
PCSK9 as a Target for Development of a New Generation of Hypolipidemic Drugs

 ,
,
Abstract
:1. Introduction
2. Physiological Role of PCSK9
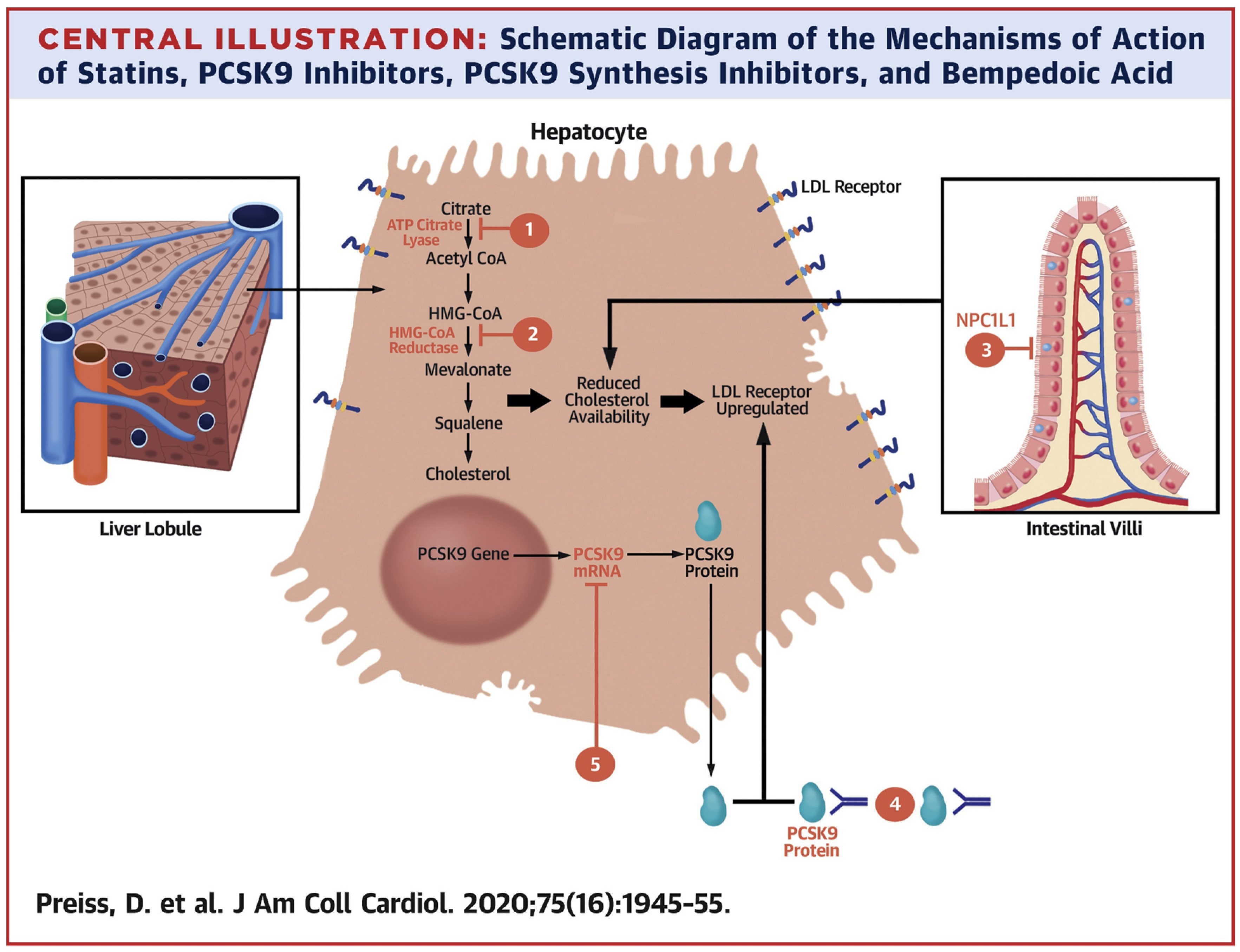
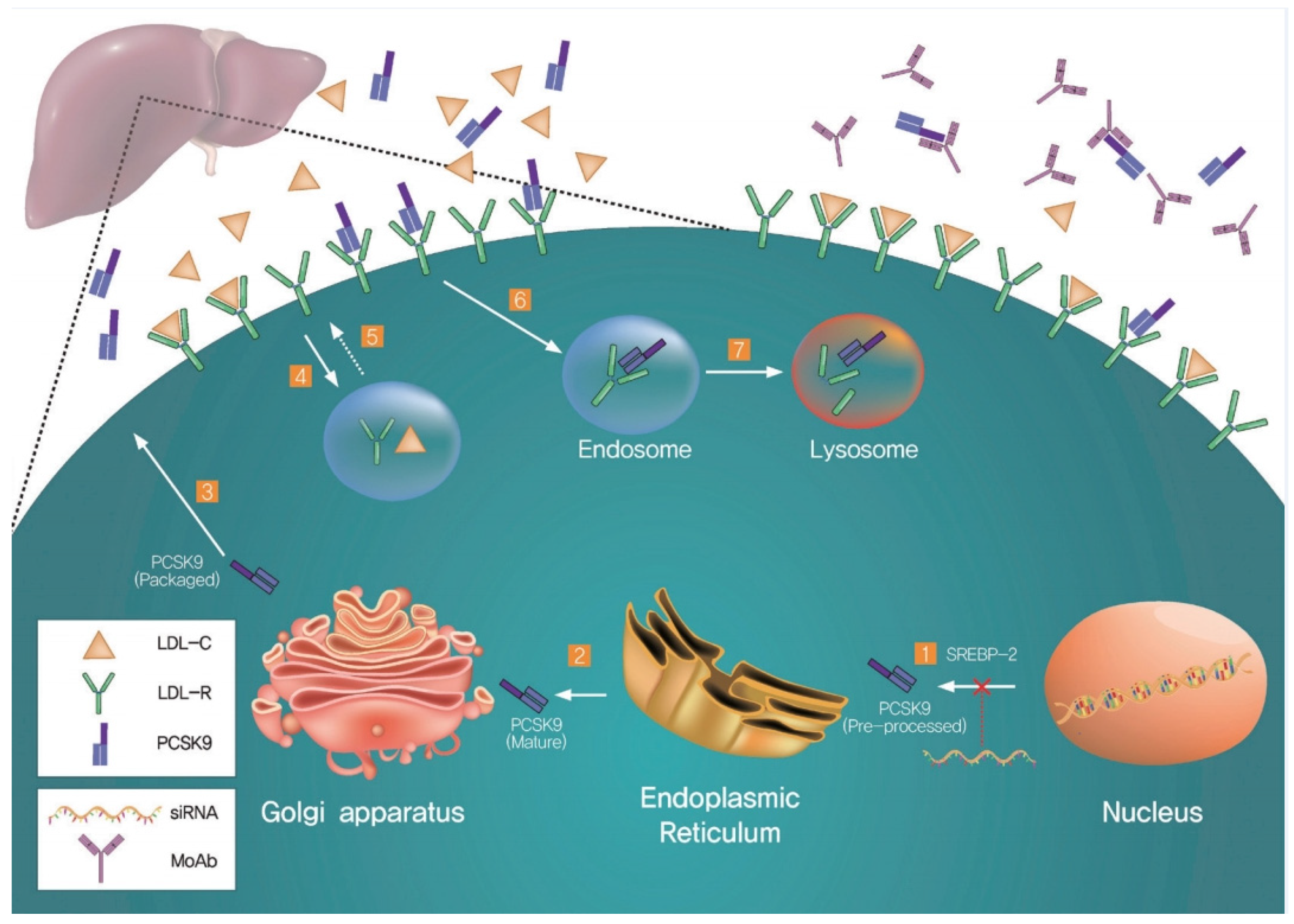
3. Mechanism of Regulation of PCSK9-Mediated LDLR Expression in Hepatocytes
4. PCSK9-Dependent LDLR Degradation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Holmes, M.V.; Millwood, I.Y.; Kartsonaki, C.; Hill, M.R.; Bennett, D.A.; Boxall, R.; Guo, Y.; Xu, X.; Bian, Z.; Hu, R.; et al. Lipids, Lipoproteins, and Metabolites and Risk of Myocardial Infarction and Stroke. J. Am. Coll. Cardiol 2018, 71, 620–632. [Google Scholar] [CrossRef]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016, 388, 2532–2561. [Google Scholar] [CrossRef] [Green Version]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39, 119–177. [Google Scholar]
- Kotseva, K.; Wood, D.; De Bacquer, D.; De Backer, G.; Ryden, L.; Jennings, C.; Gyberg, V.; Amouyel, P.; Bruthans, J.; Castro Conde, A.; et al. EUROASPIRE IV: A European Society of Cardiology survey on the lifestyle, risk factor and therapeutic management of coronary patients from 24 European countries. Eur. J. Prev. Cardiol. 2016, 23, 636–648. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019, 290, 140–205. [Google Scholar] [CrossRef] [Green Version]
- Emerging Risk Factors Collaboration; Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009, 302, 412–423. [Google Scholar]
- Denis, M.; Marcinkiewicz, J.; Zaid, A.; Gauthier, D.; Poirier, S.; Lazure, C.; Seidah, N.G.; Prat, A. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation 2012, 125, 894–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikido, T.; Ray, K.K. Non-antibody Approaches to Proprotein Convertase Subtilisin Kexin 9 Inhibition: siRNA, Antisense Oligonucleotides, Adnectins, Vaccination, and New Attempts at Small-Molecule Inhibitors Based on New Discoveries. Front. Cardiovasc. Med. 2018, 5, 199. [Google Scholar] [CrossRef] [Green Version]
