis. 2009, 33, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Muresan, Z.; Muresan, V. The Amyloid-β Precursor Protein Is Phosphorylated via Distinct Pathways during Differentiation, Mitosis, Stress, and Degeneration. Mol. Biol. Cell 2007, 18, 3835–3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, T.; Ichijo, H. ASK1-MAP kinase signaling pathway as a therapeutic target for human diseases. Nihon Rinsho 2014, 72, 957–965. [Google Scholar] [PubMed]
- Peel, A.L.; Sorscher, N.; Kim, J.Y.; Galvan, V.; Chen, S.; Bredesen, D.E. Tau phosphorylation in Alzheimer’s disease: Potential involvement of an APP-MAP kinase complex. Neuromolecular Med. 2004, 5, 205–218. [Google Scholar] [CrossRef]
- Galvan, V.; Banwait, S.; Spilman, P.; Gorostiza, O.F.; Peel, A.; Ataie, M.; Crippen, D.; Huang, W.; Sidhu, G.; Ichijo, H.; et al. Interaction of ASK1 and the β-amyloid precursor protein in a stress-signaling complex. Neurobiol. Dis. 2007, 28, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Tamagno, E.; Guglielmotto, M.; Giliberto, L.; Vitali, A.; Borghi, R.; Autelli, R.; Danni, O.; Tabaton, M. JNK and ERK1/2 pathways have a dual opposite effect on the expression of BACE1. Neurobiol. Aging 2009, 30, 1563–1573. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Chiba, T.; Yamada, M.; Nawa, M.; Kanekura, K.; Suzuki, H.; Terashita, K.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Transforming Growth Factor β2 Is a Neuronal Death-Inducing Ligand for Amyloid-β Precursor Protein. Mol. Cell. Biol. 2005, 25, 9304–9317. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Boldyrev, A.A. Significance of reactive oxygen species for neuronal function. In Free Radicals, NO, and Inflammation: Molecular, Biochemical and Clinical Aspects; Tomasi, A., Ed.; IOS Press: Harvard, MA, USA, 2003; pp. 153–169. [Google Scholar]
- Kishida, K.T.; Klann, E. Sources and Targets of Reactive Oxygen Species in Synaptic Plasticity and Memory. Antioxidants Redox Signal. 2006, 9, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Chalisova, N.I.; Lin’Kova, N.S.; Nichik, T.E.; Ryzhak, A.P.; Dudkov, A.V.; Ryzhak, G.A. Peptide Regulation of Cells Renewal Processes in Kidney Tissue Cultures from Young and Old Animals. Bull. Exp. Biol. Med. 2015, 159, 124–127. [Google Scholar] [CrossRef]
