
Design, Synthesis and Antiparasitic Evaluation of Click Phospholipids

 , ,
, ,  , , , , and
, , , , and
Abstract
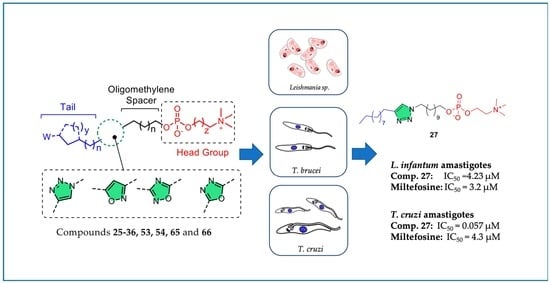
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. General Procedure for the Synthesis of Alcohols 4–6
5-bromopentan-1-ol (4)
6-bromohexan-1-ol (5)
11-bromoundecan-1-ol (6)
3.1.3. General Procedure for the Synthesis of Azides 7–9
5-azidopentan-1-ol (7)
6-azidohexan-1-ol (8)
11-azidoundecan-1-ol (9)
3.1.4. Benzyl undec-10-ynoate (15)
3.1.5. Benzyl dec-9-ynylcarbamate (16)
3.1.6. General Procedure for the Synthesis of Triazoles 17–24
11-(4-pentyl-1H-1,2,3-triazol-1-yl)undecan-1-ol (17)
6-(4-decyl-1H-1,2,3-triazol-1-yl)hexan-1-ol (18)
11-(4-decyl-1H-1,2,3-triazol-1-yl)undecan-1-ol (19)
11-(4-(cyclopentylmethyl)-1H-1,2,3-triazol-1-yl)undecan-1-ol (20)
11-(4-(cyclohexylmethyl)-1H-1,2,3-triazol-1-yl)undecan-1-ol (21)
Benzyl 9-(1-(11-hydroxyundecyl)-1H-1,2,3-triazol-4-yl)nonanoate (22)
Benzyl 8-(1-(5-hydroxypentyl)-1H-1,2,3-triazol-4-yl)octylcarbamate (23)
Benzyl 8-(1-(11-hydroxyundecyl)-1H-1,2,3-triazol-4-yl)octylcarbamate (24)
3.1.7. General Procedure for the Synthesis of Ether Phospholipids (Method A)
11-(4-pentyl-1H-1,2,3-triazol-1-yl)undecyl (2-(trimethylammonio)ethyl) phosphate inner salt (25)
6-(4-decyl-1H-1,2,3-triazol-1-yl)hexyl (2-(trimethylammonio)ethyl) phosphate inner salt (26)
11-(4-decyl-1H-1,2,3-triazol-1-yl)undecyl (2-(trimethylammonio)ethyl) phosphate inner salt (27)
11-(4-(cyclopentylmethyl)-1H-1,2,3-triazol-1-yl)undecyl (2-(trimethylammo nio)ethyl) phosphate inner salt (28)
11-(4-(cyclohexylmethyl)-1H-1,2,3-triazol-1-yl)undecyl (2-(trimethylammonio) ethyl) phosphate inner salt (29)
11-(4-(9-(benzyloxy)-9-oxononyl)-1H-1,2,3-triazol-1-yl)undecyl (2-(trimethyl ammonio)ethyl) phosphate inner salt (30)
5-(4-(8-(((benzyloxy)carbonyl)amino)octyl)-1H-1,2,3-triazol-1-yl)pentyl (2-(trimethylammonio)ethyl) phosphate inner salt (31)
11-(4-(8-(((benzyloxy)carbonyl)amino)octyl)-1H-1,2,3-triazol-1-yl)undecyl (2-(trimethylammonio)ethyl) phosphate inner salt (32)
11-(4-(cyclohexylmethyl)-1H-1,2,3-triazol-1-yl)undecyl (3-(trimethylammonio) propyl) phosphate inner salt (33)
3.1.8. General Procedure for the Hydrogenolysis (Compounds 34–36)
11-(4-(8-carboxyoctyl)-1H-1,2,3-triazol-1-yl)undecyl (2-(trimethylammonio) ethyl) phosphate inner salt (34)
5-(4-(8-aminooctyl)-1H-1,2,3-triazol-1-yl)pentyl (2-(trimethylammonio)ethyl) phosphate inner salt (35)
11-(4-(8-aminooctyl)-1H-1,2,3-triazol-1-yl)undecyl (2-(trimethylammonio) ethyl) phosphate inner salt (36)
3.1.9. 11-bromoundecanal oxime (38)
3.1.10. 3-(10-bromodecyl)-5-decylisoxazole (39)
3.1.11. 3-(10-hydroxydecyl)-5-decylisoxazole (40)
3.1.12. Nonanenitrile (43)
3.1.13. N’-hydroxynonanimidamide (44)