- Strom, T.B.; Tveten, K.; Leren, T.P. PCSK9 acts as a chaperone for the LDL receptor in the endoplasmic reticulum. Biochem. J. 2014, 457, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Benjannet, S.; Rhainds, D.; Hamelin, J.; Nassoury, N.; Seidah, N.G. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: Functional consequences of natural mutations and post-translational modifications. J. Biol. Chem. 2006, 281, 30561–30572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, J.; Holla, O.L.; Laerdahl, J.K.; Kulseth, M.A.; Berge, K.E.; Leren, T.P. Mutation S462P in the PCSK9 gene reduces secretion of mutant PCSK9 without affecting the autocatalytic cleavage. Atherosclerosis 2009, 203, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Bernier, L.; Dubuc, G.; Nuel, G.; Rabes, J.P.; Bonneau, J.; Marques, A.; Marduel, M.; Devillers, M.; Munnich, A.; et al. A PCSK9 variant and familial combined hyperlipidaemia. J. Med. Genet. 2008, 45, 780–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abifadel, M.; Guerin, M.; Benjannet, S.; Rabes, J.P.; Le Goff, W.; Julia, Z.; Hamelin, J.; Carreau, V.; Varret, M.; Bruckert, E.; et al. Identification and characterization of new gain-of-function mutations in the PCSK9 gene responsible for autosomal dominant hypercholesterolemia. Atherosclerosis 2012, 223, 394–400. [Google Scholar] [CrossRef]
- Abifadel, M.; Rabes, J.P.; Devillers, M.; Munnich, A.; Erlich, D.; Junien, C.; Varret, M.; Boileau, C. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum. Mutat. 2009, 30, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabes, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.W.; Wang, H.; Bajaj, K.; Zhang, P.; Meng, Z.X.; Ma, D.; Bai, Y.; Liu, H.H.; Adams, E.; Baines, A.; et al. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. Elife 2013, 2, e00444. [Google Scholar] [CrossRef]
