
Diketopiperazine-Based, Flexible Tadalafil Analogues: Synthesis, Crystal Structures and Biological Activity Profile

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results
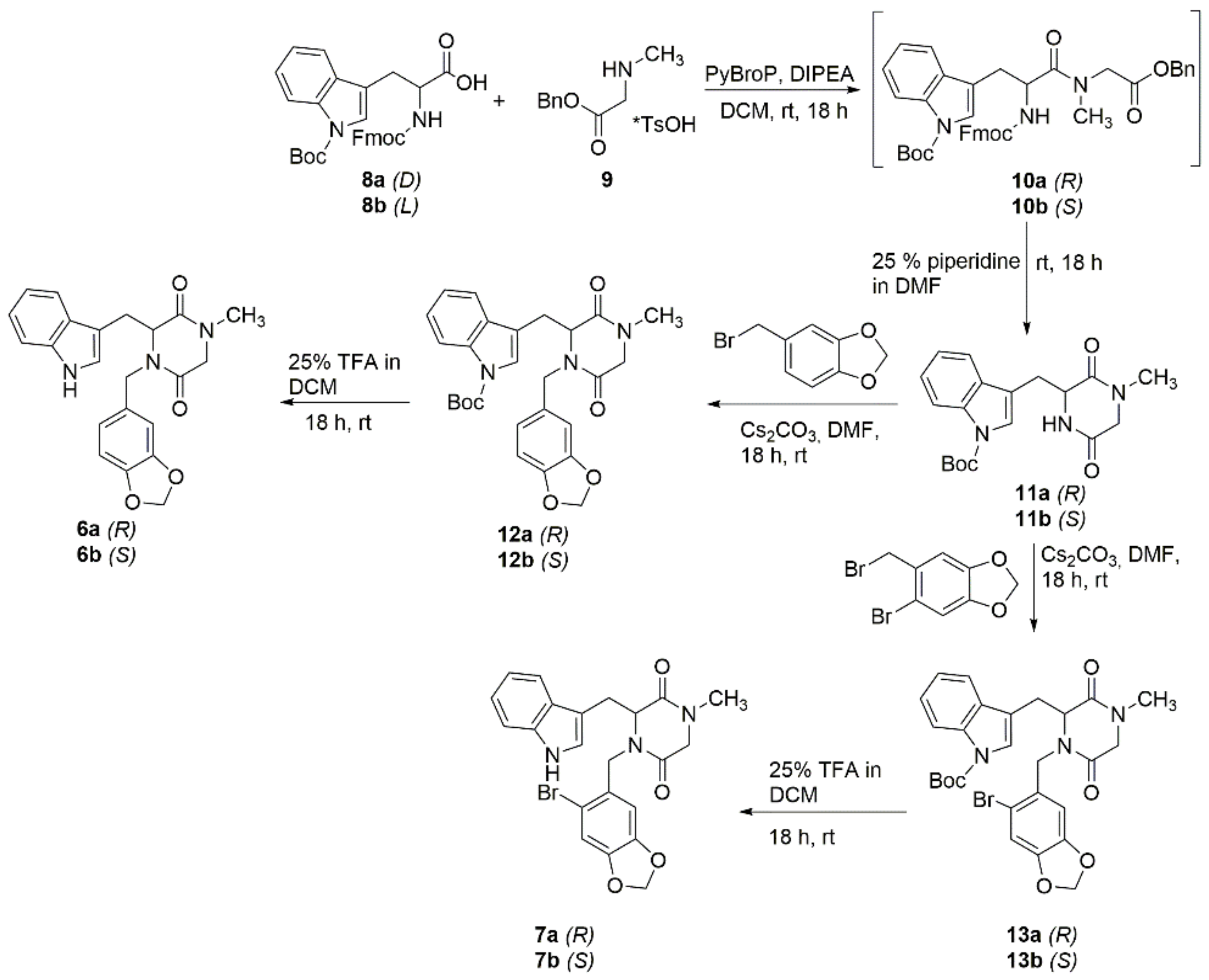
2.1. Chemistry
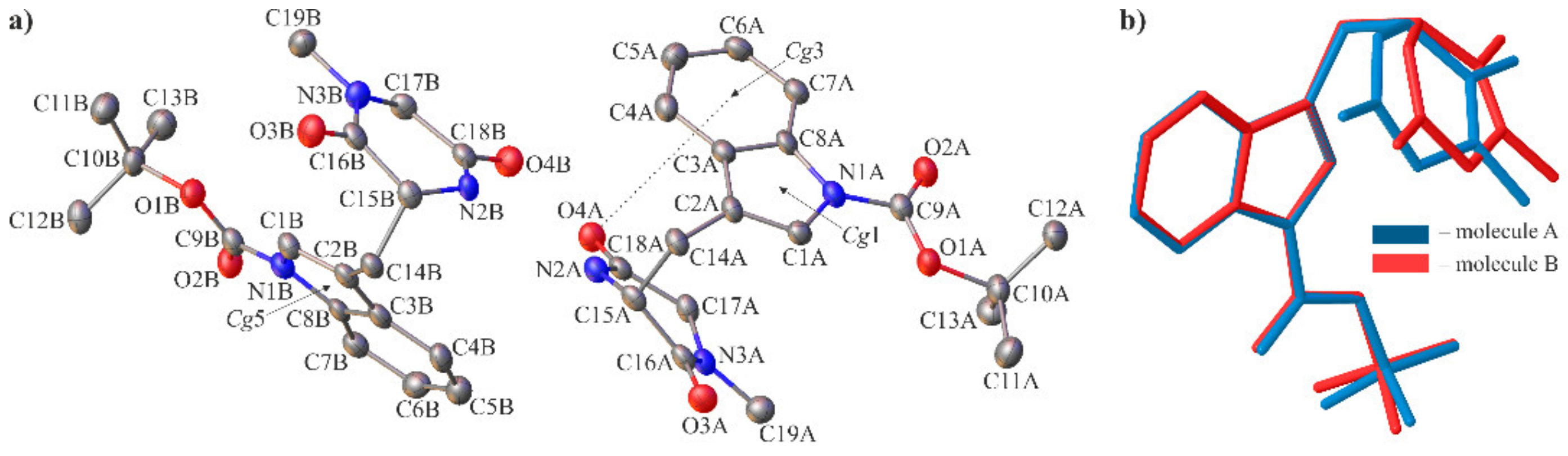
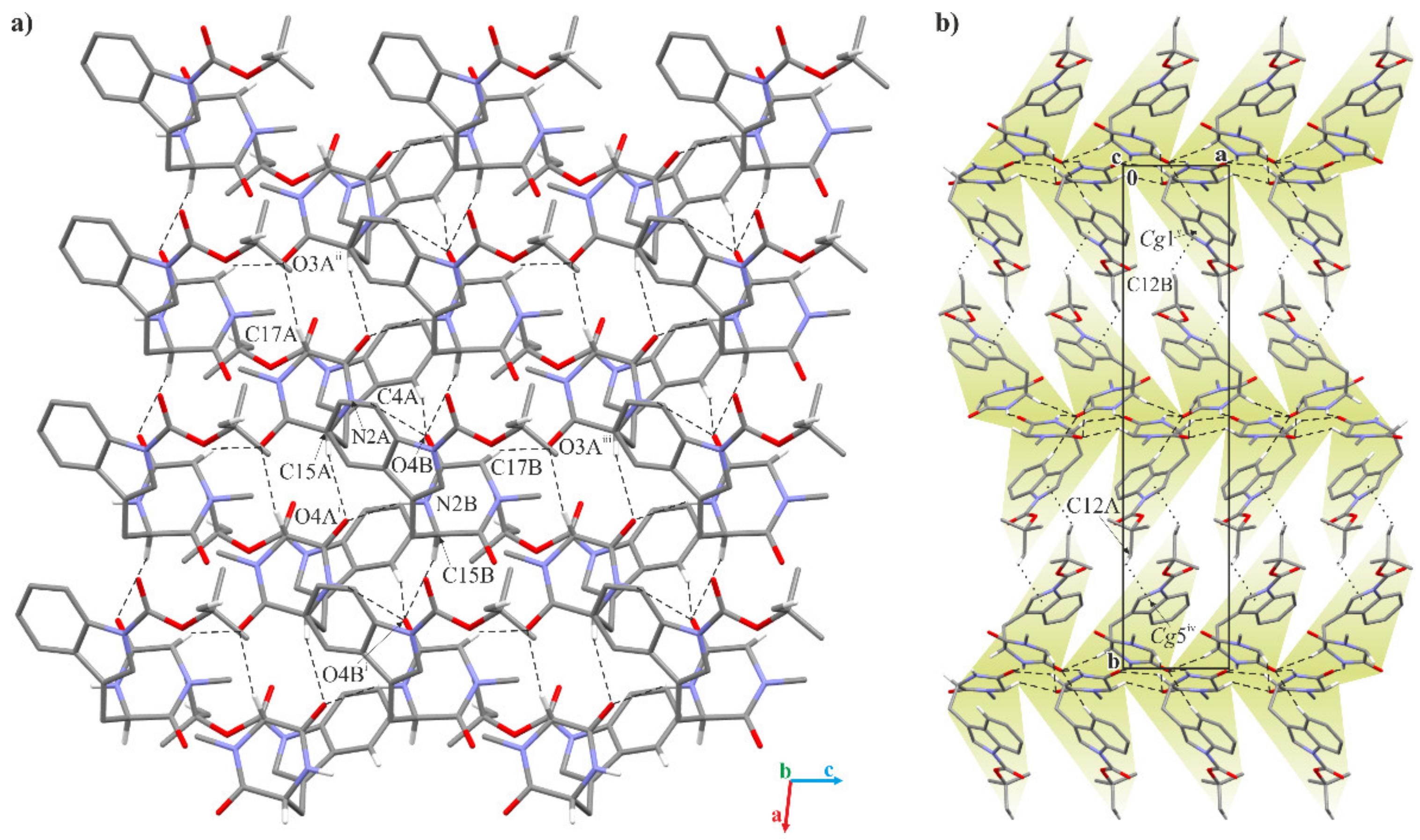
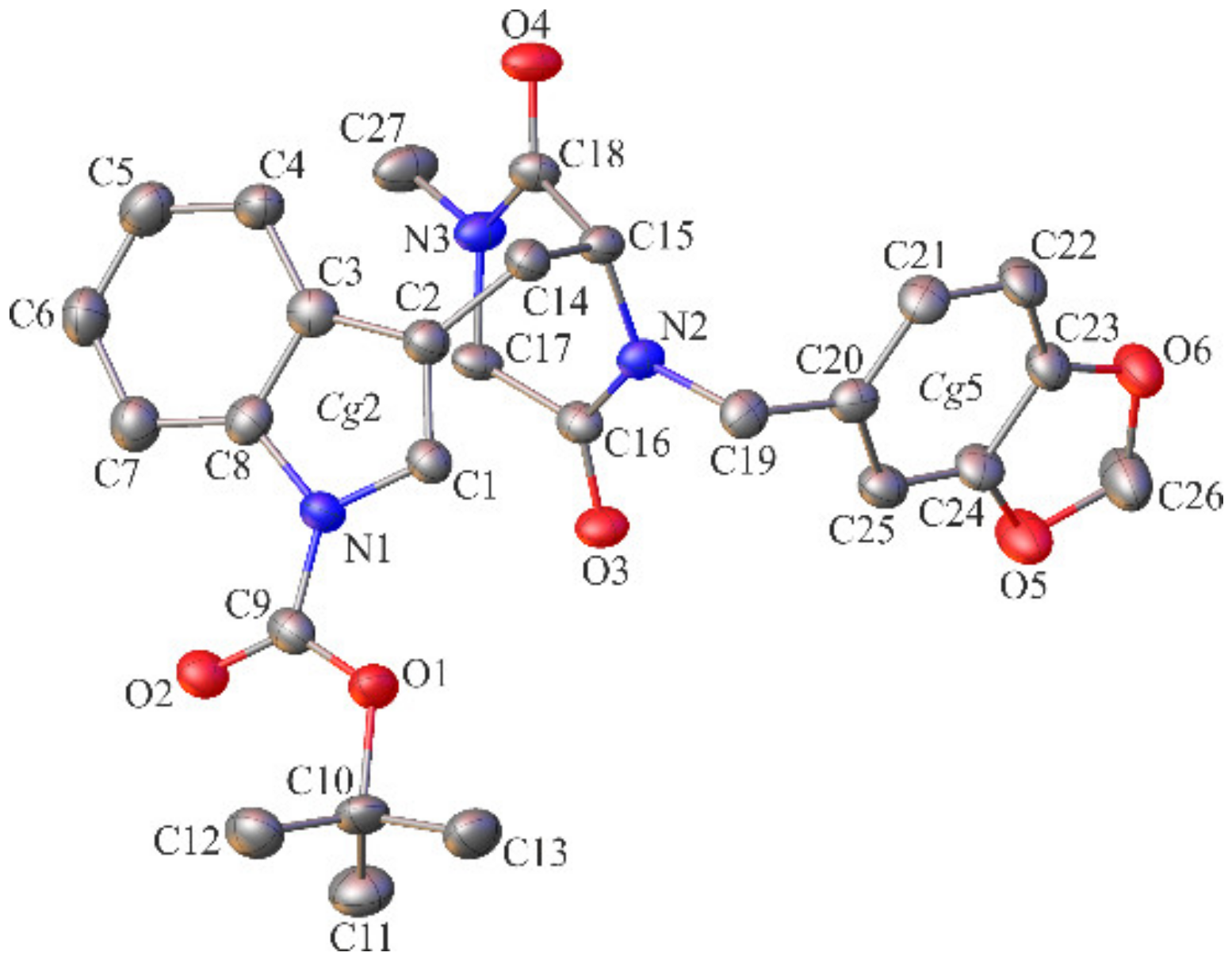
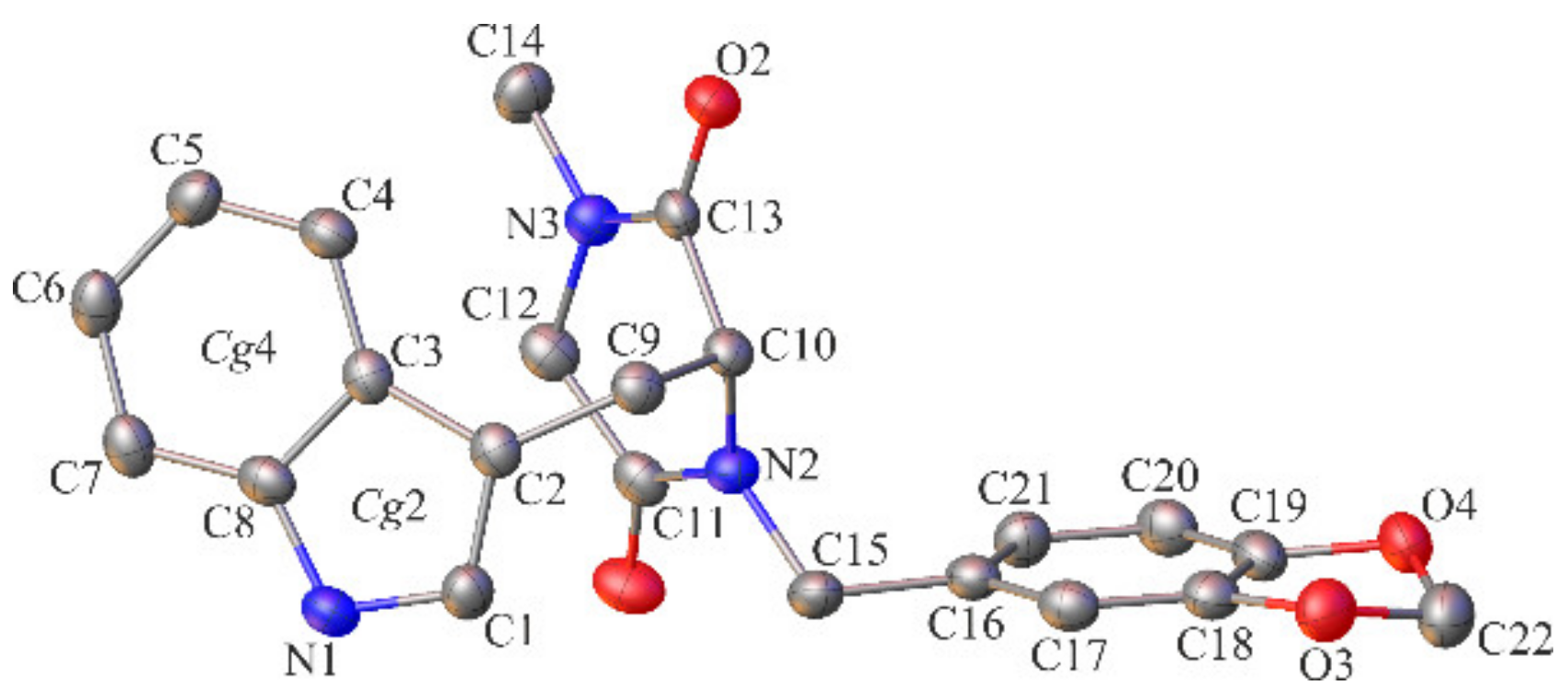
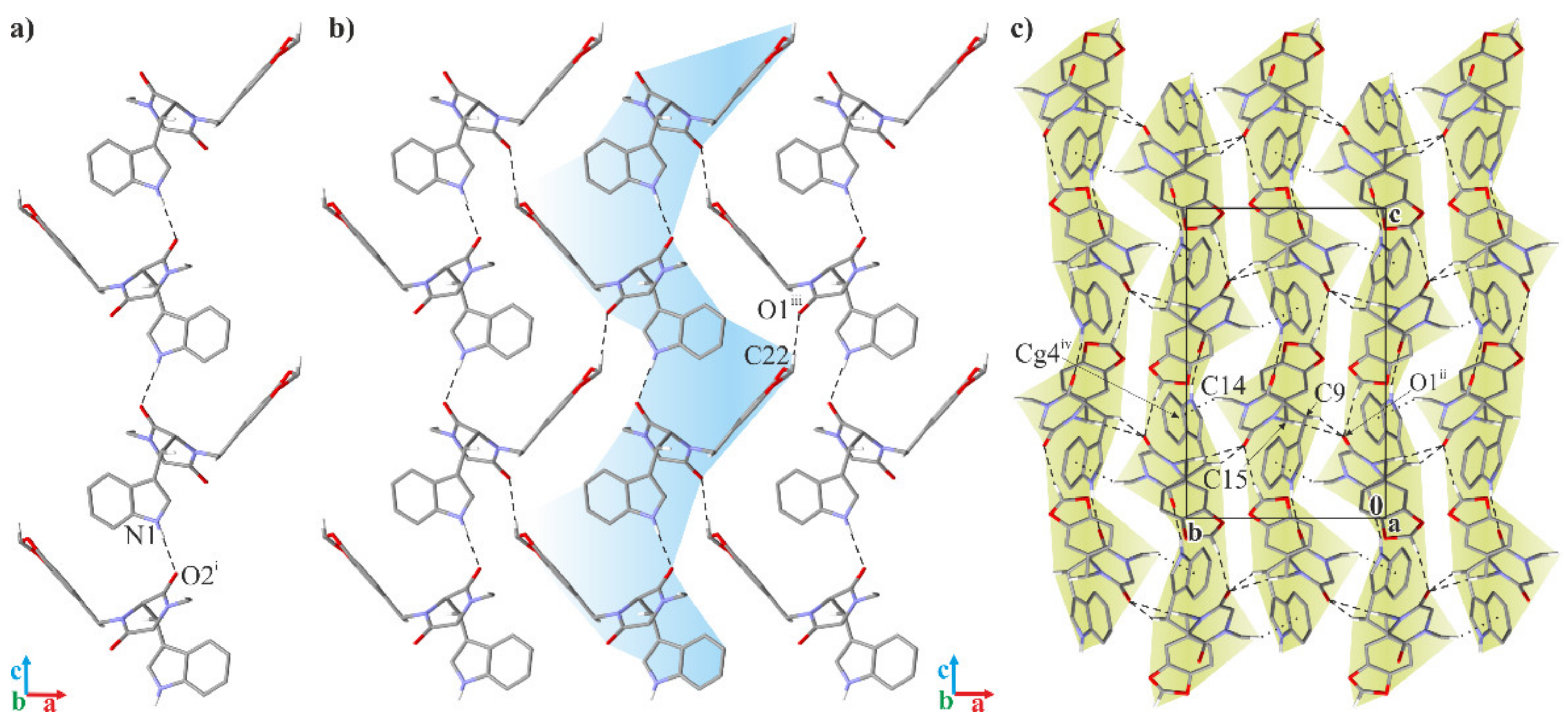
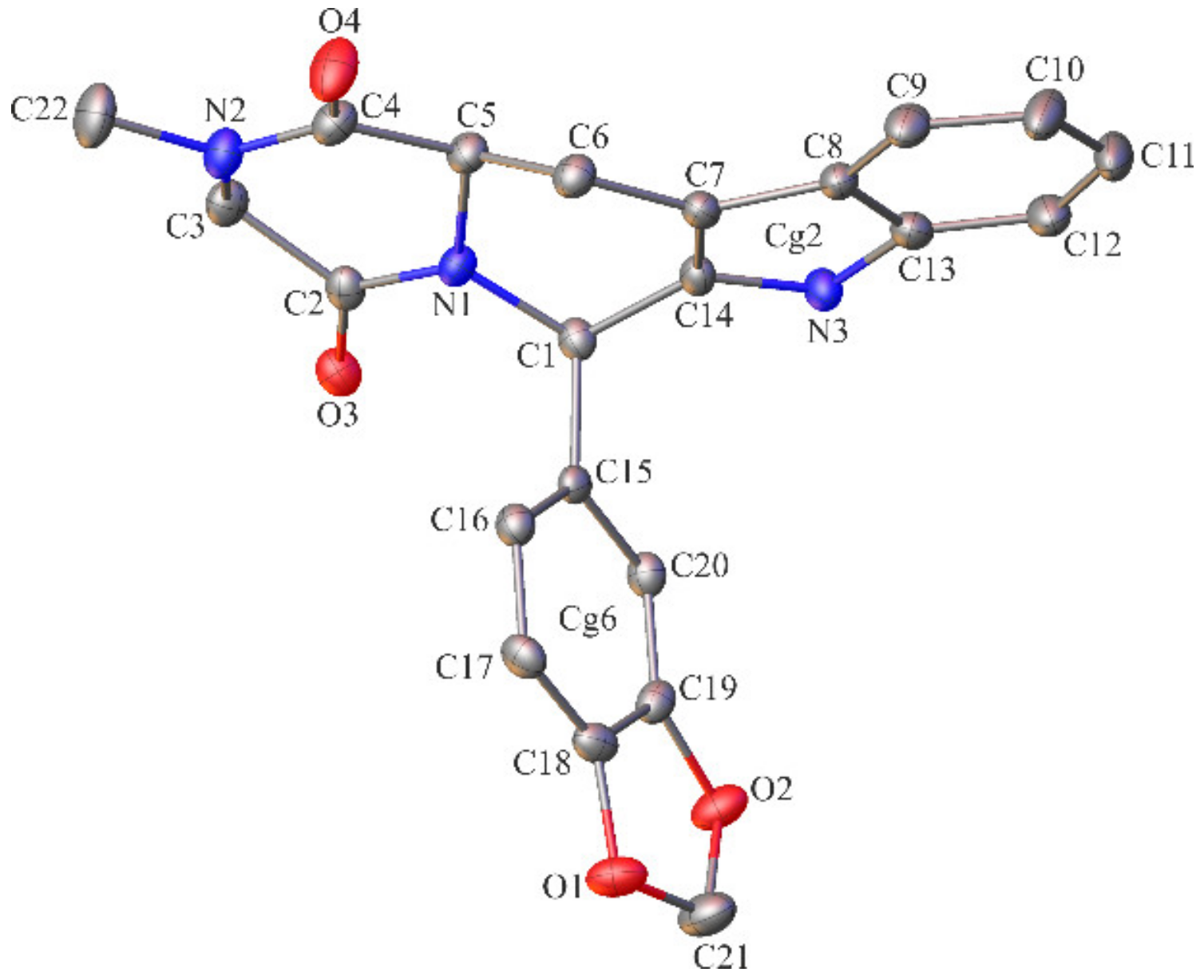
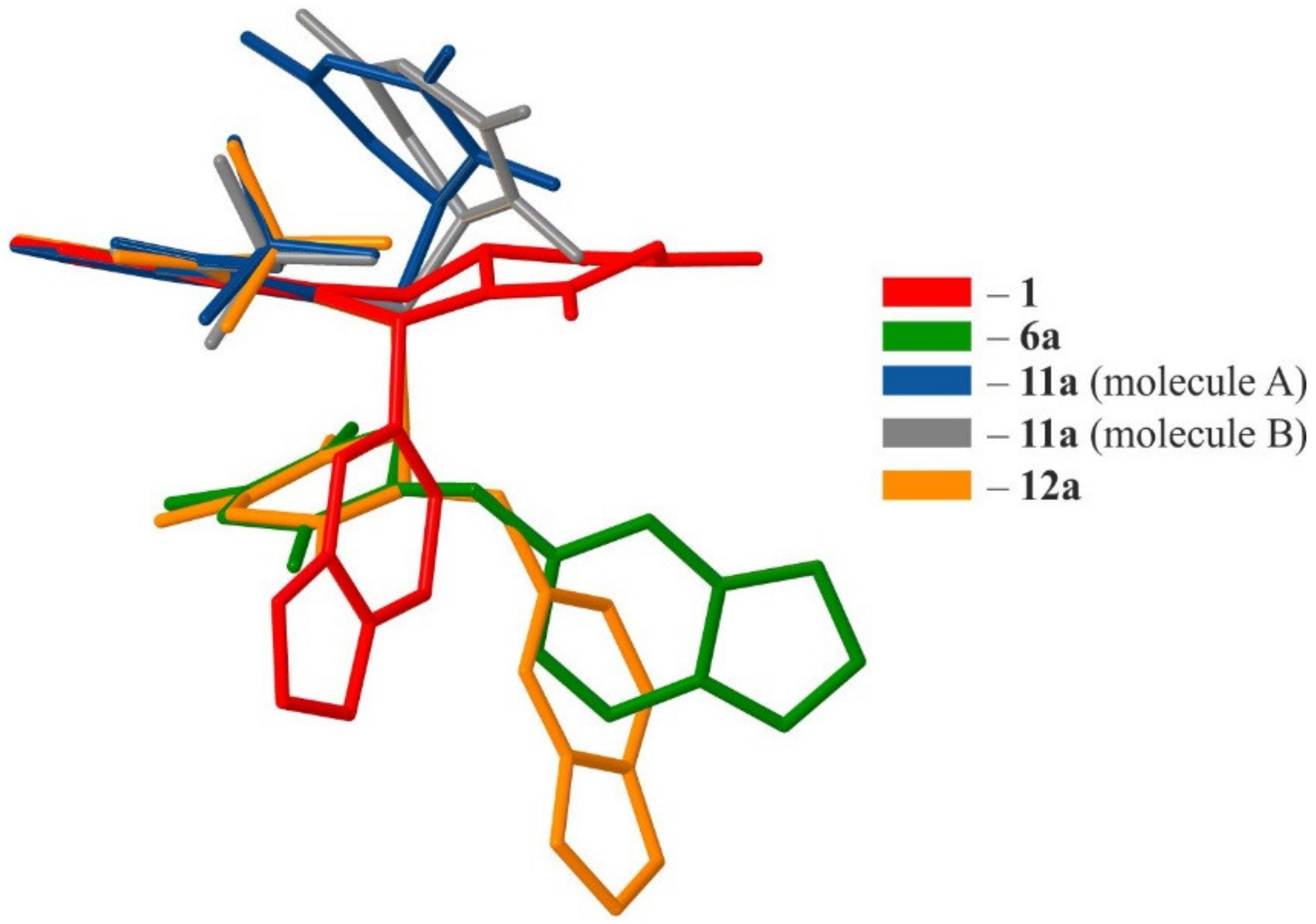
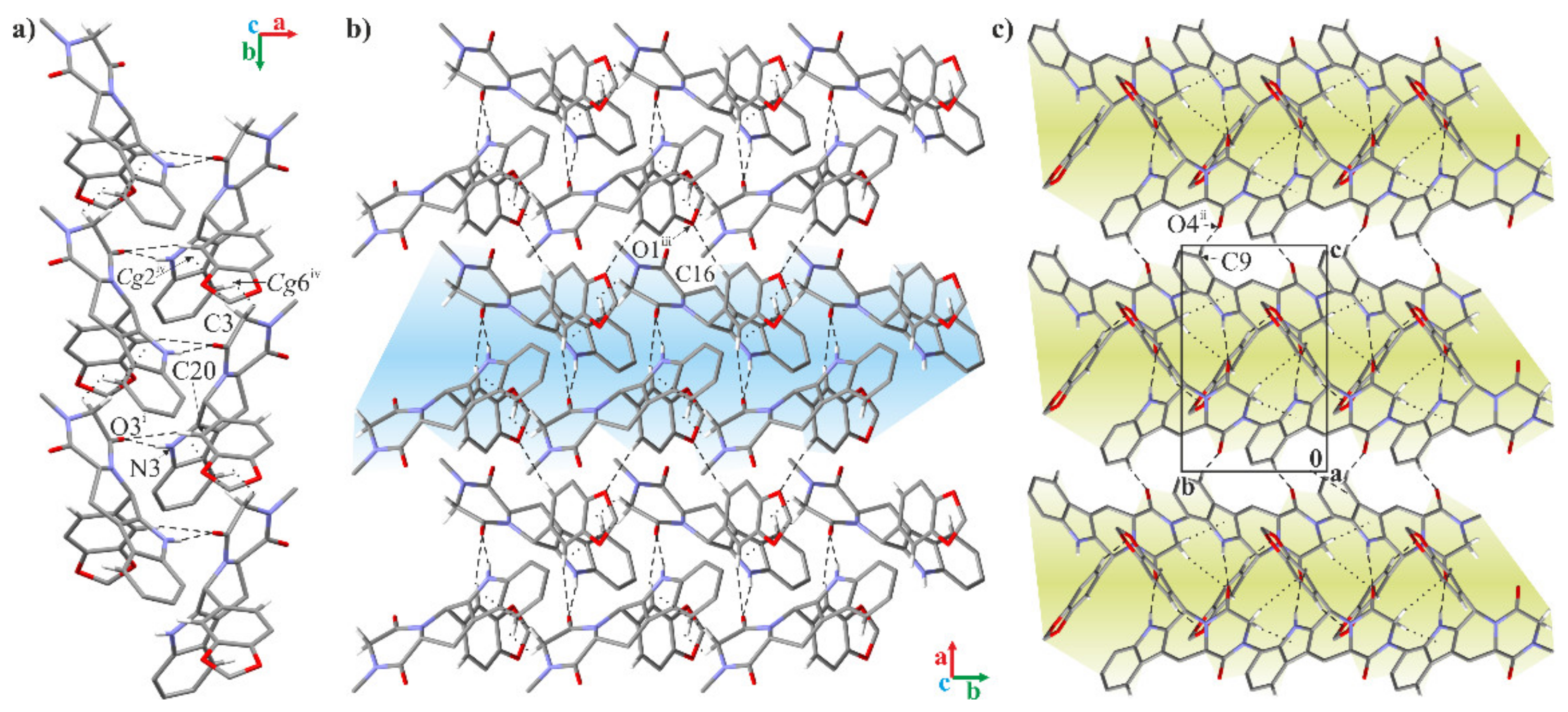
2.2. Crystallographic Studies
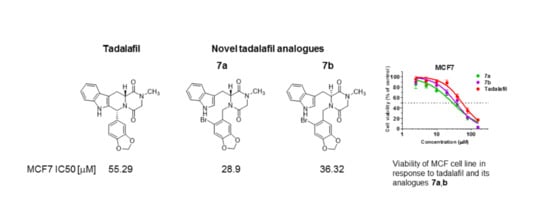
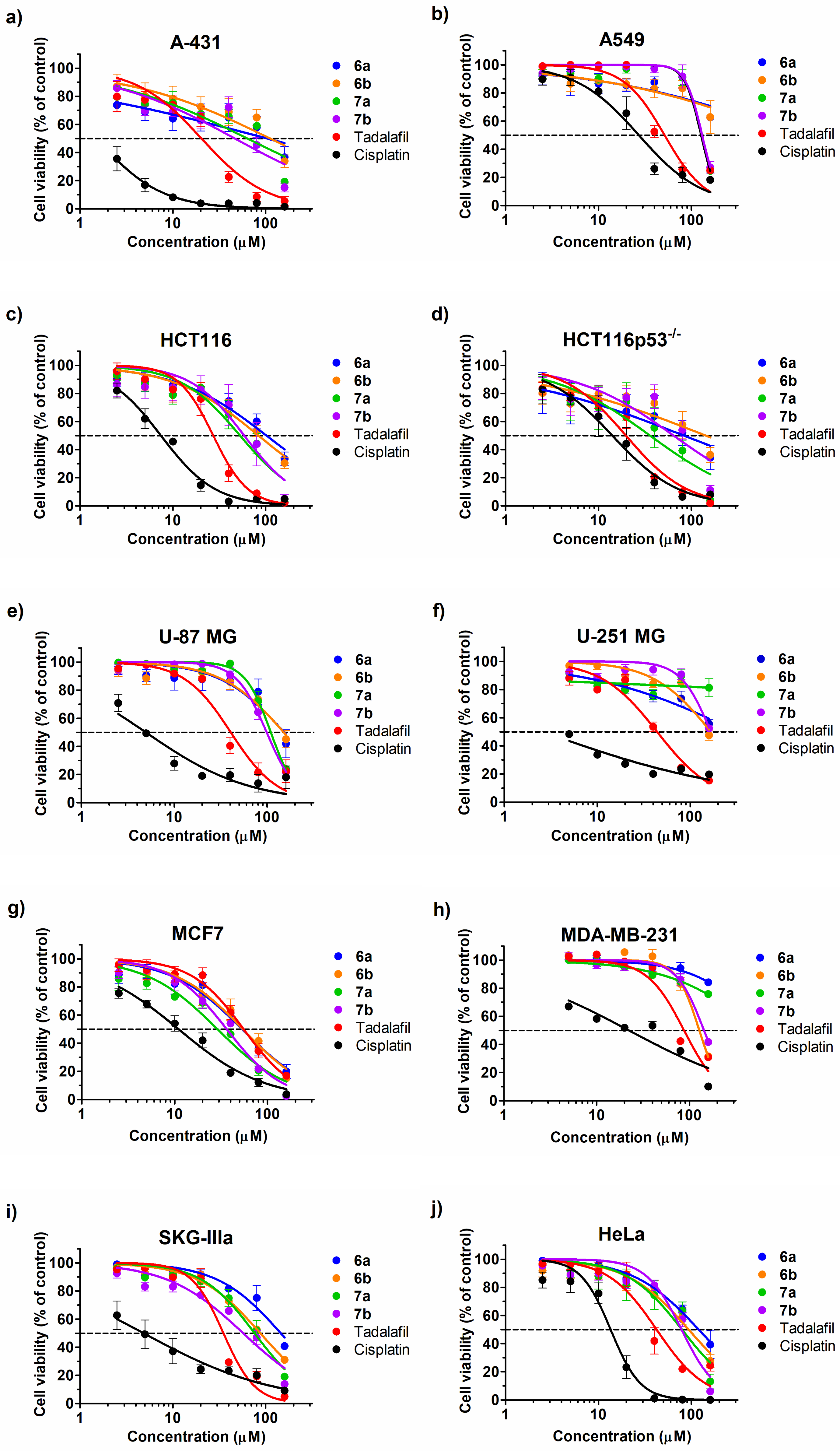
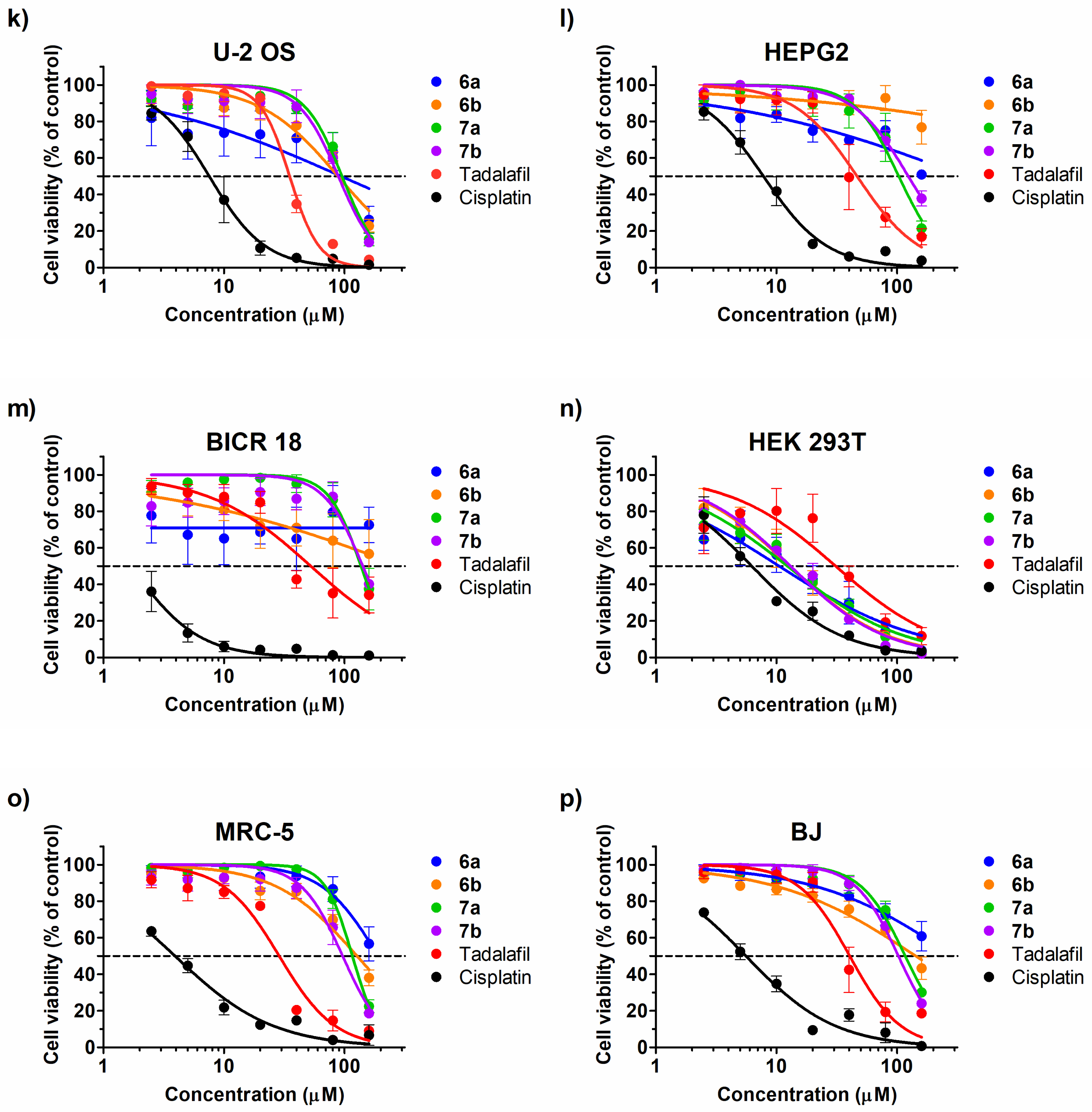
2.3. Cytotoxic Effect
2.4. Enzyme Inhibition
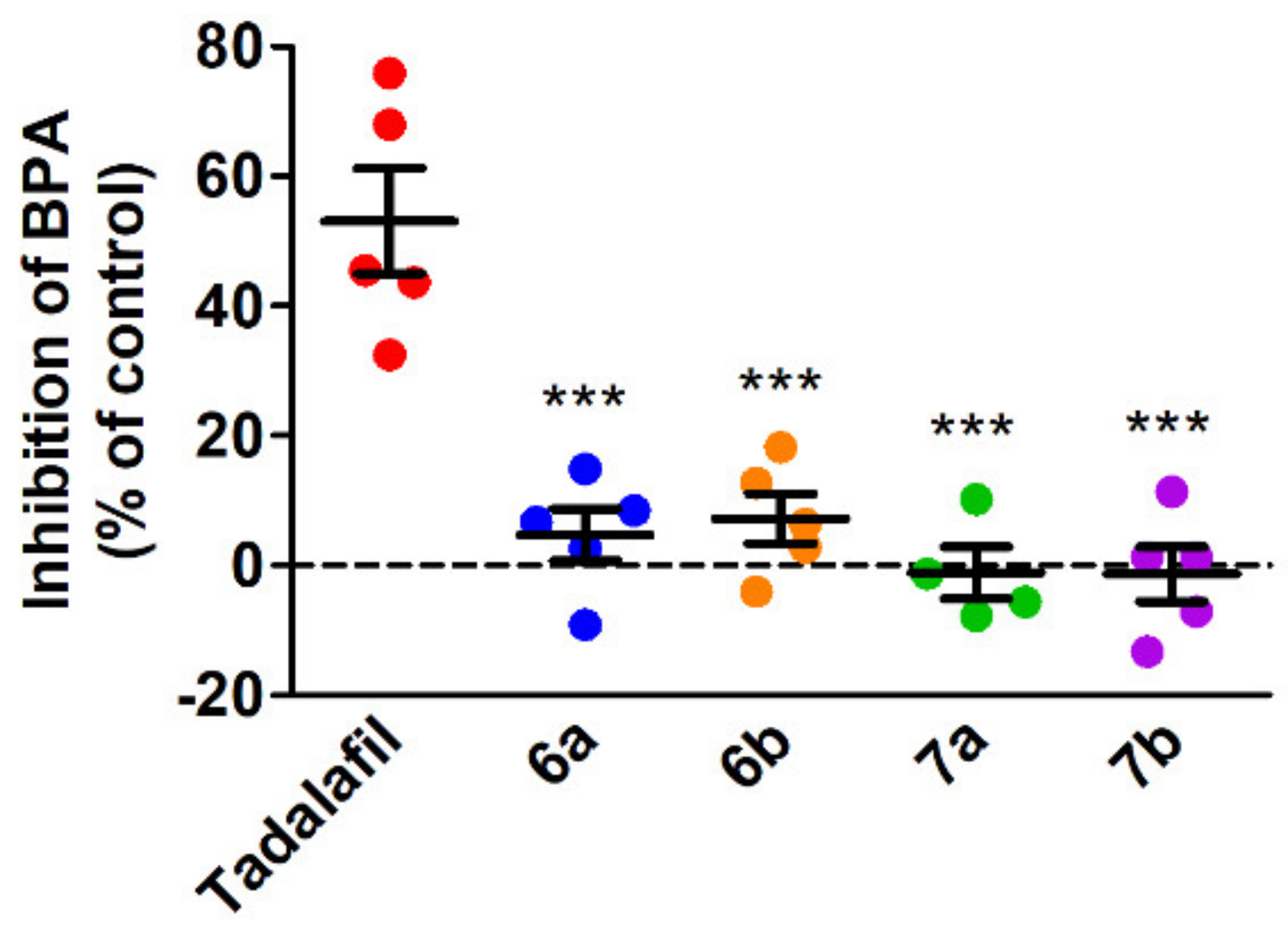
2.5. Antiplatelet Activity of the Tested Compounds
2.6. Toxicity against Caenorhabditis Elegans
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Synthetic Procedures and Analytical Data
The Synthesis of tert-butyl (R)-(11a) and (S)-3-((4-methyl-3,6-dioxopiperazin-2-yl)methyl)-1H-indole-1-carboxylate (11b)
The Synthesis of tert-butyl (R)-(12a) and (S)-3-((1-(benzo[d][1,3]dioxol-5-ylmethyl)-4-methyl-3,6-dioxopiperazin-2-yl)methyl)-1H-indole-1-carboxylate (12b)
The Synthesis of tert-butyl (R)-(13a) and (S)-3-((1-((6-bromobenzo[d][1,3]dioxol-5-yl)methyl)-4-methyl-3,6-dioxopiperazin-2-yl) methyl)-1H-indole-1-carboxylate (13b)
The Synthesis of (R)-(6a) and (S)-3-((1H-indol-3-yl)methyl)-4-(benzo[d][1,3]dioxol-5-ylmethyl)-1-methylpiperazine-2,5-dione (6b)
The Synthesis of (R)-(7a) and (S)-3-((1H-indol-3-yl)methyl)-4-((6-bromobenzo[d][1,3]dioxol-5-yl)methyl)-1-methyl-piperazine-2,5-dione (7b)
3.1.3. Crystallographic Studies
3.2. Biology
3.2.1. Cell Culture
3.2.2. Cell Viability Assay
3.2.3. Enzyme Inhibition
3.2.4. Platelet Aggregation in Platelet Rich Plasma
3.2.5. Patients and Blood Collection
3.2.6. Caenorhabditis Elegans
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Daugan, A.; Grondin, P.; Ruault, C.; Le Monnier de Gouville, A.C.; Coste, H.; Kirilovsky, J.; Hyafil, F.; Labaudiniere, R. The discovery of tadalafil: A novel and highly selective PDE5 inhibitor. 1: 5,6,11,11a-tetrahydro-1H-imidazo[1′,5′:1,6]pyrido[3,4-b]indole-1,3(2H)-dione analogues. J. Med. Chem. 2003, 46, 4525–4532. [Google Scholar] [CrossRef] [PubMed]