3.1.14. General Procedure for the Synthesis of Compounds 47 and 48
5-(5-bromopentyl)-3-octyl-1,2,4-oxadiazole (47)
5-(10-bromodecyl)-3-octyl-1,2,4-oxadiazole (48)
3.1.15. General Procedure for the Synthesis of Compounds 51 and 52
5-(3-octyl-1,2,4-oxadiazol-5-yl)pentan-1-ol (51)
10-(3-octyl-1,2,4-oxadiazol-5-yl)decan-1-ol (52)
3.1.16. General Procedure for the Synthesis of Compounds 57 and 58
n-octanoic Acid Hydrazide (57)
n-decanoic Acid Hydrazide (58)
3.1.17. General Procedure for the Synthesis of Compounds 59 and 60
2-(10-bromodecyl)-5-heptyl-1,3,4-oxadiazole (59)
2-(10-bromodecyl)-5-undecyl-1,3,4-oxadiazole (60)
3.1.18. General Procedure for the Synthesis of 63 and 64
10-(5-heptyl-1,3,4-oxadiazol-2-yl)decan-1-ol (63)
10-(5-undecyl-1,3,4-oxadiazol-2-yl)decan-1-ol (64)
3.1.19. General Procedure for the Synthesis of Ether Phospholipids (Method B)
10-(5-decylisoxazol-3-yl)decyl (2-(trimethylammonio)ethyl) phosphate inner salt (41)
5-(3-octyl-1,2,4-oxadiazol-5-yl)pentyl (2-(trimethylammonio)ethyl) phosphate inner salt (53)
10-(3-octyl-1,2,4-oxadiazol-5-yl)decyl (2-(trimethylammonio)ethyl) phosphate inner salt (54)
10-(5-heptyl-1,3,4-oxadiazol-2-yl)decyl (2-(trimethylammonio)ethyl) phosphate inner salt (65)
2-(trimethylammonio)ethyl (10-(5-undecyl-1,3,4-oxadiazol-2-yl)decyl) phosphate inner salt (66)
3.2. Biological Evaluation
3.2.1. Parasites
3.2.2. In-Vitro Evaluation of Activity Against L. donovani Intramacrophage Amastigotes
3.2.3. In-Vitro Evaluation of Activity Against L. infantum MHOM/TN/80/LEM235 Intramacrophage Amastigotes
3.2.4. In-Vitro Evaluation of Activity Against L. infantum MHOM/MA/67/ITMAP-263 Intramacrophage Amastigotes
3.2.5. In-Vitro Evaluation of Activity Against T. brucei Bloodstream Forms
3.2.6. In-Vitro Evaluation of Antitrypanosomal Activity
3.2.7. Cytotoxicity Assessment against THP-1 Macrophages
3.2.8. Early ADMET Profiling
3.2.9. Electron Microscopy
Scanning Electron Microscopy
Transmission Electron Microscopy
3.2.10. Flow Cytometry and Fluorescence Microscopy for Apoptosis Detection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Houweling, T.A.J.; Karim-Kos, H.E.; Kulik, M.C.; Stolk, W.A.; Haagsma, J.A.; Lenk, E.J.; Richardus, J.H.; de Vlas, S.J. Socioeconomic Inequalities in Neglected Tropical Diseases: A Systematic Review. PLoS Negl. Trop. Dis. 2016, 10, e0004546. [Google Scholar] [CrossRef] [Green Version]
- Hotez, P.J.; Aksoy, S.; Brindley, P.J.; Kamhawi, S. What constitutes a neglected tropical disease? PLoS Negl. Trop. Dis. 2020, 14, e0008001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.who.int/news/item/30-01-2021-neglected-tropical-diseases-who-launches-new-road-map-to-end-suffering-by-2030page (accessed on 4 May 2021).