- Norata, G.D.; Tavori, H.; Pirillo, A.; Fazio, S.; Catapano, A.L. Biology of proprotein convertase subtilisin kexin 9: Beyond low-density lipoprotein cholesterol lowering. Cardiovasc. Res. 2016, 112, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Eacho, P.I.; Knierman, M.D.; Troutt, J.S.; Konrad, R.J.; Yu, X.; Schroeder, K.M. Isolation and characterization of the circulating truncated form of PCSK9. J. Lipid Res. 2014, 55, 1505–1514. [Google Scholar] [CrossRef] [Green Version]
- Essalmani, R.; Susan-Resiga, D.; Chamberland, A.; Abifadel, M.; Creemers, J.W.; Boileau, C.; Seidah, N.G.; Prat, A. In vivo evidence that furin from hepatocytes inactivates PCSK9. J. Biol. Chem. 2011, 286, 4257–4263. [Google Scholar] [CrossRef] [Green Version]
- Lakoski, S.G.; Lagace, T.A.; Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Genetic and metabolic determinants of plasma PCSK9 levels. J. Clin. Endocrinol. Metab. 2009, 94, 2537–2543. [Google Scholar] [CrossRef]
- Krysa, J.A.; Ooi, T.C.; Proctor, S.D.; Vine, D.F. Nutritional and Lipid Modulation of PCSK9: Effects on Cardiometabolic Risk Factors. J. Nutr. 2017, 147, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsen, C.; Kjolby, M.; Nyegaard, M.; Mattheisen, M.; Lundhede, J.; Buttenschon, H.; Mors, O.; Bentzon, J.F.; Madsen, P.; Nykjaer, A.; et al. The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab. 2014, 19, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Kathiresan, S.; Melander, O.; Guiducci, C.; Surti, A.; Burtt, N.P.; Rieder, M.J.; Cooper, G.M.; Roos, C.; Voight, B.F.; Havulinna, A.S.; et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 2008, 40, 189–197. [Google Scholar] [CrossRef]
- Musunuru, K.; Strong, A.; Frank-Kamenetsky, M.; Lee, N.E.; Ahfeldt, T.; Sachs, K.V.; Li, X.; Li, H.; Kuperwasser, N.; Ruda, V.M.; et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 2010, 466, 714–719. [Google Scholar] [CrossRef]