- Daugan, A.; Grondin, P.; Ruault, C.; Le Monnier de Gouville, A.C.; Coste, H.; Linget, J.M.; Kirilovsky, J.; Hyafil, F.; Labaudiniere, R. The discovery of tadalafil: A novel and highly selective PDE5 inhibitor. 2: 2,3,6,7,12,12a-hexahydropyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione analogues. J. Med. Chem. 2003, 46, 4533–4542. [Google Scholar] [CrossRef]
- Gong, B.; Ma, M.; Xie, W.; Yang, X.; Huang, Y.; Sun, T.; Luo, Y.; Huang, J. Direct comparison of tadalafil with sildenafil for the treatment of erectile dysfunction: A systematic review and meta-analysis. Int. Urol. Nephrol. 2017, 49, 1731–1740. [Google Scholar] [CrossRef]
- Peng, Z.; Yang, L.; Dong, Q.; Wei, Q.; Liu, L.; Yang, B. Efficacy and safety of Tadalafil once-a-day versus Tadalafil on-demand in patients with erectile dysfunction: A systematic review and meta-analyses. Urol. Int. 2017, 99, 343–352. [Google Scholar] [CrossRef]
- Washington, S.L., 3rd; Shindel, A.W. A once-daily dose of tadalafil for erectile dysfunction: Compliance and efficacy. Drug Des. Devel. 2010, 4, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Ventimiglia, E.; Capogrosso, P.; Montorsi, F.; Salonia, A. The safety of phosphodiesterase type 5 inhibitors for erectile dysfunction. Expert Opin. Drug Saf. 2016, 15, 141–152. [Google Scholar] [CrossRef]
- Curran, M.; Keating, G. Tadalafil. Drugs 2003, 63, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Forgue, S.T.; Patterson, B.E.; Bedding, A.W.; Payne, C.D.; Phillips, D.L.; Wrishko, R.E.; Mitchell, M.I. Tadalafil pharmacokinetics in healthy subjects. Br. J. Clin. Pharm. 2006, 61, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, E.B. Tadalafil for the treatment of pulmonary arterial hypertension. Expert Opin. Pharm. 2010, 11, 127–132. [Google Scholar] [CrossRef]
- Klinger, J.R. Tadalafil for the treatment of pulmonary arterial hypertension. Expert Rev. Respir. Med. 2011, 5, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Arif, S.A.; Poon, H. Tadalafil: A long-acting phosphodiesterase-5 inhibitor for the treatment of pulmonary arterial hypertension. Clin. Ther. 2011, 33, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K.; Lang, I. Tadalafil for the treatment of pulmonary arterial hypertension. Expert Opin. Pharm. 2012, 13, 747–755. [Google Scholar] [CrossRef]
- Cantrell, M.A.; Baye, J.; Vouri, S.M. Tadalafil: A phosphodiesterase-5 inhibitor for benign prostatic hyperplasia. Pharmacotherapy 2013, 33, 639–649. [Google Scholar] [CrossRef]