- Savoia, D. Recent updates and perspectives on leishmaniasis. J. Infect. Dev. Ctries. 2015, 9, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Dujardin, J.C.; Decuypere, S. Epidemiology of leishmaniasis in the time of drug resistance (the Miltefosine era). In Drug Resistance in Leishmania Parasites: Consequences, Molecular Mechanisms and Possible Treatments; Ponte-Sucre, A., Diaz, E., Padron-Nieves, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 65–83. [Google Scholar]
- Chulay, J.D.; Fleckenstein, L.; Smith, D.H. Pharmacokinetics of antimony during treatment of visceral leishmaniasis with sodium stibogluconate or meglumine antimoniate. Trans. R. Soc. Trop. Med. Hyg. 1988, 82, 69–72. [Google Scholar] [CrossRef]
- Croft, S.L.; Coombs, G.H. Leishmaniasis—Current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003, 19, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Ahmed, A.H.A.; Brocchini, S.; Croft, S.L. Recent advances in development of amphotericin B formulations for the treatment of visceral leishmaniasis. Curr. Opin. Infect. Dis. 2012, 25, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Sands, M.; Kron, M.A.; Brown, R.B. Pentamidine: A review. Rev. Infect. Dis. 1985, 7, 625–634. [Google Scholar] [CrossRef]
- Wispelwey, B.; Pearson, R.D. Pentamidine: A Review. Infect. Control Hosp. Epidemiol. 1991, 12, 375–382. [Google Scholar] [CrossRef]
- Sundar, S.; Olliaro, P.L. Miltefosine in the treatment of leishmaniasis: Clinical evidence for informed clinical risk management. Ther. Clin. Risk. Manag. 2007, 3, 733–740. [Google Scholar] [PubMed]
- Sundar, S.; Singh, A.; Rai, M.; Prajapati, V.K.; Singh, A.K.; Ostyn, B.; Boelaert, M.; Dujardin, J.-C.; Chakravarty, J. Efficacy of Miltefosine in the Treatment of Visceral Leishmaniasis in India After a Decade of Use. Clin. Infect. Dis. 2012, 55, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.K.; Sinha, P.K.; Sundar, S.; Thakur, C.P.; Jha, T.K.; Pandey, K.; Das, V.R.; Kumar, N.; Lal, C.; Singh, N.V.V.P.; et al. Phase 4 trial of miltefosine for the treatment of Indian visceral leishmaniasis. J. Infect. Dis. 2007, 196, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://www.who.int/chagas/disease/en/ (accessed on 4 May 2021).