- Kathiresan, S.; Willer, C.J.; Peloso, G.M.; Demissie, S.; Musunuru, K.; Schadt, E.E.; Kaplan, L.; Bennett, D.; Li, Y.; Tanaka, T.; et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009, 41, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, A.; Ding, Q.; Edmondson, A.C.; Millar, J.S.; Sachs, K.V.; Li, X.; Kumaravel, A.; Wang, M.Y.; Ai, D.; Guo, L.; et al. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J. Clin. Investig. 2012, 122, 2807–2816. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, J.; Matye, D.J.; Wang, Y.; Li, T. Hepatocyte sortilin 1 knockout and treatment with a sortilin 1 inhibitor reduced plasma cholesterol in Western diet-fed mice. J. Lipid Res. 2019, 60, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Gao, A.; Cayabyab, F.S.; Chen, X.; Yang, J.; Wang, L.; Peng, T.; Lv, Y. Implications of Sortilin in Lipid Metabolism and Lipid Disorder Diseases. DNA Cell Biol. 2017, 36, 1050–1061. [Google Scholar] [CrossRef]
- Rymer, J.A.; Mues, K.E.; Monda, K.L.; Bratton, E.W.; Wirtz, H.S.; Okerson, T.; Overman, R.A.; Brookhart, M.A.; Muntner, P.; Wang, T.Y. Use of Low-Density Lipoprotein-Lowering Therapies Before and After PCSK9 Inhibitor Initiation. J. Am. Heart Assoc. 2020, 9, e014347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Lagace, T.A.; Garuti, R.; Zhao, Z.; McDonald, M.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 2007, 282, 18602–18612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Prat, A.; Chretien, M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Lagace, T.A.; Curtis, D.E.; Garuti, R.; McNutt, M.C.; Park, S.W.; Prather, H.B.; Anderson, N.N.; Ho, Y.K.; Hammer, R.E.; Horton, J.D. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Investig. 