
The Effect of Vicinal Difluorination on the Conformation and Potency of Histone Deacetylase Inhibitors


 and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Conformational Analysis
2.3. HDAC Inhibition
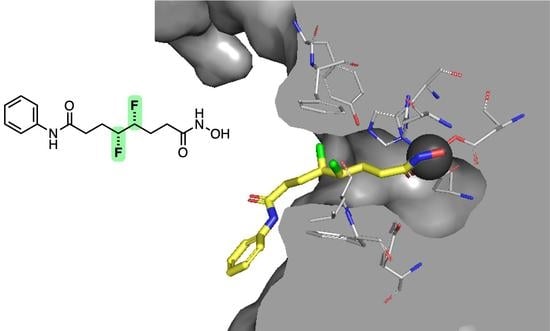
2.4. Docking Studies
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic Protein Families: A New Frontier for Drug Discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W. HDAC Expression and Clinical Prognosis in Human Malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl Sulfoxide to Vorinostat: Development of this Histone Deacetylase Inhibitor as an Anticancer Drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Su, G.H.; Sohn, T.A.; Ryu, B.; Kern, S.E. A Novel Histone Deacetylase Inhibitor Identified by High-Throughput Transcriptional Screening of a Compound Library. Cancer Res. 2000, 60, 3137–3142. [Google Scholar]
- Micelli, C.; Rastelli, G. Histone Deacetylases: Structural Determinants of Inhibitor Selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Melesina, J.; Simoben, C.V.; Praetorius, L.; Bülbül, E.F.; Robaa, D.; Sippl, W. Strategies to Design Selective Histone Deacetylase Inhibitors. ChemMedChem 2021. [Google Scholar] [CrossRef]
- Porter, N.J.; Osko, J.D.; Diedrich, D.; Kurz, T.; Hooker, J.M.; Hansen, F.K.; Christianson, D.W. Histone Deacetylase 6-Selective Inhibitors and the Influence of Capping Groups on Hydroxamate-Zinc Denticity. J. Med. Chem. 2018, 61, 8054–8060. [Google Scholar] [CrossRef] [PubMed]
- Linciano, P.; Benedetti, R.; Pinzi, L.; Russo, F.; Chianese, U.; Sorbi, C.; Altucci, L.; Rastelli, G.; Brasili, L.; Franchini, S. Investigation of the Effect of Different Linker Chemotypes on the Inhibition of Histone Deacetylases (HDACs). Bioorg. Chem. 2021, 106, 104462. [Google Scholar] [CrossRef] [PubMed]
- Thiehoff, C.; Rey, Y.P.; Gilmour, R. The Fluorine Gauche Effect: A Brief History. Israel J. Chem. 2017, 57, 92–100. [Google Scholar] [CrossRef]
- Fox, S.J.; Gourdain, S.; Coulthurst, A.; Fox, C.; Kuprov, I.; Essex, J.W.; Skylaris, C.-K.; Linclau, B. A Computational Study of Vicinal Fluorination in 2,3-Difluorobutane: Implications for Conformational Control in Alkane Chains. Chem. Eur. J. 2015, 21, 1682–1691. [Google Scholar] [CrossRef]
- Tavasli, M.; O’Hagan, D.; Pearson, C.; Petty, M.C. The Fluorine Gauche Effect. Langmuir Isotherms Report the Relative Conformational Stability of (±)-Erythro- and (±)-Threo-9,10-Difluorostearic Acids. Chem. Commun. 2002, 1226–1227. [Google Scholar] [CrossRef]
- Fischer, S.; Huwyler, N.; Wolfrum, S.; Carreira, E.M. Synthesis and Biological Evaluation of Bromo- and Fluorodanicalipin A. Angew. Chem. Int. Ed. 2016, 55, 2555–2558. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Jordan, M.J.T.; Gavande, N.; Doddareddy, M.R.; Chebib, M.; Hunter, L. The Enantiomers of Syn-2,3-Difluoro-4-Aminobutyric Acid Elicit Opposite Responses at the GABAC Receptor. Chem. Commun. 2012, 48, 829–831. [Google Scholar] [CrossRef]
- Hu, X.-G.; Thomas, D.S.; Griffith, R.; Hunter, L. Stereoselective Fluorination Alters the Geometry of a Cyclic Peptide: Exploration of Backbone-Fluorinated Analogs of Unguisin A. Angew. Chem. Int. Ed. 2014, 53, 6176–6179. [Google Scholar] [CrossRef]
- Au, C.; Gonzalez, C.; Leung, Y.C.; Mansour, F.; Trinh, J.; Wang, Z.; Hu, X.-G.; Griffith, R.; Pasquier, E.; Hunter, L. Tuning the Properties of a Cyclic RGD-Containing Tetrapeptide through Backbone Fluorination. Org. Biomol. Chem. 2019, 17, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Lizarme-Salas, Y.; Ariawan, A.D.; Ratnayake, R.; Luesch, H.; Finch, A.; Hunter, L. Vicinal Difluorination as a C=C Surrogate: An Analog of Piperine with Enhanced Solubility, Photostability, and Acetylcholinesterase Inhibitory Activity. Beilstein J. Org. Chem. 2020, 16, 2663–2670. [Google Scholar] [CrossRef]
- Linclau, B.; Arda, A.; Reichardt, N.-C.; Sollogoub, M.; Unione, L.; Vincent, S.P.; Jimenez-Barbero, J. Fluorinated Carbohydrates as Chemical Probes for Molecular Recognition Studies. Current Status and Perspectives. Chem. Soc. Rev. 2020, 49, 3863–3888. [Google Scholar] [CrossRef]
- Hofman, G.-J.; Ottoy, E.; Light, M.E.; Kieffer, B.; Kuprov, I.; Martins, J.C.; Sinnaeve, D.; Linclau, B. Minimising Conformational Bias in Fluoroprolines through Vicinal Difluorination. Chem. Commun. 2018, 54, 5118–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, T.J.; Lipiński, R.M. Interplay of Cascade Oxidative Cyclization and Hydride Shifts in the Synthesis of the ABC Spiroketal Ring System of Pectenotoxin-4. Angew. Chem. Int. Ed. 2013, 52, 2491–2494. [Google Scholar] [CrossRef]
- Nicoletti, M.; O’Hagan, D.; Slawin, A.M.Z. α,β,γ-Trifluoroalkanes: A Stereoselective Synthesis Placing Three Vicinal Fluorines along a Hydrocarbon Chain. J. Am. Chem. Soc. 2005, 127, 482–483. [Google Scholar] [CrossRef]
- CCDC 2084780 and 2084781 Contain the Crystallographic Information for Compounds 14 and 18 Respectively. Available online: www.ccdc.cam.ac.uk (accessed on 24 May 2021).