- Khavinson, V.; Lin’Kova, N.S.; Polyakova, V.O.; Durnova, A.O.; Nichik, T.E.; Kvetnoi, I.M. Peptides Regulate Expression of Signaling Molecules in Kidney Cell Cultures during In Vitro Aging. Bull. Exp. Biol. Med. 2014, 157, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Khavinson, V.; Lin’Kova, N.S.; Evlashkina, E.V.; Durnova, A.O.; Kozlov, K.L.; Gutop, O.E. Molecular Aspects of Anti-Atherosclerotic Effects of Short Peptides. Bull. Exp. Biol. Med. 2014, 158, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Linkova, N.S.; Polyakova, V.O.; Trofimov, A.V.; Kvetnoy, I.M.; Khavinson, V.K. Peptidergic Regulation of Thymocyte Differentiation, Proliferation, and Apoptosis during Aging of the Thymus. Bull. Exp. Biol. Med. 2011, 151, 239–242. [Google Scholar] [CrossRef]
- Lin’Kova, N.S.; O Drobintseva, A.; Orlova, O.A.; Kuznetsova, E.P.; Polyakova, V.O.; Kvetnoy, I.M.; Khavinson, V. Peptide Regulation of Skin Fibroblast Functions during Their Aging In Vitro. Bull. Exp. Biol. Med. 2016, 161, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, R.; Jeong, N.Y. Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases. Int. J. Med. Sci. 2019, 16, 1386–1396. [Google Scholar] [CrossRef] [Green Version]
- Apelt, J.; Bigl, M.; Wunderlich, P.; Schliebs, R. Aging-related increase in oxidative stress correlates with developmental pattern of beta-secretase activity and beta-amyloid plaque formation in transgenic Tg2576 mice with Alzheimer-like pathology. Int. J. Dev. Neurosci. 2004, 22, 475–484. [Google Scholar] [CrossRef]
- Lynn, S.; Huang, E.J.; Elchuri, S.; Naeemuddin, M.; Nishinaka, Y.; Yodoi, J.; Ferriero, D.M.; Epstein, C.J.; Huang, T.-T. Selective neuronal vulnerability and inadequate stress response in superoxide dismutase mutant mice. Free Radic. Biol. Med. 2005, 38, 817–828. [Google Scholar] [CrossRef]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.-Q.; Bien-Ly, N.; Puoliväli, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in Mitochondrial Superoxide Dismutase Modulates Alzheimer’s Disease-Like Pathology and Accelerates the Onset of Behavioral Changes in Human Amyloid Precursor Protein Transgenic Mice. J. Neurosci. 2006, 26, 5167–5179. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Hoeffer, C.A.; Wong, H.; Massaad, C.A.; Zhou, P.; Iadecola, C.; Murphy, M.P.; Pautler, R.G.; Klann, E. Amyloid -Induced Impairments in Hippocampal Synaptic Plasticity Are Rescued by Decreasing Mitochondrial Superoxide. J. Neurosci. 2011, 31, 5589–5595. [Google Scholar] [CrossRef]
- Melov, S.; Adlard, P.A.; Morten, K.; Johnson, F.; Golden, T.R.; Hinerfeld, D.; Schilling, B.; Mavros, C.; Masters, C.L.; Volitakis, I.; et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE 2007, 2, e536. [Google Scholar] [CrossRef]