- Carson, C.C.; Rosenberg, M.; Kissel, J.; Wong, D.G. Tadalafil—A therapeutic option in the management of BPH-LUTS. Int. J. Clin. Pr. 2014, 68, 94–103. [Google Scholar] [CrossRef]
- Koka, S.; Das, A.; Zhu, S.G.; Durrant, D.; Xi, L.; Kukreja, R.C. Long-acting phosphodiesterase-5 inhibitor tadalafil attenuates doxorubicin-induced cardiomyopathy without interfering with chemotherapeutic effect. J. Pharm. Exp. 2010, 334, 1023–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, P.R.; Tiwari, A.K.; Ohnuma, S.; Lee, J.W.; An, X.; Dai, C.L.; Lu, Q.S.; Singh, S.; Yang, D.H.; Talele, T.T.; et al. The phosphodiesterase-5 inhibitor vardenafil is a potent inhibitor of ABCB1/P-glycoprotein transporter. PLoS ONE 2011, 6, e19329. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Cruickshanks, N.; Tavallai, S.; Webb, T.; Samuel, P.; Conley, A.; Binion, B.; Young, H.F.; Poklepovic, A.; et al. PDE5 inhibitors enhance celecoxib killing in multiple tumor types. J. Cell Physiol. 2015, 230, 1115–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Strada, S.J. The novel functions of cGMP-specific phosphodiesterase 5 and its inhibitors in carcinoma cells and pulmonary/cardiovascular vessels. Curr. Top. Med. Chem. 2007, 7, 437–454. [Google Scholar] [CrossRef]
- Pantziarka, P.; Sukhatme, V.; Crispino, S.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)-selective PDE5 inhibitors as anti-cancer agents. Ecancermedicalscience 2018, 12, 824. [Google Scholar] [CrossRef] [Green Version]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef]
- Capuano, G.; Rigamonti, N.; Grioni, M.; Freschi, M.; Bellone, M. Modulators of arginine metabolism support cancer immunosurveillance. BMC Immunol. 2009, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, T.R.; Mierzwa, M.L.; Wells, S.I.; Fox, S.R.; Ben-Jonathan, N. The cyclic GMP/protein kinase G pathway as a therapeutic target in head and neck squamous cell carcinoma. Cancer Lett. 2016, 370, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Sponziello, M.; Verrienti, A.; Rosignolo, F.; De Rose, R.F.; Pecce, V.; Maggisano, V.; Durante, C.; Bulotta, S.; Damante, G.; Giacomelli, L.; et al. PDE5 expression in human thyroid tumors and effects of PDE5 inhibitors on growth and migration of cancer cells. Endocrine 2015, 50, 434–441. [Google Scholar] [CrossRef]
