
Interactions between Rhodamine Dyes and Model Membrane Systems—Insights from Molecular Dynamics Simulations

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results
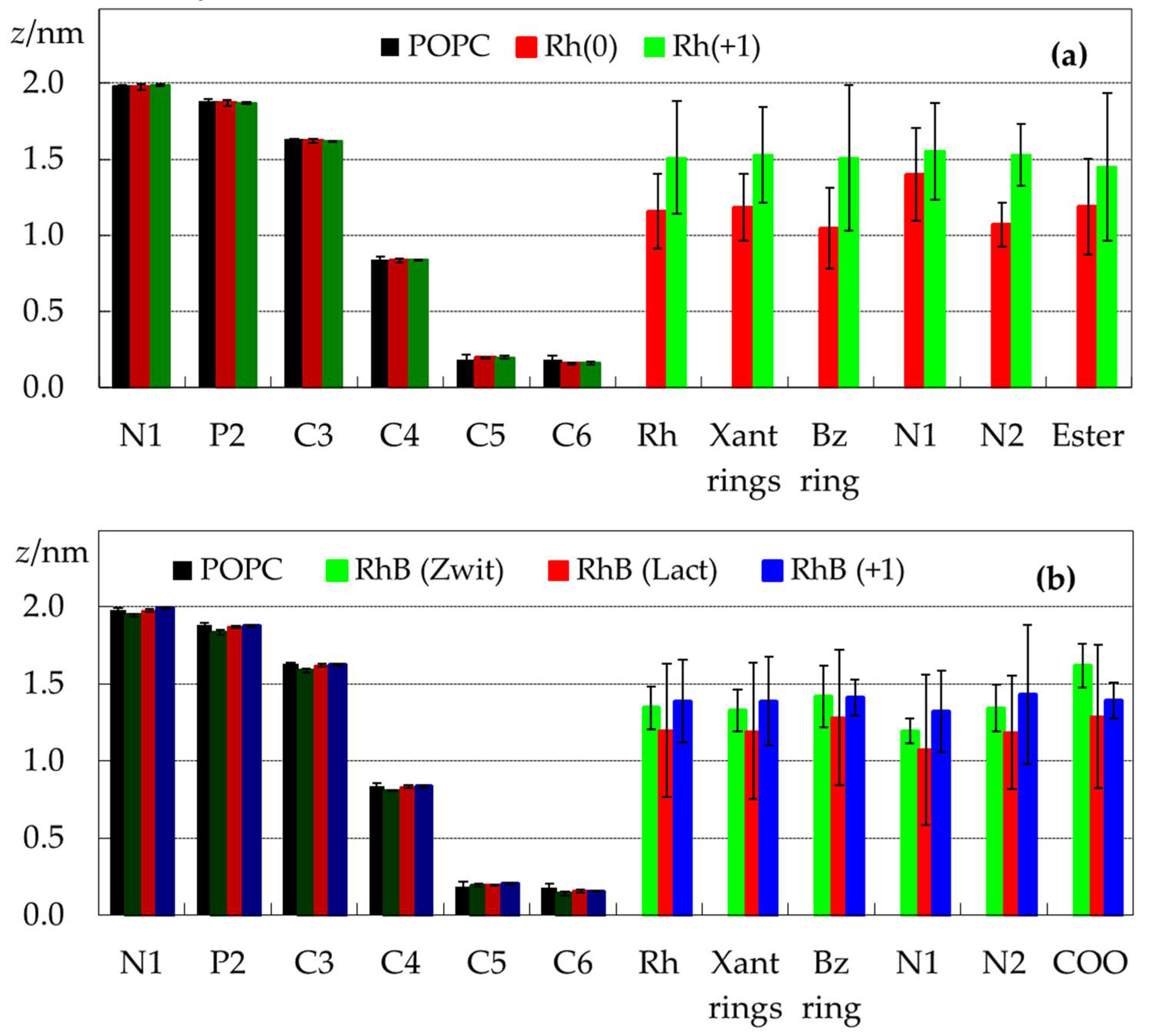
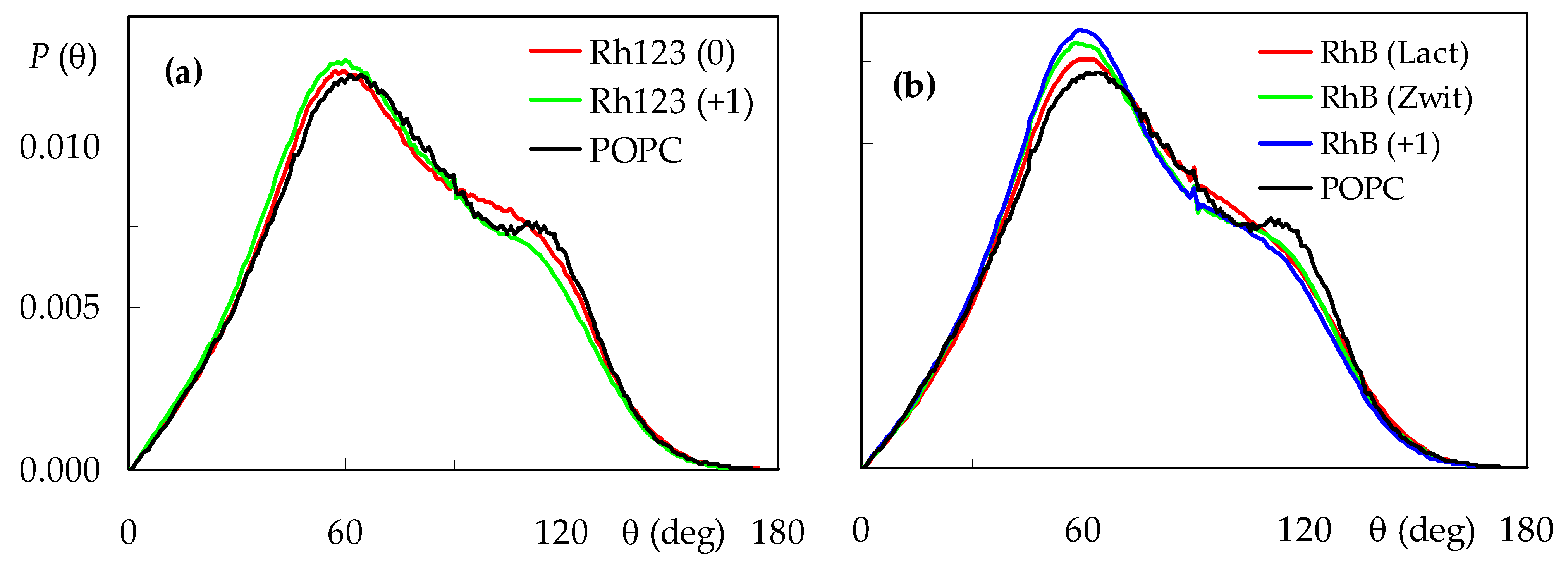
2.1. Location and Orientation of Rhodamine Dyes
2.2. Hydrogen Bonding
2.3. Effects on Selected Lipid Properties
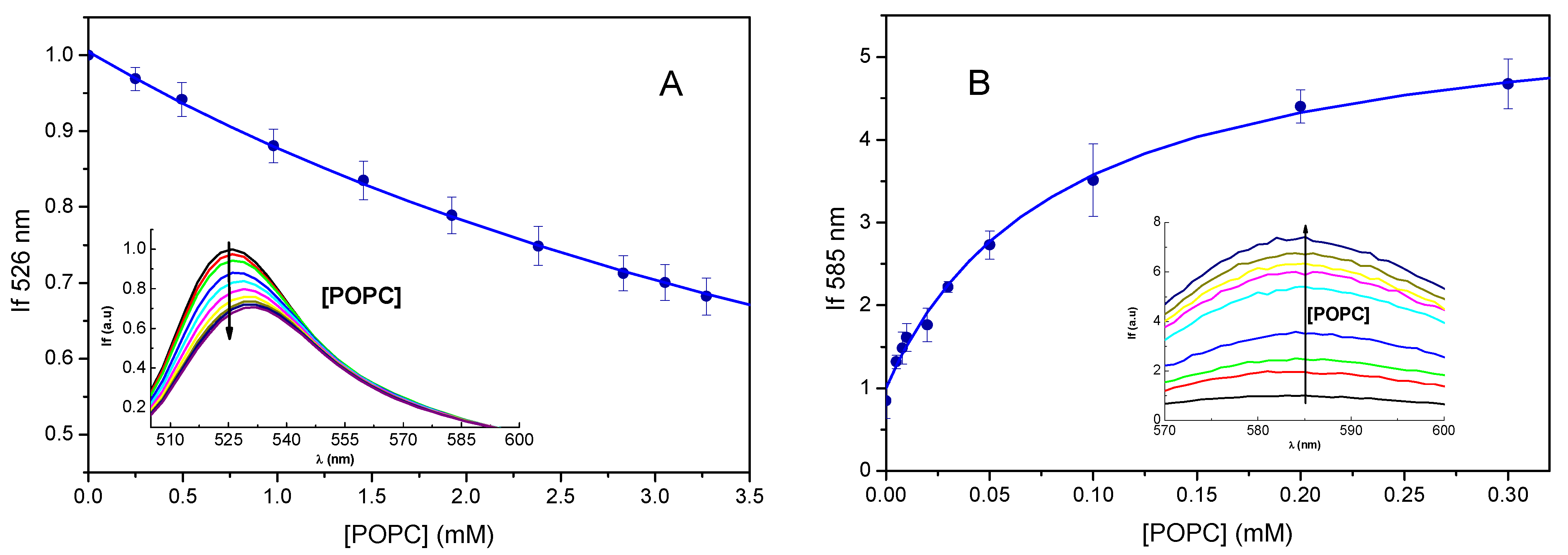
2.4. Free Energy Profiles across the Bilayer and Relation to Experimental Partition
3. Materials and Methods
3.1. Quantum Chemical Calculations
3.2. MD Parameters and System Set-Up
3.3. Unrestrained Membrane Simulations
3.4. Umbrella Sampling Membrane Simulations and PMF Calculation
3.5. Materials
3.6. Preparation of Large Unilamelar Vesicles (LUVs)
3.7. Association of Rh123 and RhB with Model Membranes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beija, M.; Afonso, C.A.M.; Martinho, J.M.G. Synthesis and applications of rhodamine derivatives as fluorescent probes. Chem. Soc. Rev. 2009, 38, 2410–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavis, L.D. Teaching Old Dyes New Tricks: Biological Probes Built from Fluoresceins and Rhodamines. Annu. Rev. Biochem. 2017, 86, 825–843. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Artursson, P.; Avdeef, A.; Di, L.; Ecker, G.F.; Faller, B.; Houston, J.B.; Kansy, M.; Kerns, E.H.; Krämer, S.D.; et al. Passive lipoidal diffusion and carrier-mediated cell uptake are both important mechanisms of membrane permeation in drug disposition. Mol. Pharm. 2014, 11, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Seelig, A. P-Glycoprotein: One Mechanism, Many Tasks and the Consequences for Pharmacotherapy of Cancers. Front. Oncol. 2020, 10, 1989. [Google Scholar] [CrossRef] [PubMed]