- Gore, V.G.; Patil, M.S.; Bhalerao, R.A.; Mande, H.M.; Sandeep, G. Process for Preparation of Vorinostat. U.S. Patent 9162974B2, 20 October 2015. [Google Scholar]
- Mai, A.; Esposito, M.; Sbardella, G.; Massa, S. A New Facile and Expeditious Synthesis of N-Hydroxy-N′-Phenyloctanediamide, a Potent Inducer of Terminal Cytodifferentiation. Org. Prep. Proced. Int. 2001, 33, 391–394. [Google Scholar] [CrossRef]
- Barco, A.; Benetti, S.; Pollini, G.P.; Taddia, R. Synthesis of Cis-4-Octene-1,8-Dioic Acid and its Esters. Org. Prep. Proc. Int. 1974, 6, 217–220. [Google Scholar] [CrossRef]
- Banik, S.M.; Medley, J.W.; Jacobsen, E.N. Catalytic, Diastereoselective 1,2-Difluorination of Alkenes. J. Am. Chem. Soc. 2016, 138, 5000–5003. [Google Scholar] [CrossRef] [Green Version]
- Olszewska, B.; Kryczka, B.; Zawisza, A. Asymmetric Synthesis of Optically Active 2-Vinylpyrrolidines and 2-Vinylpiperidines by Palladium-Catalysed Cyclisation of Amino Allylic Carbonates. Tetrahedron Lett. 2012, 53, 6826–6829. [Google Scholar] [CrossRef]
- Patel, A.R.; Liu, F. Diastereospecific Fluorination of Substituted Azepanes. Tetrahedron 2013, 69, 744–752. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Banks, J.W.; Batsanov, A.S.; Howard, J.A.K.; O’Hagan, D.; Rzepa, H.S.; Martin-Santamaria, S. The Preferred Conformation of α-Fluoroamides. J. Perkin Soc. Perkin Trans. 2 1999, 2409–2411. [Google Scholar] [CrossRef]
- Müller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.-J.; Dietel, M.; Weichert, W.; Denkert, C. Differential Expression of Histone Deacetylases HDAC1, 2 and 3 in Human Breast Cancer—Overexpression of HDAC2 and HDAC3 is Associated with Clinicopathological Indicators of Disease Progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janaki, R.M.; Naushad, S.M.; Lavanya, A.; Srinivas, C.; Anjana Devi, T.; Sampathkumar, S.; Dharan, D.B.; Bhadra, M.P. Scriptaid Cause Histone Deacetylase Inhibition and Cell Cycle Arrest in HeLa Cancer Cells: A Study on Structural and Functional Aspects. Gene 2017, 627, 379–386. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Structure | Conformations | Calculated (Black) vs. Experimental (Grey) NMR Coupling Constants |
|---|---|---|
 |  |  |
 |  |  |
 |  | n/a * |
 |  |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ariawan, A.D.; Mansour, F.; Richardson, N.; Bhadbhade, M.; Ho, J.; Hunter, L. The Effect of Vicinal Difluorination on the Conformation and Potency of Histone Deacetylase Inhibitors. Molecules 2021, 26, 3974. https://doi.org/10.3390/molecules26133974
Ariawan AD, Mansour F, Richardson N, Bhadbhade M, Ho J, Hunter L. The Effect of Vicinal Difluorination on the Conformation and Potency of Histone Deacetylase Inhibitors. Molecules. 2021; 26(13):3974. https://doi.org/10.3390/molecules26133974
Chicago/Turabian StyleAriawan, A. Daryl, Flora Mansour, Nicole Richardson, Mohan Bhadbhade, Junming Ho, and Luke Hunter. 2021. "The Effect of Vicinal Difluorination on the Conformation and Potency of Histone Deacetylase Inhibitors" Molecules 26, no. 13: 3974. https://doi.org/10.3390/molecules26133974