- Wang, R.; Chen, W.; Zhang, Q.; Liu, Y.; Qiao, X.; Meng, K.; Mao, Y. Phosphodiesterase type 5 inhibitor Tadalafil increases Rituximab treatment efficacy in a mouse brain lymphoma model. J. Neurooncol. 2015, 122, 35–42. [Google Scholar] [CrossRef]
- Hassel, J.C.; Jiang, H.; Bender, C.; Winkler, J.; Sevko, A.; Shevchenko, I.; Halama, N.; Dimitrakopoulou-Strauss, A.; Haefeli, W.E.; Jager, D.; et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology 2017, 6, e1326440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.N.; Li, L.; Kee, C.L.; Ge, X.; Low, M.Y.; Koh, H.L. Screening of synthetic PDE-5 inhibitors and their analogues as adulterants: Analytical techniques and challenges. J. Pharm. Biomed. Anal. 2014, 87, 176–190. [Google Scholar] [CrossRef]
- Kee, C.L.; Ge, X.; Gilard, V.; Malet-Martino, M.; Low, M.Y. A review of synthetic phosphodiesterase type 5 inhibitors (PDE-5i) found as adulterants in dietary supplements. J. Pharm. Biomed. Anal. 2018, 147, 250–277. [Google Scholar] [CrossRef]
- Ahmed, N.S.; Gary, B.D.; Tinsley, H.N.; Piazza, G.A.; Laufer, S.; Abadi, A.H. Design, synthesis and structure-activity relationship of functionalized tetrahydro-beta-carboline derivatives as novel PDE5 inhibitors. Arch. Pharm. (Weinh.) 2011, 344, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Elhady, A.K.; Sigler, S.C.; Noureldin, N.; Canzoneri, J.C.; Ahmed, N.S.; Piazza, G.A.; Abadi, A.H. Structure-based design of novel tetrahydro-beta-carboline derivatives with a hydrophilic side chain as potential phosphodiesterase inhibitors. Sci. Pharm. 2015, 84, 428–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.S.; Ali, A.H.; El-Nashar, S.M.; Gary, B.D.; Fajardo, A.M.; Tinsley, H.N.; Piazza, G.A.; Negri, M.; Abadi, A.H. Exploring the PDE5 H-pocket by ensemble docking and structure-based design and synthesis of novel beta-carboline derivatives. Eur. J. Med. Chem. 2012, 57, 329–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abadi, A.H.; Gary, B.D.; Tinsley, H.N.; Piazza, G.A.; Abdel-Halim, M. Synthesis, molecular modeling and biological evaluation of novel tadalafil analogues as phosphodiesterase 5 and colon tumor cell growth inhibitors, new stereochemical perspective. Eur. J. Med. Chem. 2010, 45, 1278–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beghyn, T.B.; Charton, J.; Leroux, F.; Laconde, G.; Bourin, A.; Cos, P.; Maes, L.; Deprez, B. Drug to genome to drug: Discovery of new antiplasmodial compounds. J. Med. Chem. 2011, 54, 3222–3240. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Wang, H.; Ni, W.; Zheng, X.; Wang, M.; Bao, K.; Ling, D.; Li, X.; Xu, Y.; Zhang, H.; et al. Design, synthesis, and biological evaluation of orally available first-generation dual-target selective inhibitors of acetylcholinesterase (AChE) and phosphodiesterase 5 (PDE5) for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 2018, 9, 328–345. [Google Scholar] [CrossRef]
- Ni, W.; Wang, H.; Li, X.; Zheng, X.; Wang, M.; Zhang, J.; Gong, Q.; Ling, D.; Mao, F.; Zhang, H.; et al. Novel Tadalafil derivatives ameliorates scopolamine-induced cognitive impairment in mice via inhibition of acetylcholinesterase (AChE) and phosphodiesterase 5 (PDE5). ACS Chem. Neurosci. 2018, 9, 1625–1636. [Google Scholar] [CrossRef]
- Borthwick, A.D. 2,5-Diketopiperazines: Synthesis, reactions, medicinal chemistry, and bioactive natural products. Chem. Rev. 2012, 112, 3641–3716. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, P.; Ma, H.; Zhu, W. Developments around the bioactive diketopiperazines: A patent review. Expert Opin. Pat. 2013, 23, 1415–1433. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Zhang, X.; Lai, D.; Zhou, L. Structural diversity and biological activities of the cyclodipeptides from fungi. Molecules 2017, 22, 2026. [Google Scholar] [CrossRef] [Green Version]
- Jurczak, J.; Mieczkowski, A.; Koźmiński, W. A Traceless, Solid-supported synthesis of β-turn mimetics based on the hexahydropyrazino[1,2-a]pyrazine-1,2-dione scaffold. Synthesis 2009, 221–232. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Jurczak, J. A traceless solid-supported synthesis of novel pyrazinediazepinedione derivatives. Tetrahedron 2010, 66, 2514–2519. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Trzybiński, D.; Wilczek, M.; Psurski, M.; Bagiński, M.; Bieszczad, B.; Mroczkowska, M.; Woźniak, K. (S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione—Synthesis and Crystallographic Studies. Molbank 2017, 2017, M964. [Google Scholar] [CrossRef] [Green Version]