- Himori, K.; Abe, M.; Tatebayashi, D.; Lee, J.; Westerblad, H.; Lanner, J.T.; Yamada, T. Superoxide dismutase/catalase mimetic EUK-134 prevents diaphragm muscle weakness in monocrotalin-induced pulmonary hypertension. PLoS ONE 2017, 12, e0169146. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.R.; Ong, T.P.; Jacob-Filho, W.; Jaluul, O.; Freitas, M.I.D.; Cominetti, C.; Cozzolino, S.M.F. Glutathione Peroxidase 1 Pro198Leu Polymorphism in Brazilian Alzheimer’s Disease Patients: Relations to the Enzyme Activity and to Selenium Status. J. Nutr. 2012, 5, 72–80. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Ong, T.P.; Jacob-Filho, W.; Jaluul, O.; Freitas, M.I.D.; Cozzolino, S.M.F. Nutritional status of selenium in Alzheimer’s disease patients. Br. J. Nutr. 2009, 103, 803–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorova, T.N.; Macletsova, M.G.; Kulikov, A.V.; Stepanova, M.S.; Boldyrev, A.A. Carnosine protects from the oxidative stress induced by prenatal hypoxia. Dokl. Biol. Sci. 2006, 408, 207–210. [Google Scholar] [CrossRef]
- Kozina, L.S.; Arutjunyn, A.V.; Stvolinskii, S.L.; Stepanova, M.S.; Makletsova, M.G.; Khavinson, V.K. Regulatory peptides protect brain neurons from hypoxia in vivo. Dokl. Biol. Sci. 2007, 418, 1–4. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015, 356, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Stanga, S.; Lanni, C.; Govoni, S.; Uberti, D.; D’Orazi, G.; Racchi, M. Unfolded p53 in the pathogenesis of Alzheimer’s disease: Is HIPK2 the link? Aging 2010, 2, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Jembrek, M.J.; Slade, N.; Hof, P.R.; Šimić, G. The interactions of p53 with tau and Aß as potential therapeutic targets for Alzheimer’s disease. Prog. Neurobiol. 2018, 168, 104–127. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.R.; Ghafouri, M.; Mukerjee, R.; Bagashev, A.; Chabrashvili, T.; Sawaya, B.E. Role of p53 in Neurodegenerative Diseases. Neurodegener. Dis. 2011, 9, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Gasiorowski, K.; Brokos, B.; Leszek, J.; Tarasov, V.V.; Ashraf, G.M.; Aliev, G. Insulin Resistance in Alzheimer Disease: p53 and MicroRNAs as Important Players. Curr. Top. Med. Chem. 2017, 17, 1429–1437. [Google Scholar] [CrossRef]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 Has a Direct Apoptogenic Role at the Mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Barone, E.; Cenini, G.; Sultana, R.; Di Domenico, F.; Fiorini, A.; Perluigi, M.; Noel, T.; Wang, C.; Mancuso, C.; Clair, D.K.S.; et al. Lack of p53 Decreases Basal Oxidative Stress Levels in the Brain Through Upregulation of Thioredoxin-1, Biliverdin Reductase-A, Manganese Superoxide Dismutase, and Nuclear Factor Kappa-B. Antioxid. Redox Signal. 