- Viotti, R.; Vigliano, C.; Lococo, B.; Alvarez, M.G.; Petti, M.; Bertocchi, G.; Armenti, A. Side effects of benznidazole as treatment of Chagas disease: Fears and realities. Expert. Rev. Anti-Infect. Ther. 2009, 7, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Ademar Sales, P., Jr.; Molina, I.; Fonseca Murta, S.M.; Sánchez-Montalvá, A.; Salvador, F.; Corrêa-Oliveira, R.; Martins Carneiro, C. Experimental and Clinical Treatment of Chagas Disease: A Review. Am. J. Trop. Med. Hyg. 2017, 97, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, V.; Dias, N.; Paiva, T.; Hagström-Bex, L.; Nitz, N.; Pratesi, R.; Hecht, M. Current trends in the pharmacological management of Chagas disease. Int. J. Parasitol. Drugs Drug. Resist. 2020, 12, 7–17. [Google Scholar] [CrossRef]
- Simarro, P.P.; Cecchi, G.; Franco, J.R.; Paone, M.; Diarra, A.; Ruiz-Postigo, J.A.; Fèvre, E.M.; Mattioli, R.C.; Jannin, J.G. Estimating and mapping the population at risk of sleeping sickness. PLOS Negl. Trop. Dis. 2012, 6, e1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairlamb, A.H.; Henderson, G.B.; Cerami, A. Trypanothione is the primary target for arsenical drugs against African trypanosomes. Proc. Natl. Acad. Sci. USA 1989, 86, 2607–2611. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, P.G.E. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2003, 12, 186–194. [Google Scholar] [CrossRef]
- Calogeropoulou, T.; Angelou, P.; Detsi, A.; Fragiadaki, I.; Scoulica, E. Design and synthesis of potent antileishmanial cycloalkylidene-substituted ether phospholipid derivatives. J. Med. Chem. 2008, 51, 897–908. [Google Scholar] [CrossRef]
- Chazapi, E.; Magoulas, G.E.; Prousis, K.C.; Calogeropoulou, T. Phospholipid analogues as chemotherapeutic agents against trypanosomatids. Curr. Pharm. Des. 2021. online ahead of print. [Google Scholar] [CrossRef]
- De Souza, W.; Godinho, J.; Barrias, E.; Roussaki, M.; Rodrigues, J.C.F.F.; Calogeropoulou, T. Effects of Phospholipid Analogues on Trypanosomatids. Molecular Biology of Kinetoplastid Parasites; Caister Academic Press: Poole, UK, 2018; pp. 221–242. [Google Scholar] [CrossRef] [Green Version]
- Godinho, J.L.P.; Georgikopoulou, K.; Calogeropoulou, T.; de Souza, W.; Rodrigues, J.C.F. A novel alkyl phosphocholine-dinitroaniline hybrid molecule exhibits biological activity in vitro against Leishmania amazonensis. Exp. Parasitol. 2013, 135, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Koufaki, M.; Polychroniou, V.; Calogeropoulou, T.; Tsotinis, A.; Drees, M.; Fiebig, H.H.; LeClerc, S.; Hendriks, H.R.; Makriyannis, A. Alkyl and Alkoxyethyl Antineoplastic Phospholipids. J. Med. Chem. 1996, 39, 2609–2614. [Google Scholar] [CrossRef]
- Avlonitis, N.; Lekka, E.; Detsi, A.; Koufaki, M.; Calogeropoulou, T.; Scoulica, E.; Siapi, E.; Kyrikou, I.; Mavromoustakos, T.; Tsotinis, A.; et al. Antileishmanial ring-substituted ether phospholipids. J. Med. Chem. 2003, 46, 755–767. [Google Scholar] [CrossRef]
- Barrias, E.; Reignault, L.C.; Calogeropoulou, T.; de Souza, W. In vitro activities of adamantylidene-substituted alkylphosphocholine TCAN26 against Trypanosoma cruzi: Antiproliferative and ultrastructural effects. Exp. Parasitol. 2019, 206, 107730. [Google Scholar] [CrossRef]