- Mieczkowski, A.; Psurski, M.; Bagiński, M.; Bieszczad, B.; Mroczkowska, M.; Wilczek, M.; Czajkowska, J.; Trzybiński, D.; Woźniak, K.; Wietrzyk, J. Novel (S)-1,3,4,12a-tetrahydropyrazino[2,1-c][1,4]benzodiazepine-6,12(2H,11H)-dione derivatives: Selective inhibition of MV-4-11 biphenotypic B myelomonocytic leukemia cells’ growth is accompanied by reactive oxygen species overproduction and apoptosis. Bioorg. Med. Chem. Lett. 2018, 28, 618–625. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Frączyk, T.; Psurski, M.; Wińska, P.; Siedlecki, P.; Dziełak, M.; Trzybiński, D.; Wilczek, M.; Bagiński, M.; Bieszczad, B.; et al. Design and in vitro characterization of tricyclic benzodiazepine derivatives as potent and selective anti-leukemic agents. Chem. Biodivers. 2021, 19, e200733. [Google Scholar] [CrossRef]
- Bieszczad, B.; Garbicz, D.; Trzybiński, D.; Mielecki, D.; Woźniak, K.; Grzesiuk, E.; Mieczkowski, A. Unsymmetrically substituted dibenzo[b,f][1,5]-diazocine-6,12(5H,11H)dione-A convenient scaffold for bioactive molecule design. Molecules 2020, 25, 906. [Google Scholar] [CrossRef] [Green Version]
- Bieszczad, B.; Garbicz, D.; Trzybiński, D.; Dudek, M.K.; Woźniak, K.; Grzesiuk, E.; Mieczkowski, A. Unsymmetrically-substituted 5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione scaffold-A useful tool for bioactive molecules design. Molecules 2020, 25, 2855. [Google Scholar] [CrossRef] [PubMed]
- Bieszczad, B.; Pawlędzio, S.; Polak, K.; Antonowicz, J.; Mieczkowski, A.; Trzybiński, D. Influence of halogen size on the supramolecular and energy landscape of the THF solvates of the halogen derivatives of dianthranilide. CrystEngComm 2020, 22, 5389–5399. [Google Scholar] [CrossRef]
- Bieszczad, B.; Siwek, A.; Wilczek, M.; Trzybiński, D.; Woźniak, K.; Satała, G.; Bojarski, A.J.; Mieczkowski, A. Synthesis, crystal structure and biological activity of novel analogues of tricyclic drugs. Bioorg. Med. Chem. Lett. 2020, 30, 127493. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharm. 1973, 22, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Poppe, H.; Rybalkin, S.D.; Rehmann, H.; Hinds, T.R.; Tang, X.B.; Christensen, A.E.; Schwede, F.; Genieser, H.G.; Bos, J.L.; Doskeland, S.O.; et al. Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 2008, 5, 277–278. [Google Scholar] [CrossRef]
- Blount, M.A.; Beasley, A.; Zoraghi, R.; Sekhar, K.R.; Bessay, E.P.; Francis, S.H.; Corbin, J.D. Binding of tritiated sildenafil, tadalafil, or vardenafil to the phosphodiesterase-5 catalytic site displays potency, specificity, heterogeneity, and cGMP stimulation. Mol. Pharm. 2004, 66, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Rosen, R.C.; Kostis, J.B. Overview of phosphodiesterase 5 inhibition in erectile dysfunction. Am. J. Cardiol. 2003, 92, 9–18. [Google Scholar] [CrossRef]