- Sajid, A.; Lusvarghi, S.; Murakami, M.; Chufan, E.E.; Abel, B.; Gottesman, M.M.; Durell, S.R.; Ambudkar, S.V. Reversing the direction of drug transport mediated by the human multidrug transporter P-glycoprotein. Proc. Natl. Acad. Sci. 2020, 117, 29609–29617. [Google Scholar] [CrossRef] [PubMed]
- Lusvarghi, S.; Robey, R.W.; Gottesman, M.M.; Ambudkar, S.V. Multidrug transporters: Recent insights from cryo-electron microscopy-derived atomic structures and animal models. F1000Research 2020, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.D.; Lee, O.W.; Brimacombe, K.R.; Chen, L.; Guha, R.; Lusvarghi, S.; Tebase, B.G.; Klumpp-Thomas, C.; Robey, R.W.; Ambudkar, S.V.; et al. A high-throughput screen of a library of therapeutics identifies cytotoxic substrates of P-glycoprotein. Mol. Pharmacol. 2019, 96, 629–640. [Google Scholar] [CrossRef]
- Al-Shawi, M.K.; Polar, M.K.; Omote, H.; Figler, R.A. Transition State Analysis of the Coupling of Drug Transport to ATP Hydrolysis by P-glycoprotein. J. Biol. Chem. 2003, 278, 52629–52640. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, A.B.; Ling, V. Positively Cooperative Sites for Drug Transport by P-Glycoprotein with Distinct Drug Specificities. Eur. J. Biochem. 1997, 250, 130–137. [Google Scholar] [CrossRef]
- Landwojtowicz, E.; Nervi, P.; Seelig, A. Real-Time Monitoring of P-Glycoprotein Activation in Living Cells. Biochemistry 2002, 41, 8050–8057. [Google Scholar] [CrossRef]