- Papanastasiou, I.; Prousis, K.C.; Georgikopoulou, K.; Pavlidis, T.; Scoulica, E.; Kolocouris, N.; Calogeropoulou, T. Design and synthesis of new adamantyl-substituted antileishmanial ether phospholipids. Bioorg. Med. Chem. Lett. 2010, 20, 5484–5487. [Google Scholar] [CrossRef]
- Fragiadaki, I.; Katogiritis, A.; Calogeropoulou, T.; Brückner, H.; Scoulica, E. Synergistic combination of alkylphosphocholines with peptaibols in targeting Leishmania infantum in vitro. Int. J. Parasitol. Drugs Drug. Resist. 2018, 8, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.M.; Kumar, A.; Dwivedi, J.; Singh, R. A review: Biological significances of heterocyclic compounds. Inter. J. Pharma Sci. Res. 2013, 4, 66–77. [Google Scholar]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.S.; Kavitha, H.P. Synthesis and biological applications of triazole derivatives-a review. Mini-Rev. Org. Chem. 2013, 10, 40–65. [Google Scholar] [CrossRef]
- Sysak, A.; Obmińska-Mrukowicz, B. Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur. J. Med. Chem. 2017, 137, 292–309. [Google Scholar] [CrossRef]
- Bora, R.O.; Dar, B.; Pradhan, V.; Farooqui, M. [1,2,4]-Oxadiazoles: Synthesis and biological applications. Mini Rev. Med. Chem. 2014, 14, 355–369. [Google Scholar] [CrossRef]
- Khalilullah, H.; Ahsan, M.J.; Hedaitullah, M.; Khan, S.; Ahmed, B. 1,3,4-Oxadiazole: A biologically active scaffold. Mini Rev. Med. Chem. 2012, 12, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Dickschat, J.S.; Bode, H.B.; Kroppenstedt, R.M.; Müller, R.; Schulz, S. Biosynthesis of iso-fatty acids in myxobacteria. Org. Biomol. Chem. 2005, 3, 2824–2831. [Google Scholar] [CrossRef] [PubMed]
- Eaton, P.E.; Ravi Shankar, B.K. Synthesis of 1,4-dinitrocubane. J. Org. Chem. 1984, 49, 185–186. [Google Scholar] [CrossRef]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar] [CrossRef]
- Houghton, S.R.; Furst, L.; Boddy, C.N. Biomimetic transannular oxa-conjugate addition approach to the 2,6-disubstituted dihydropyran of laulimalide yields an unprecedented transannular oxetane. J. Org. Chem. 2009, 74, 1454–1463. [Google Scholar] [CrossRef]
- Croft, S.L.; Yardley, V.; Kendrick, H. Drug sensitivity of Leishmania species: Some unresolved problems. Trans. R. Soc. Trop. Med. Hyg. 2002, 96, S127–S129. [Google Scholar] [CrossRef]
- Palacios, G.; Parodi, A.; Upegui, Y.A.; Montoya, A.; Pulido, S.; Vélez, I.D.; Robledo, S.M. Studies in vitro on infectivity and sensitivity to antileishmanial drugs in New World Leishmania species transfected with the green fluorescent protein [pIR3(-)-eGFP]. Parasitology 2017, 144, 1718–1725. [Google Scholar] [CrossRef]
- Bagley, R.G.; Kurtzberg, L.; Rouleau, C.; Yao, M.; Teicher, B.A. Erufosine, an alkylphosphocholine, with differential toxicity to human cancer cells and bone marrow cells. Cancer Chemother. Pharmacol. 2011, 68, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Fiegl, M.; Lindner, L.H.; Juergens, M.; Eibl, H.; Hiddemann, W.; Braess, J. Erufosine, a novel alkylphosphocholine, in acute myeloid leukemia: Single activity and combination with other antileukemic drugs. Cancer Chemother. Pharmacol. 2007, 62, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyekwelu, K.C. Life Cycle of Trypanosoma cruzi in the Invertebrate and the Vertebrate Hosts. 2019. IntechOpen. Available online: https://www.intechopen.com/books/biology-of-em-trypanosoma-cruzi-em-/life-cycle-of-em-trypanosoma-cruzi-em-in-the-invertebrate-and-the-vertebrate-hosts (accessed on 9 May 2021).