2006, 116, 2995–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Boyd, J.; Werner, J.M.; Knott, V.; Handford, P.A.; Campbell, I.D.; Downing, A.K. Solution structure of the LDL receptor EGF-AB pair: A paradigm for the assembly of tandem calcium binding EGF domains. Structure 2001, 9, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Downing, A.K.; Knott, V.; Werner, J.M.; Cardy, C.M.; Campbell, I.D.; Handford, P.A. Solution structure of a pair of calcium-binding epidermal growth factor-like domains: Implications for the Marfan syndrome and other genetic disorders. Cell 1996, 85, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Malby, S.; Pickering, R.; Saha, S.; Smallridge, R.; Linse, S.; Downing, A.K. The first epidermal growth factor-like domain of the low-density lipoprotein receptor contains a noncanonical calcium binding site. Biochemistry 2001, 40, 2555–2563. [Google Scholar] [CrossRef]
- Beglova, N.; Blacklow, S.C. The LDL receptor: How acid pulls the trigger. Trends Biochem. Sci. 2005, 30, 309–317. [Google Scholar] [CrossRef]
- Fisher, T.S.; Lo Surdo, P.; Pandit, S.; Mattu, M.; Santoro, J.C.; Wisniewski, D.; Cummings, R.T.; Calzetta, A.; Cubbon, R.M.; Fischer, P.A.; et al. Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation. J. Biol. Chem. 2007, 282, 20502–20512. [Google Scholar] [CrossRef] [Green Version]
- Cochran, J.C.; Zhao, Y.C.; Wilcox, D.E.; Kull, F.J. A metal switch for controlling the activity of molecular motor proteins. Nat. Struct. Mol. Biol. 2011, 19, 122–127. [Google Scholar] [CrossRef]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrie, A.; Cuchel, M.; et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef]
- Santos, R.D.; Ruzza, A.; Hovingh, G.K.; Wiegman, A.; Mach, F.; Kurtz, C.E.; Hamer, A.; Bridges, I.; Bartuli, A.; Bergeron, J.; et al. Evolocumab in Pediatric Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhuravleva, A.; Gierasch, L.M. Exploring weak, Transient protein-protein interactions in crowded in vivo environments by in-cell nuclear magnetic resonance spectroscopy. Biochemistry 2011, 50, 9225–9236. [Google Scholar] [CrossRef] [Green Version]