2012, 16, 1407–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, E.; Cenini, G.; Di Domenico, F.; Noel, T.; Wang, C.; Perluigi, M.; Clair, D.K.S.; Butterfield, D.A. Basal brain oxidative and nitrative stress levels are finely regulated by the interplay between superoxide dismutase 2 and p53. J. Neurosci. Res. 2015, 93, 1728–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorini, A.; Sultana, R.; Barone, E.; Cenini, G.; Perluigi, M.; Mancuso, C.; Cai, J.; Klein, J.B.; Clair, D.S.; Butterfield, D.A. Lack of p53 Affects the Expression of Several Brain Mitochondrial Proteins: Insights from Proteomics into Important Pathways Regulated by p53. PLoS ONE 2012, 7, e49846. [Google Scholar] [CrossRef]
- Nixon, R.A.; Cataldo, A.M. Lysosomal system pathways: Genes to neurodegeneration in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 277–289. [Google Scholar] [CrossRef]
- Kudryashova, I.V.; Kudryashov, I.E.; Gulyaeva, N.V. Long-term potentiation in the hippocampus in conditions of inhibition of caspase-3: Analysis of facilitation in paired-pulse stimulation. Neurosci. Behav. Physiol. 2006, 36, 817–824. [Google Scholar] [CrossRef]
- Lu, D.C.; Rabizadeh, S.; Chandra, S.; Shayya, R.F.; Ellerby, L.M.; Ye, X.; Salvesen, G.S.; Koo, E.H.; Bredesen, D.E. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat. Med. 2000, 6, 397–404. [Google Scholar] [CrossRef]
- Zhao, M.; Su, J.; Head, E.; Cotman, C.W. Accumulation of caspase cleaved amyloid precursor protein represents an early neurodegenerative event in aging and in Alzheimer’s disease. Neurobiol. Dis. 2003, 14, 391–403. [Google Scholar] [CrossRef]
- Su, J.H.; Zhao, M.; Anderson, A.J.; Srinivasan, A.; Cotman, C.W. Activated caspase-3 expression in Alzheimer’s and aged control brain: Correlation with Alzheimer pathology. Brain Res. 2001, 898, 350–357. [Google Scholar] [CrossRef]
- D’Amelio, M.; Cavallucci, V.; Middei, S.; Marchetti, C.; Pacioni, S.; Ferri, A.; Diamantini, A.; De Zio, D.; Carrara, P.; Battistini, L.; et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 14, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.; Lauretti, E.; Praticò, D. Caspase-3-dependent cleavage of Akt modulates tau phosphorylation via GSK3β kinase: Implications for Alzheimer’s disease. Mol. Psychiatry 2017, 22, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Mendzheritski, A.M.; Karantysh, G.V.; Abramchuk, V.A.; Ryzhak, G.A. Effect of peptide geroprotectors on the navigation system learning and caspase-3 in brain structures in rats of different age. Adv. Gerontol. 2013, 26, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Ferenczi, S.; Kovacs, K. Systemic inflammatory challenges compromise survival after experimental stroke via augmenting brain inflammation, blood-brain barrier damage and brain oedema independently of infarct size. J. Neuroinflammation 2011, 8, 164–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, T.