- Niwa, M.; Higashizaki, T.; Higashi, N. Aggregation properties of oligo(methacrylic acid)-shelled dendrimer and its microenvironment in aqueous solutions. Tetrahedron 2003, 59, 4011–4015. [Google Scholar] [CrossRef]
- Popovitz-Biro, R.; Hill, K.; Shavit, E.; Hung, D.J.; Lahav, M.; Leiserowitz, L.; Sagiv, J.; Hsiung, H.; Meredith, G.R.; Vanherzeele, H. A new series of amphiphilic molecules forming stable Z-type (polar) Langmuir-Blodgett films. J. Am. Chem. Soc. 1990, 112, 2498–2506. [Google Scholar] [CrossRef]
- Moreno, P.; Quéléver, G.; Peng, L. Synthesis of poly(aminoester) dendrimers via ‘click’ chemistry in combination with the divergent and convergent strategies. Tetrahedron Lett. 2015, 56, 4043–4046. [Google Scholar] [CrossRef]
- Kataoka, K.; Nishiyama, N.; Lee, Y.; Ishii, T. Protein charge regulator and protein-encapsulating polymer micelle complex. WO2010/106700 A1, 23 September 2010. [Google Scholar]
- Andersen, S.M.; Ling, C.-C.; Zhang, P.; Townson, K.; Willison, H.J.; Bundle, D.R. Synthesis of gangliosideepitopes for oligosaccharide specific immunoadsorption therapy of Guillian-Barré syndrome. Org. Biomol. Chem. 2004, 2, 1199–1212. [Google Scholar] [CrossRef] [PubMed]
- Hähsler, M.; Behrens, S. Dendritic Ligands for Magnetic Suspensions in Liquid Crystals. Eur. J. Org. Chem. 2019, 7820–7830. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, M.; Joshi, K.S.; Aware, V.S.; Sarde, A.G.; Bagul, S.M.; Manohar, S.M. Oxadiazole compounds, their preparation and use. WO2011/104680 A1, 1 September 2011. [Google Scholar]
- Saha, A.; Kumar, R.; Kumar, R.; Devakumar, C. Development and assessment of green synthesis of hydrazides. Indian, J. Org. Chem. Sect. B. Org. Med. Chem. 2010, 49, 526–531. [Google Scholar]
- Pore, V.S.; Agalave, S.G.; Pharande, S.G.; Patilb, P.A.; Kotmale, A.S. Bile acid hydrazides: Gelation, structural, physical and spectroscopic properties. New J. Chem. 2015, 39, 453–460. [Google Scholar] [CrossRef]
- Decuypere, S.; Rijal, S.; Yardley, V.; De Doncker, S.; Laurent, T.; Khanal, B.; Chappuis, F.; Dujardin, J.C. Gene expression analysis of the mechanism of natural Sb(V) resistance in Leishmania donovani isolates from Nepal. Antimicrob. Agents Chemother. 2005, 49, 4616–4621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bifeld, E.; Nevado, T.P.; Bartsch, J.; Eick, J.; Clos, J. A versatile qPCR assay to quantify trypanosomatidic infections of host cells and tissues. Med. Microbiol. Immunol. 2016, 205, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Bifeld, E. Generation of bone marrow-derived macrophages for in vitro infection experiments. In Leishmania: Methods and Protocols; Clos, J., Ed.; Springer: New York, NY, USA, 2019; pp. 237–248. [Google Scholar]
- Bifeld, E. Quantification of Intracellular Leishmania spp. Using Real-Time Quantitative PCR (qPCR). In Leishmania: Methods and Protocols; Clos, J., Ed.; Springer: New York, NY, USA, 2019; pp. 249–263. [Google Scholar]