- Wen, T.; Wang, Z.; Chen, X.; Ren, Y.; Lu, X.; Xing, Y.; Lu, J.; Chang, S.; Zhang, X.; Shen, Y.; et al. Structural basis for activation and allosteric modulation of full-length calcium-sensing receptor. Sci. Adv. 2021, 7, 1483. [Google Scholar] [CrossRef]
- Beglova, N.; Jeon, H.; Fisher, C.; Blacklow, S.C. Cooperation between fixed and low pH-inducible interfaces controls lipoprotein release by the LDL receptor. Mol. Cell 2004, 16, 281–292. [Google Scholar] [CrossRef]
- Nozue, T. Lipid Lowering Therapy and Circulating PCSK9 Concentration. J Atheroscler. Thromb. 2017, 24, 895–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preiss, D.; Tobert, J.A.; Hovingh, G.K.; Reith, C. Lipid-Modifying Agents, From Statins to PCSK9 Inhibitors: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 1945–1955. [Google Scholar] [CrossRef]
- Jia, L.; Betters, J.L.; Yu, L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu. Rev. Physiol. 2011, 73, 239–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.C.; Piper, D.E.; Cao, Q.; Liu, D.; King, C.; Wang, W.; Tang, J.; Liu, Q.; Higbee, J.; Xia, Z.; et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. USA 2009, 106, 9820–9825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Revkin, J.; Amarenco, P.; Brunell, R.; Curto, M.; Civeira, F.; Flather, M.; Glynn, R.J.; Gregoire, J.; Jukema, J.W.; et al. Cardiovascular Efficacy and Safety of Bococizumab in High-Risk Patients. N. Engl. J. Med. 2017, 376, 1527–1539. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.G.; Bessac, L.; Berdan, L.G.; Bhatt, D.L.; Bittner, V.; Diaz, R.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; Jukema, J.W.; et al. Effect of alirocumab, a monoclonal antibody to PCSK9, on long-term cardiovascular outcomes following acute coronary syndromes: Rationale and design of the ODYSSEY outcomes trial. Am. Heart J. 2014, 168, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Investigators, Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Investigators, Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.L.