-T.; Tsay, H.-J.; Chang, L.; Hsu, C.-L.; Lai, T.-H.; Huang, F.-L.; Shiao, Y.-J. Caspase 3 involves in neuroplasticity, microglial activation and neurogenesis in the mice hippocampus after intracerebral injection of kainic acid. J. Biomed. Sci. 2013, 20, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory Mechanisms in Alzheimer’s Disease: Inhibition of β-Amyloid-Stimulated Proinflammatory Responses and Neurotoxicity by PPARγ Agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar] [CrossRef]
- Heun, R.; Kölsch, H.; Ibrahim-Verbaas, C.A.; Combarros, O.; Aulchenko, Y.S.; Breteler, M.; Schuur, M.; van Duijn, C.M.; Hammond, N.; Belbin, O.; et al. Interactions between PPAR-α and inflammation-related cytokine genes on the development of Alzheimer’s disease, observed by the Epistasis Project. Int. J. Mol. Epidemiol. Genet 2012, 3, 39–47. [Google Scholar]
- Combs, C.K.; Bates, P.; Karlo, J.; E Landreth, G. Regulation of β-amyloid stimulated proinflammatory responses by peroxisome proliferator-activated receptor α. Neurochem. Int. 2001, 39, 449–457. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The inflammatory response system of brain: Implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 1995, 21, 195–218. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef]
- Corbett, G.T.; Gonzalez, F.J.; Pahan, K. Activation of peroxisome proliferator-activated receptor α stimulates ADAM10-mediated proteolysis of APP. Proc. Natl. Acad. Sci. USA 2015, 112, 8445–8450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayeux, R.; Honig, L.S.; Tang, M.-X.; Manly, J.; Stern, Y.; Schupf, N.; Mehta, P.D. Plasma A 40 and A 42 and Alzheimer’s disease: Relation to age, mortality, and risk. Neurology 2003, 61, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guan, L.; Luo, D.; Liu, J.; Lin, H.; Li, X.; Liu, X. Gene- gene interaction between PPARG and APOE gene on late-onset Alzheimer’s disease: A case- control study in Chinese han population. J. Nutr. Health Aging 2016, 21, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, A.M.; Helisalmi, S.; Pihlajamäki, J.; Hiltunen, M.; Koivisto, K.; Moilanen, L.; Kuusisto, J.; Helkala, E.-L.; Hänninen, T.; Kervinen, K.; et al. Association Analysis of Peroxisome Proliferator-Activated Receptor Gamma Polymorphisms and Late Onset Alzheimer’s Disease in the Finnish Population. Dement. Geriatr. Cogn. Disord. 2006, 22, 449–453. [Google Scholar] [CrossRef]