- Gresele, P.; Momi, S.; Falcinelli, E. Anti-platelet therapy: Phosphodiesterase inhibitors. Br. J. Clin. Pharm. 2011, 72, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajraktari, G.; Burhenne, J.; Bugert, P.; Haefeli, W.E.; Weiss, J. Cyclic guanosine monophosphate modulates accumulation of phosphodiesterase 5 inhibitors in human platelets. Biochem. Pharm. 2017, 145, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Apostoli, G.L.; Solomon, A.; Smallwood, M.J.; Winyard, P.G.; Emerson, M. Role of inorganic nitrate and nitrite in driving nitric oxide-cGMP-mediated inhibition of platelet aggregation in vitro and in vivo. J. Thromb. Haemost. 2014, 12, 1880–1889. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in nultifaceted crystals. Acta Cryst. A 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. No. | 6a | 6b | 7a | 7b | Tadalafil | Cisplatin |
|---|---|---|---|---|---|---|
| Cell Line | IC50 Values (μM) | |||||
| A-431 | 104.1 (29.58–366.3) * | 111.8 (52.68–237.3) * | 63.56 (37.34–108.2) * | 46.56 (28.02–77.39) * | 20.36 (15.13–27.39) * | 1.51 (1.02–2.22) * |
| A549 | [62.67] ** | [62.85] ** | 127.6 (116–140.3) * | 130.6 (119.7–142.5) * | 51.51 (41.69–63.65) * | 27.22 (20.9–35.46) * |
| HCT116 | 102 (73.02–142.4) * | 83.74 (67.19–104.4) * | 52.99 (39.18–71.68) * | 58.73 (39.9–86.44) * | 27.25 (22.29–33.31) * | 7.44 (6.44–8.6) * |
| HCT116p53−/− | 83.21 (21.27–325.5) * | 143 (48.92–417.9) * | 35.93 (20.05–64.39) * | 64.42 (36.06–115.1) * | 19.77 (15.89–24.59) * | 14.12 (10.44–19.09) * |
| U-87 MG | 153.2 (97.54–240.6) * | 151.2 (116.6–196) * | 109.7 (101.4–118.7) * | 98.99 (91.26–107.4) * | 40.66 (33.27–49.7) * | 4.96 (3.39–7.25) * |
| U-251 MG | [55.76] ** | [47.58] ** | [81.34] ** | [52.48] ** | 44.13 (37.43–52.04) * | 2.61 (1.57–4.34) * |
| MCF7 | 53.20 (41.97–67.44) * | 54.05 (44.27–65.99) * | 28.9 (23.15–36.07) * | 36.32 (30.85–42.77) * | 55.29 (46.56–65.64) * | 10.99 (9.22–13.11) * |
| MDA-MB-231 | [84.18] ** | 128 (118.7–138) * | [75.86] ** | 143.6 (132.6–155.5) * | 87.6 (76.56–100.2) * | 22.3 (16.37–30.38) * |
| SKGIIIa | 145.5 (108.9–194.6) * | 87.58 (77.73–98.68) * | 75.94 (63.71–90.52) * | 57.28 (46.02–71.29) * | 35.08 (29.88–41.19) * | 4.84 (2.96–7.93) * |