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Lintner, N.G.; McClure, K.F.; Petersen, D.; Londregan, A.T.; Piotrowski, D.W.; Wei, L.; Xiao, J.; Bolt, M.; Loria, P.M.; Maguire, B.; et al. Selective stalling of human translation through small-molecule engagement of the ribosome nascent chain. PLoS Biol. 2017, 15, e2001882. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.H.; Hong, Y.J. Proprotein convertase subtilisin/kexin type 9 inhibition in cardiovascular disease: Current status and future perspectives. Korean J. Intern. Med. 2020, 35, 1045–1058. [Google Scholar] [CrossRef]
- McClure, K.F.; Piotrowski, D.W.; Petersen, D.; Wei, L.; Xiao, J.; Londregan, A.T.; Kamlet, A.S.; Dechert-Schmitt, A.M.; Raymer, B.; Ruggeri, R.B.; et al. Liver-Targeted Small-Molecule Inhibitors of Proprotein Convertase Subtilisin/Kexin Type 9 Synthesis. Angew Chem. Int. Ed. Engl. 2017, 56, 16218–16222. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Smith, D.A.; Memarzadeh, S.; Lowell, C.A.; Cooper, J.A.; Witte, O.N. Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 6579–6584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
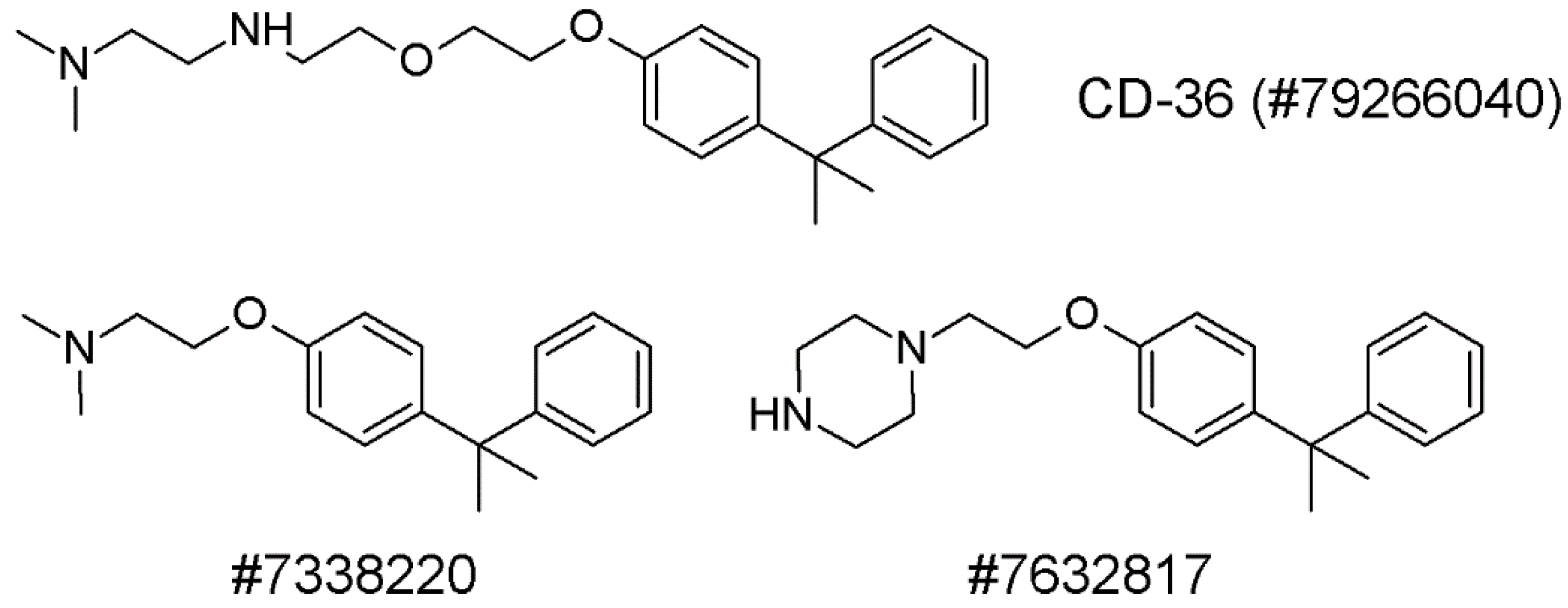
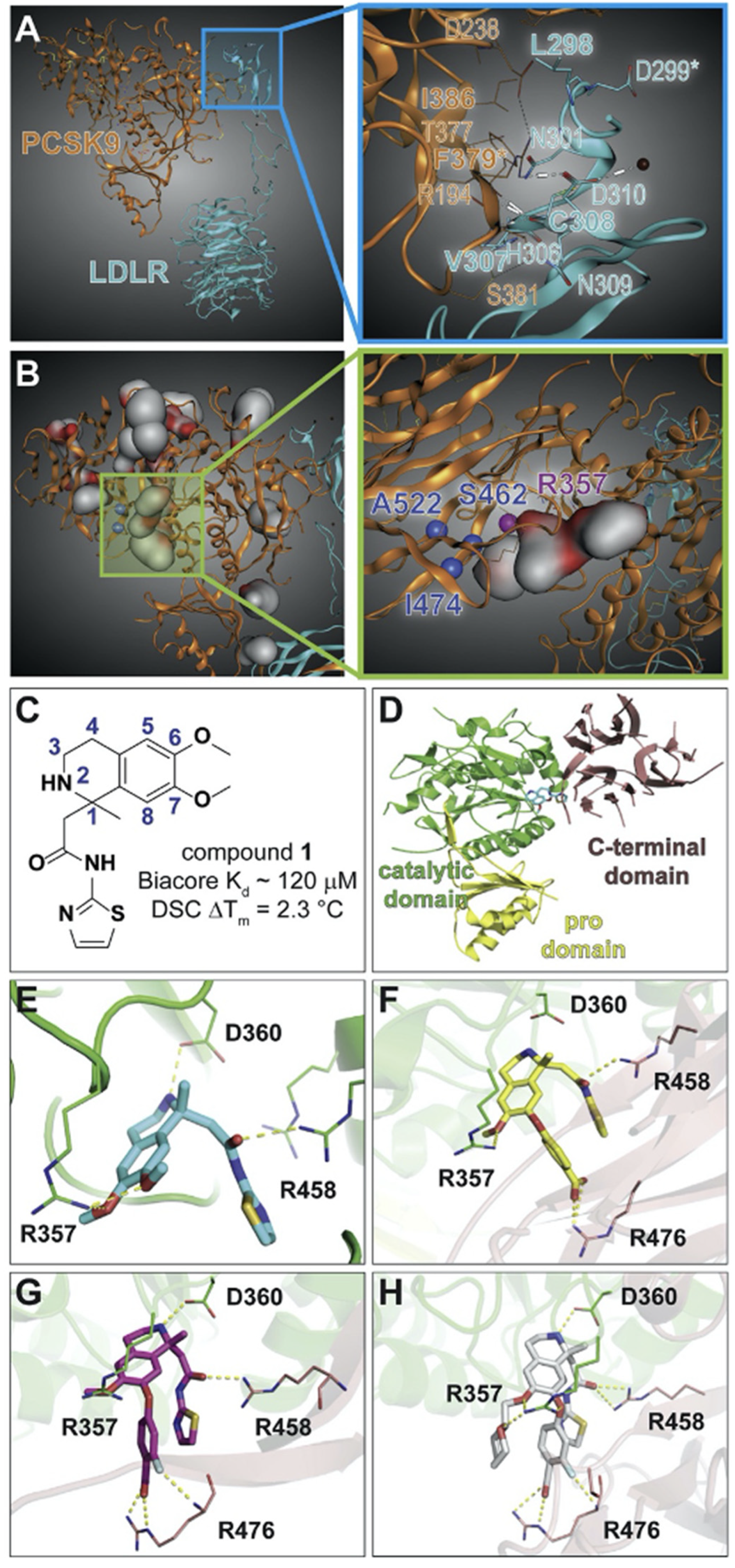
- Min, D.K.; Lee, H.S.; Lee, N.; Lee, C.J.; Song, H.J.; Yang, G.E.; Yoon, D.; Park, S.W. In silico Screening of Chemical Libraries to Develop Inhibitors That Hamper the Interaction of PCSK9 with the LDL Receptor. Yonsei Med. J. 2015, 56, 1251–1257. [Google Scholar] [CrossRef] [Green Version]
- Petrilli, W.L.; Adam, G.C.; Erdmann, R.S.; Abeywickrema, P.; Agnani, V.; Ai, X.; Baysarowich, J.; Byrne, N.; Caldwell, J.P.; Chang, W.; et al. From Screening to Targeted Degradation: Strategies for the Discovery and Optimization of Small Molecule Ligands for PCSK9. Cell Chem. Biol. 2020, 27, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Konuma, T.; Sakurai, K.; Goto, Y. Promiscuous binding of ligands by beta-lactoglobulin involves hydrophobic interactions and plasticity. J. Mol. Biol. 2007, 368, 209–218. [Google Scholar] [CrossRef]