- Sereno, D.; Cavaleyra, M.; Zemzoumi, K.; Maquaire, S.; Ouaissi, A.; Lemesre, J.L. Axenically grown amastigotes of Leishmania infantum used as an in vitro model to investigate the pentavalent antimony mode of action. Antimicrob. Agents Chemother. 1998, 42, 3097–3102. [Google Scholar] [CrossRef] [Green Version]
- Bowling, T.; Mercer, L.; Don, R.; Jacobs, R.; Nare, B. Application of a resazurin-based high-throughput screening assay for the identification and progression of new treatments for human African trypanosomiasis. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 262–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L. infantum (MHOM/MA/67/ITMAP-263) Intracellular Amastigotes | Toxicity CC50 ± SD Or CC50 Interval Estimation (μM) | Selectivity Index CC50/IC50 | ||
|---|---|---|---|---|
| Compound | Mean % Inhibition ± SD at 10μM or (1 μM) | IC50 ± SD (μM) | ||
| 25 | 32 ± 19 | 34.3 ± 19.4 | >100 | >3 |
| 26 | 44 ± 17 | >12.5 | ||
| 27 | 78 ± 21 | 4.23 ± 0.94 | 50–100 | 11.8–23.6 |
| 28 | N.A. | >100 | ||
| 29 | 43 ± 1 | >100 | ||
| 30 | N.A. | >100 | ||
| 31 | N.A. | >100 | ||
| 32 | 5 ± 18 | >100 | ||
| 33 | 12 ± 5 | > 10 | ||
| 34 | 10 ± 9 | 50–100 | ||
| 35 | N.A. | >100 | ||
| 36 | N.A. | >100 | ||
| 41 | 93.7 ± 10 | 12.5–25 | ||
| 53 | 98.4 ± 1 (34.6 ± 19) | 12.5–25 | ||
| 54 | 100.4 ± 1 (67.1 ± 11) | 0.8 ± 0.18 | 10.85 | 13.6 |
| 65 | 92.9 ± 5 (4.8 ± 25) | 12.5–25 | ||
| 66 | 94.2 ± 3 (3.3 ± 20) | 25–50 | ||
| Miltefosine | 3.2 ± 1.4 | 15.9 ± 1.2 | 4.9 | |
| Compound | IC50 (μM) 24 h | IC50 (μM) 48 h | IC50 (μM) 72 h |
|---|---|---|---|
| 25 | 7.8 | 3.85 | 2.16 |
| 27 | 1.27 | 0.89 | 0.67 |
| 29 | >10 | >10 | >10 |
| Miltefosine | 18.4 ± 1.2 | 9.5 ± 0.9 | 8.34 ± 0.0 |
| Compound | IC50 (μM) 24 h | IC50 (μM) 48 h | IC50 (μM) 72 h |
|---|---|---|---|
| 27 | 0.1 | 0.087 | 0.057 |
| Miltefosine | 12.4 ± 0.6 | 4.59 ± 0.1 | 4.3 ± 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magoulas, G.E.; Afroudakis, P.; Georgikopoulou, K.; Roussaki, M.; Borsari, C.; Fotopoulou, T.; Santarem, N.; Barrias, E.; Tejera Nevado, P.; Hachenberg, J.; et al. Design, Synthesis and Antiparasitic Evaluation of Click Phospholipids. Molecules 2021, 26, 4204. https://doi.org/10.3390/molecules26144204
Magoulas GE, Afroudakis P, Georgikopoulou K, Roussaki M, Borsari C, Fotopoulou T, Santarem N, Barrias E, Tejera Nevado P, Hachenberg J, et al. Design, Synthesis and Antiparasitic Evaluation of Click Phospholipids. Molecules. 2021; 26(14):4204. https://doi.org/10.3390/molecules26144204
Chicago/Turabian StyleMagoulas, George E., Pantelis Afroudakis, Kalliopi Georgikopoulou, Marina Roussaki, Chiara Borsari, Theano Fotopoulou, Nuno Santarem, Emile Barrias, Paloma Tejera Nevado, Julia Hachenberg, and et al. 2021. "Design, Synthesis and Antiparasitic Evaluation of Click Phospholipids" Molecules 26, no. 14: 4204. https://doi.org/10.3390/molecules26144204