| HeLa | 119.9 (96.9–148.5) * | 93.49 (74.63–117.1) * | 79.19 (57.88–108.4) * | 78.39 (65.68–93.57) * | 42.29 (33.45–53.46) * | 13.52 (11.46–15.96) * |
| U-2 OS | 93.52 (28.41–307.8) * | 88.26 (70.31–110.8) * | 95.39 (83.27–109.3) * | 88.83 (74.16–106.4) * | 35.06 (31.77–38.7) * | 7.55 (6.36–8.96) * |
| HEPG2 | [50.9] ** | [76.76] ** | 103.3 (83.65–127.5) * | 123.9 (109.7–139.9) * | 46.56 (35.22–61.55) * | 7.8 (6.77–8.99) * |
| BICR 18 | [72.57] ** | [56.72] ** | 137.1 (119.8–156.9) * | 141.9 (108.6–185.5) * | 52.53 (34.89–79.08) * | 1.69 (1.15–2.48) * |
| HEK 293T | 10.52 (6.64–16.68) * | 12.95 (9.9–16.93) * | 12.34 (9.59–15.87) * | 13.06 (10.98–15.55) * | 30.96 (19.52–49.12) * | 6.25 (5.17–7.55) * |
| MRC-5 | [56.68] ** | 127 (103.7–155.6) * | 116.3 (109.1–124) * | 96.57 (81.85–113.9) * | 28.72 (23.33–35.37) * | 3.92 (3.23–4.76) * |
| BJ | [60.84] ** | 142.8 (105.1–194.1) * | 117 (103.9–131.7) * | 102.6 (95.38–110.4) * | 40.48 (33.11–49.50) * | 5.58 (4.64–6.71) * |
| Compound No. | IC50 (μM) | Ki (μM) |
|---|---|---|
| 6a | 137 ± 47 | 1.4 ± 0.8 |
| 6b | 260 ± 210 | 2.6 ± 2.7 |
| 7a | 22 ± 12 | 0.22 ± 0.17 |
| 7b | 39 ± 27 | 0.39 ± 0.36 |
| tadalafil | 0.072 ± 0.031 | (7.1 ± 4.8) × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mieczkowski, A.; Speina, E.; Trzybiński, D.; Winiewska-Szajewska, M.; Wińska, P.; Borsuk, E.M.; Podsiadła-Białoskórska, M.; Przygodzki, T.; Drabikowski, K.; Stanczyk, L.; et al. Diketopiperazine-Based, Flexible Tadalafil Analogues: Synthesis, Crystal Structures and Biological Activity Profile. Molecules 2021, 26, 794. https://doi.org/10.3390/molecules26040794
Mieczkowski A, Speina E, Trzybiński D, Winiewska-Szajewska M, Wińska P, Borsuk EM, Podsiadła-Białoskórska M, Przygodzki T, Drabikowski K, Stanczyk L, et al. Diketopiperazine-Based, Flexible Tadalafil Analogues: Synthesis, Crystal Structures and Biological Activity Profile. Molecules. 2021; 26(4):794. https://doi.org/10.3390/molecules26040794
Chicago/Turabian StyleMieczkowski, Adam, Elżbieta Speina, Damian Trzybiński, Maria Winiewska-Szajewska, Patrycja Wińska, Ewelina M. Borsuk, Małgorzata Podsiadła-Białoskórska, Tomasz Przygodzki, Krzysztof Drabikowski, Lidia Stanczyk, and et al. 2021. "Diketopiperazine-Based, Flexible Tadalafil Analogues: Synthesis, Crystal Structures and Biological Activity Profile" Molecules 26, no. 4: 794. https://doi.org/10.3390/molecules26040794