- Lange, D.C.; Kothari, R.; Patel, R.C.; Patel, S.C. Retinol and retinoic acid bind to a surface cleft in bovine beta-lactoglobulin: A method of binding site determination using fluorescence resonance energy transfer. Biophys. Chem. 1998, 74, 45–51. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
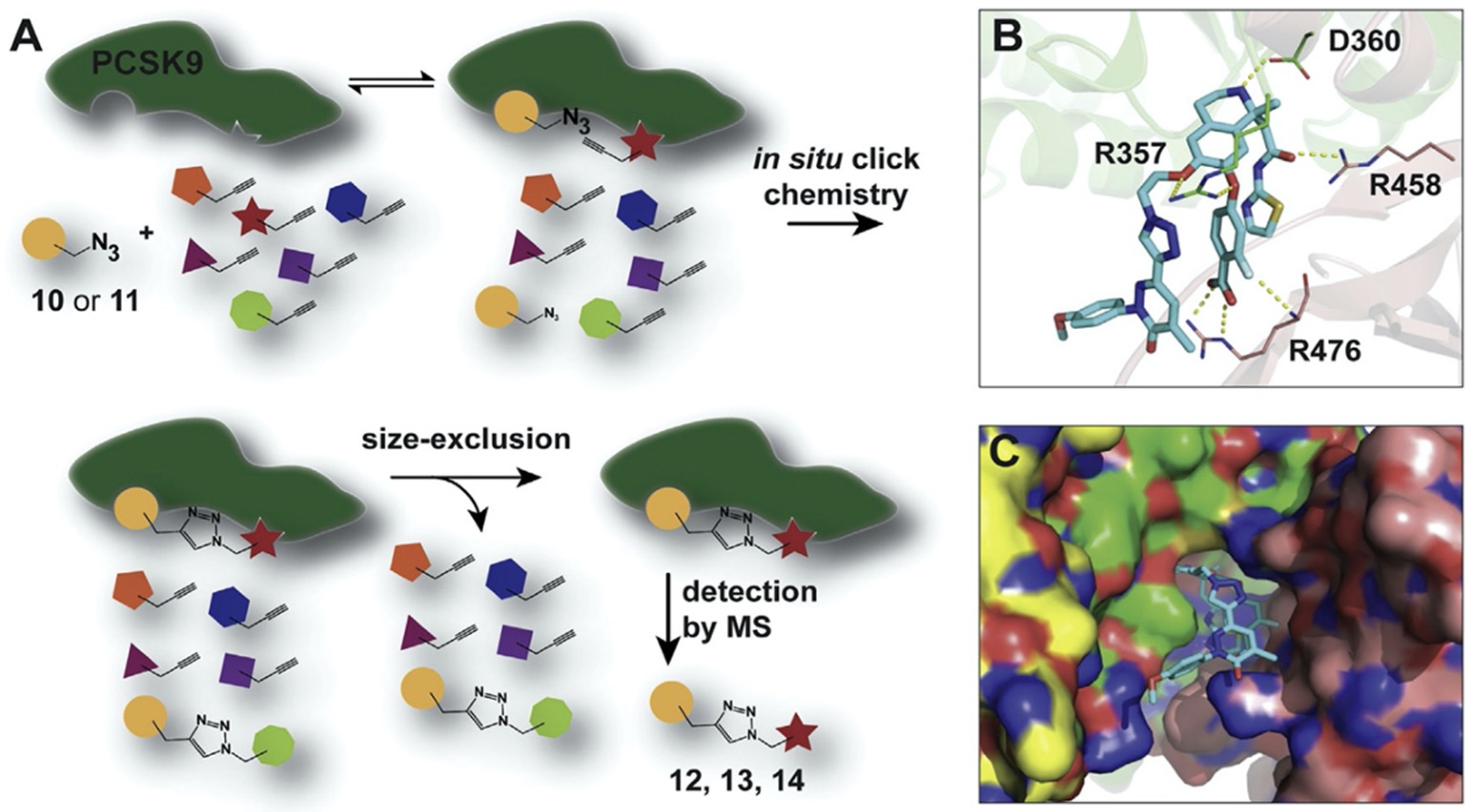{kind=link}
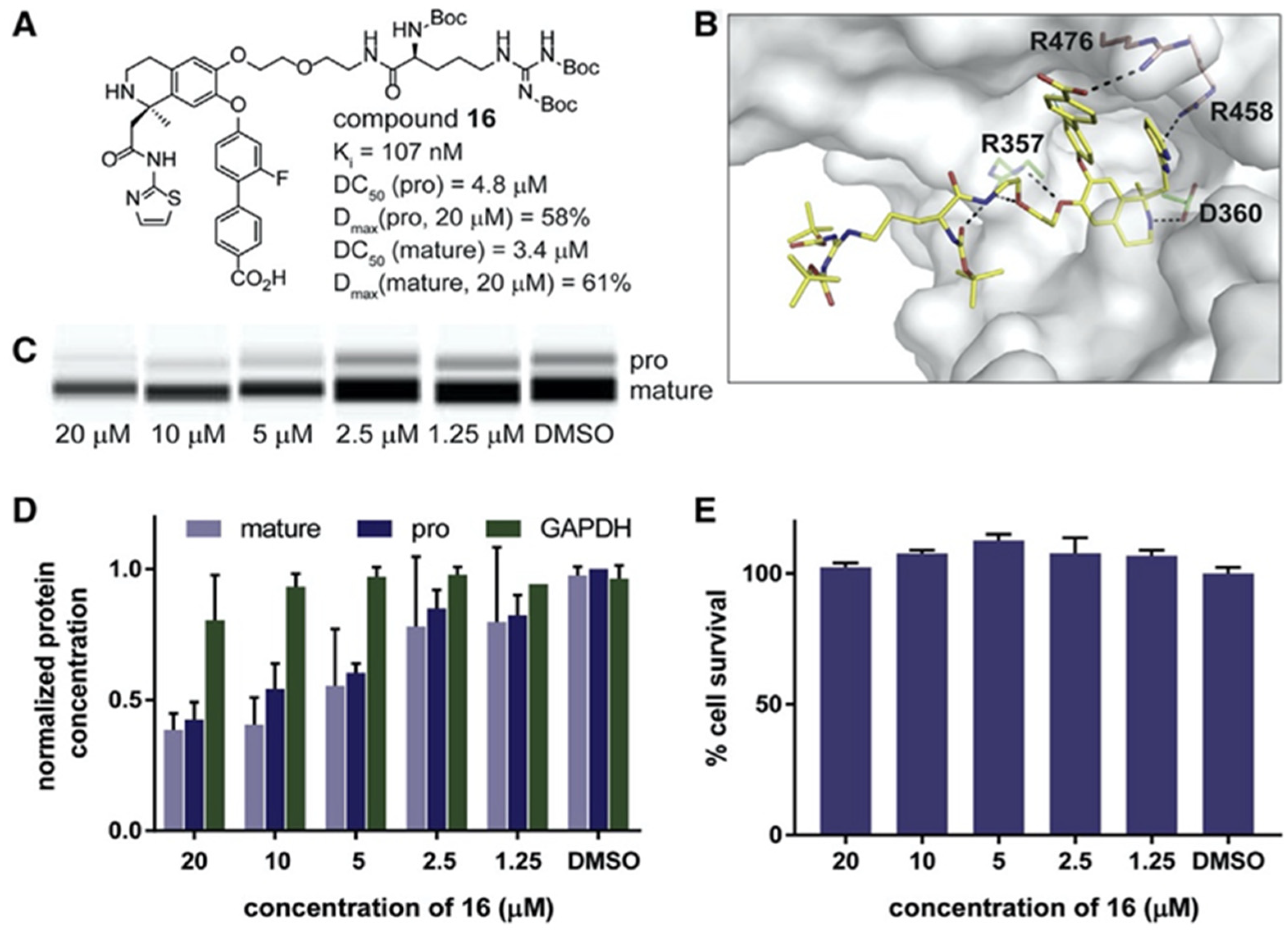{kind=link}
| Mechanism of Action | Examples |
|---|---|
| Blocking of PCSK9/LDLR complex formation | Peptides |
| Adnectins | |
| Monoclonal antibodies | |
| (alirocumab, evolocumab, bocozicumab) | |
| Suppression of PCSK9 expression | CRISPR/Cas9 technology |
| Small molecules: | |
| berberine, oleanolic acid | |
| Antisense nucleotides | |
| siRNA | |
| Interference with PCSK9 secretion | Sortilin |
| Sec24a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuzmich, N.; Andresyuk, E.; Porozov, Y.; Tarasov, V.; Samsonov, M.; Preferanskaya, N.; Veselov, V.; Alyautdin, R. PCSK9 as a Target for Development of a New Generation of Hypolipidemic Drugs. Molecules 2022, 27, 434. https://doi.org/10.3390/molecules27020434
Kuzmich N, Andresyuk E, Porozov Y, Tarasov V, Samsonov M, Preferanskaya N, Veselov V, Alyautdin R. PCSK9 as a Target for Development of a New Generation of Hypolipidemic Drugs. Molecules. 2022; 27(2):434. https://doi.org/10.3390/molecules27020434
Chicago/Turabian StyleKuzmich, Nikolay, Elena Andresyuk, Yuri Porozov, Vadim Tarasov, Mikhail Samsonov, Nina Preferanskaya, Valery Veselov, and Renad Alyautdin. 2022. "PCSK9 as a Target for Development of a New Generation of Hypolipidemic Drugs" Molecules 27, no. 2: 434. https://doi.org/10.3390/molecules27020434