- Sastre, M.; Dewachter, I.; Rossner, S.; Bogdanovic, N.; Rosen, E.; Borghgraef, P.; Evert, B.O.; Dumitrescu-Ozimek, L.; Thal, D.R.; Landreth, G.; et al. Nonsteroidal anti-inflammatory drugs repress -secretase gene promoter activity by the activation of PPAR. Proc. Natl. Acad. Sci. USA 2006, 103, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Khavinson, V.; Linkova, N.S.; Tarnovskaya, S.I.; Umnov, R.S.; Elashkina, E.V.; Durnova, A.O. Short Peptides Stimulate Serotonin Expression in Cells of Brain Cortex. Bull. Exp. Biol. Med. 2014, 157, 77–80. [Google Scholar] [CrossRef]
- Khavinson, V.; Linkova, N.; Kukanova, E.; Bolshakova, A.; Gainullina, A.; Tendler, S.; Morozova, E.; Tarnovskaya, S.; Vinski, D.S.P.; Bakulev, V.; et al. Neuroprotective effect of EDR peptide in mouse model of huntington’s disease. J. Neurol. Neurosci. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Vigliante, I.; Mannino, G.; Maffei, M.E. Chemical Characterization and DNA Fingerprinting of Griffonia simplicifolia Baill. Molecules 2019, 24, 1032. [Google Scholar] [CrossRef] [Green Version]
- Lukiw, W.J.; Rogaev, E.I. Genetics of Aggression in Alzheimer’s Disease (AD). Front. Aging Neurosci. 2017, 9, 87. [Google Scholar] [CrossRef]
- Maitre, M.; Klein, C.; Patte-Mensah, C.; Mensah-Nyagan, A.-G. Tryptophan metabolites modify brain Aβ peptide degradation: A role in Alzheimer’s disease? Prog. Neurobiol. 2020, 190, 101800. [Google Scholar] [CrossRef]
- Hornedo-Ortega, R.; Da Costa, G.; Cerezo, A.B.; Troncoso, A.M.; Richard, T.; García-Parrilla, M.C. In Vitro Effects of Serotonin, Melatonin, and Other Related Indole Compounds on Amyloid-β Kinetics and Neuroprotection. Mol. Nutr. Food Res. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Bostancıklıoğlu, M. Optogenetic stimulation of serotonin nuclei retrieve the lost memory in Alzheimer’s disease. J. Cell. Physiol. 2019, 235, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.J.; Park, D.H.; Chung, H.D.; Marshall, G.L.; Haring, J.H.; Joh, T.H. Evidence for decreased transport of tryptophan hydroxylase in Alzheimer’s disease. Brain Res. 1990, 537, 83–87. [Google Scholar] [CrossRef]
- Wirth, A.; Holst, K.; Ponimaskin, E. How serotonin receptors regulate morphogenic signalling in neurons. Prog. Neurobiol. 2017, 151, 35–56. [Google Scholar] [CrossRef]
- Butzlaff, M.; Ponimaskin, E. The role of serotonin receptors in Alzheimer’s disease. Opera Med. Physiol. 2016, 2, 77–86. [Google Scholar]
- McClam, T.D.; Marano, C.M.; Rosenberg, P.B.; Lyketsos, C.G. Interventions for Neuropsychiatric Symptoms in Neurocognitive Impairment Due to Alzheimer’s Disease. Harv. Rev. Psychiatry 2015, 23, 377–393. [Google Scholar] [CrossRef]
- Schneider, L.S.; Frangakis, C.; Drye, L.T.; Devanand, D.; Marano, C.M.; Mintzer, J.; Mulsant, B.H.; Munro, C.A.; Newell, J.A.; Pawluczyk, S.; et al. Heterogeneity of Treatment Response to Citalopram for Patients With Alzheimer’s Disease With Aggression or Agitation: The CitAD Randomized Clinical Trial. Am. J. Psychiatry 2016, 173, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Sheline, Y.I.; West, T.; Yarasheski, K.; Swarm, R.; Jasielec, M.S.; Fisher, J.R.; Ficker, W.D.; Yan, P.; Xiong, C.; Frederiksen, C.; et al. An Antidepressant Decreases CSF A Production in Healthy Individuals and in Transgenic AD Mice. Sci. Transl. Med. 2014, 6, 236re4. [Google Scholar] [CrossRef] [Green Version]
- Khavinson, V.; Tarnovskaya, S.I.; Linkova, N.S. Short Peptides Regulate Gene Expression. Bull. Exp. Biol. Med. 2016, 162, 288–292. [Google Scholar] [CrossRef]
- Kuznik, B.I.; Davydov, S.O.; Popravka, E.S.; Lin’kova, N.S.; Kozina, L.S.; Khavinson, V.K. Epigenetic mechanisms of peptide-driven regulation and neuroprotective protein FKBP1b. Mol. Biol. 2019, 53, 299–307. [Google Scholar] [CrossRef]
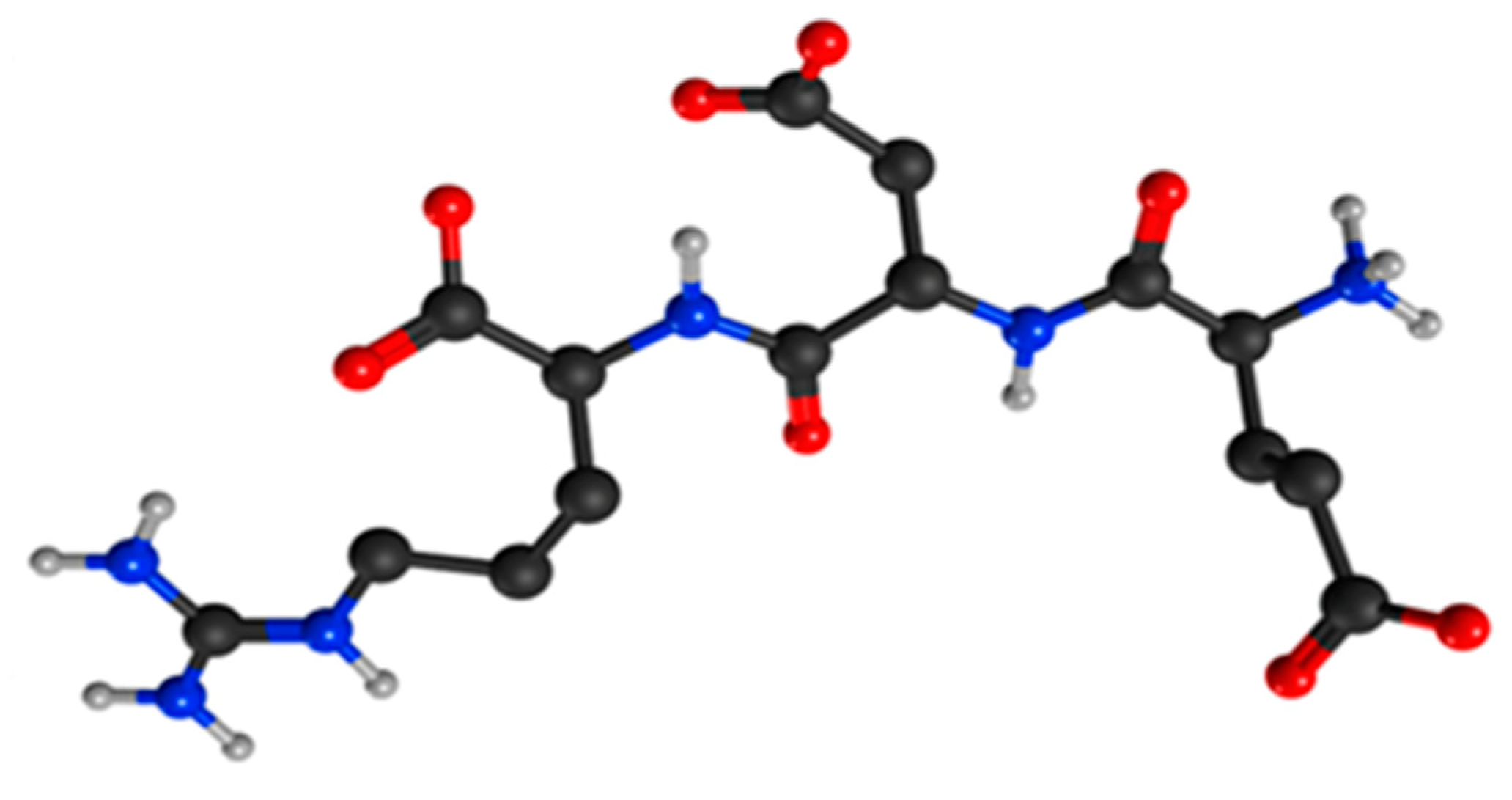
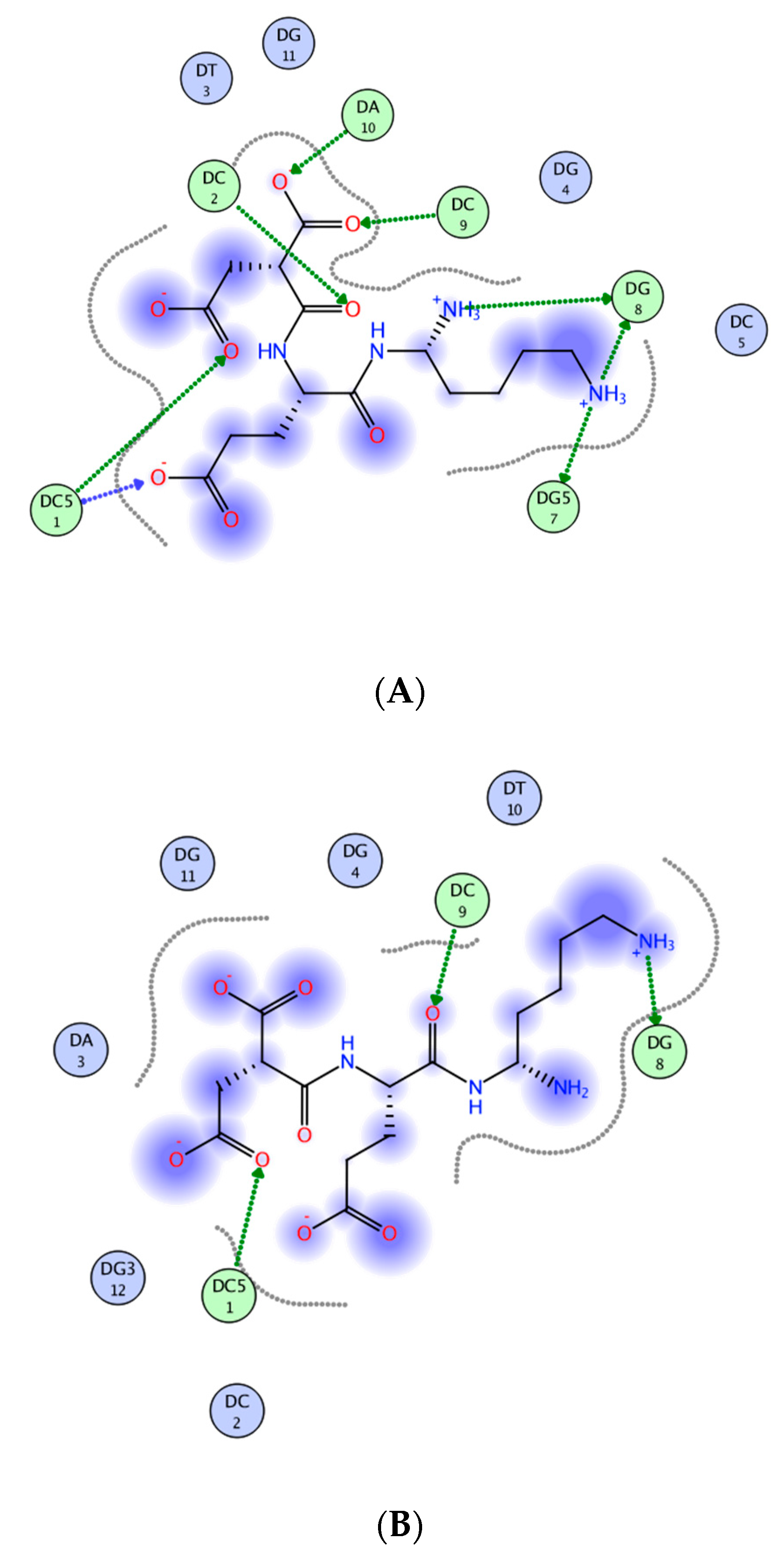
- Kolchina, N.; Khavinson, V.; Linkova, N.; Yakimov, A.; Baitin, D.; Afanasyeva, A.; Petukhov, M. Systematic search for structural motifs of peptide binding to double-stranded DNA. Nucleic Acids Res. 2019, 47, 10553–10563. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Khavinson, V.K. Peptide bioregulation of aging: Results and prospects. Biogerontology 2010, 11, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Khavinson, V.; Linkova, N.S.; Dyatlova, A.; Kuznik, B.I.; Umnov, R. Peptides: Prospects for use in the treatment of COVID-19. J. Mol. Spec. Issue 2020, 25, 4389. [Google Scholar] [CrossRef]
- Fedoreyeva, L.I.; Kireev, I.I.; Khavinson, V.; Vanyushin, B.F. Penetration of short fluorescence labeled peptides into the nucleus in HeLa cells and in vitro specific interaction of the peptides with deoxyribooligonucleotides and DNA. Biochemistry 2011, 76, 1210–1219. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Regulatory Site, Range -499 to 100 bp (cDNA 5′→3′) | Gen Bank NO |
|---|---|---|
| PPARA Homo sapiens | ACCGGCTCATCGCACAGAGTAGCAGAGCCGGGCTCATCGAGGAGGCAGGAGGGGCTCGCCAGCGTGGCACGGGCGCCCGGCGGGAACCTCCACCCGCCCCGCGGCCGCGCGTCCCCGCCTCGAATTCAGCCCCGCCCCGGTGCGCCGGGCTGGAGGGGCGCTGACGCTCAGCGGTGTCCCATCGGTGACCTTGGACGGTCCCTCCACCTCTCCGGCCTCAGTTTCCCTTGGCTGCAGCGGCCGCGGGGCGCTAGGTGGGAGCCGCTGAGCGCTCCCGGGGCCCCGCCCACCGCGAGCAGCCAATCGGGCGCCGCCCTCCGGGGGGTGTGTCCCGGGGCCGAGGCCCGGGGCCCGGAGGGCGCGCGGGGCGGGCGGGGCTTCCGGGTCGGGCCTCGGGACACTGGCTCGCGCGGACCGGGGCAGGGGGCGGGCCGAGGGGCGGTGCGTGTCGCGGGGGCGCGGCTGGCACGGACGCGCGGAGGCGGCGCCGGGCATGGGCCGTGGACGCGGCGGCCCCGCGGCGGGGGCAGCGGGCGGCGGGGGCGGAGGCGGCCGCTAGCGCCCTGCCCGGCGCCGCCTCCTTCGGCGTTCGCCCCACGG | NM_005036.4 |
| PPARG Homo sapiens | ACCAAGGGACCCGAAATATGCTTTAATTAAATTTTCTTTTAAAATGTCACTGGAAAGAACATCTTGGGAAGACGGCCTGGCCGATCGCCGTGTGAAGGGCAAGCCACTCTGGCCGAGAGGGAGCCCCACACCTCGGTCTCCCCAGACCGGCCCTGGCCGGGGGCATCCCCCTAAACTTCGGATCCCTCCTCGGAAATGGGACCCTCTCTGGGCCGCCTCCCAGCGGTGGTGGCGAGGAGCAAACGACACCAGGTAGCCTGCCGCGGGGCAGAGAGTGGACGCGGGAAAGCCGGTGGCTCCCGCCGTGGGCCCTACTGTGCGCGGGCGGCGGCCGAGCCCGGGCCGCTCCCTCCCAGTCGCGCGCCGCCGCCCCCGCCCCCGCCCCCGCCCCCGCCCCCACCCCCACCCCCACCCCCACCCCCAGCCGGCGCCCGCGCCCGCCCCCGCGCCGGGCCCGGCTCGGCCCGACCCGGCTCCGCCGCGGGCAGGCGGGGCCCAGCGCACTCGGAGCCCGAGCCCGAGCCGCAGCCGCCGCCTGGGGCGCTTGGGTCGGCCTCGAGGACACCGGAGAGGGGCGCCACGCCGCCGTGGCCGCAGGTC | NM_138712.3 |
| TPH1 Rattus norvegicus | GCTTCTCCTATAAGAGGCGGCAGCTCCCGTCCGCAGGTGACCCTCTGAACTCCAGTGGCTTTGAGGTCCTCTTTCCAGTGCCGGATCCTGCCCACTGGGTCATCTTCATTCAGATTCACCATGATTGAAGACAACAAGGAGAACAAAGACCATTCCTCAGAAAGGGGGAGAGTGACTCTCATCTTTTCCTTGAAGAATGAAGTTGGAGGACTCATAAAAG | X53501.1 |
| GPX1 Homo sapiens | GACTCTGCCCGGTTAGAAAACCCGCACGAGGGCGGTGCCGCTTTGGAGACAGGGAGGAGGGAGACCGGAAGCCTAGATCCCTCTGGCTGTCCCCTGCACTGCCGGTAACATGGCACAGGAGAGGAGGGCTGTTTGTGCACGGGCAGCTCCTGCAGCTGCTGCCGTCGCCCACCAGCCTCCTATGCCAAACCCCACATCCTAACTCAGGAACCTCTGAGAAAAAACGGAGCCCTCGAGGGCCCCAGCCCTTGGAAGGGTAACCTGGACCGCTGCCGCCTGGTTGCCTGGGCCAGACCAGACATGCCTGCTGCTCCTTCCGGCTTAGGAGGAGCACGCGTCCCGCTCGGGCGCACTCTCCAGCCTTTTCCTGGCTGAGGAGGGGCCGAGCCCTCCGGGTAGGGCGGGGGCCGGATGAGGCGGGACCCTCAGGCCCGGAAAACTGCCTGTGCCACGTGACCCGCCGCCGGCCAGTTAAAAGGAGGCGCCTGCTGGCCTCCCCTTACAGTGCTTGTTCGGGGCGCTCCGCTGGCTTCTTGGACAATTGCGCCATGTGTGCTGCTCGGCTAGCGGCGGCGGCGGCGGCGGCCCAGTCGGTGTATG | NM_000581 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khavinson, V.; Linkova, N.; Kozhevnikova, E.; Trofimova, S. EDR Peptide: Possible Mechanism of Gene Expression and Protein Synthesis Regulation Involved in the Pathogenesis of Alzheimer’s Disease. Molecules 2021, 26, 159. https://doi.org/10.3390/molecules26010159
Khavinson V, Linkova N, Kozhevnikova E, Trofimova S. EDR Peptide: Possible Mechanism of Gene Expression and Protein Synthesis Regulation Involved in the Pathogenesis of Alzheimer’s Disease. Molecules. 2021; 26(1):159. https://doi.org/10.3390/molecules26010159
Chicago/Turabian StyleKhavinson, Vladimir, Natalia Linkova, Ekaterina Kozhevnikova, and Svetlana Trofimova. 2021. "EDR Peptide: Possible Mechanism of Gene Expression and Protein Synthesis Regulation Involved in the Pathogenesis of Alzheimer’s Disease" Molecules 26, no. 1: 159. https://doi.org/10.3390/molecules26010159