- Chufan, E.E.; Kapoor, K.; Sim, H.M.; Singh, S.; Talele, T.T.; Durell, S.R.; Ambudkar, S.A. Multiple Transport-Active Binding Sites Are Available for a Single Substrate on Human P-Glycoprotein (ABCB1). PLoS ONE 2013, 8, e82463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eytan, G.D.; Regev, R.; Oren, G.; Hurwitz, C.D.; Assaraf, Y.G. Efficiency of P-glycoprotein–Mediated Exclusion of Rhodamine Dyes from Multidrug-Resistant Cells is Determined by their Passive Transmembrane Movement Rate. Eur. J. Biochem. 1997, 248, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emaus, R.K.; Grunwald, R.; Lemasters, J.J. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: Spectral and metabolic properties. Biochim. Biophys. Acta Bioenerg. 1986, 850, 436–448. [Google Scholar] [CrossRef]
- Baracca, A.; Sgarbi, G.; Solaini, G.; Lenaz, G. Rhodamine 123 as a probe of mitochondrial membrane potential: Evaluation of proton flux through F0 during ATP synthesis. Biochim. Biophys. Acta Bioenerg. 2003, 1606, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Scaduto, R.C.; Grotyohann, L.W. Measurement of Mitochondrial Membrane Potential Using Fluorescent Rhodamine Derivatives. Biophys. J. 1999, 76, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.; Kansy, M.; Avdeef, A.; Senner, F. Permeation of permanently positive charged molecules through artificial membranes—Influence of physico-chemical properties. Eur. J. Pharm. Sci. 2007, 31, 32–42. [Google Scholar] [CrossRef]
- Seelig, A.; Landwojtowicz, E. Structure–activity relationship of P-glycoprotein substrates and modifiers. Eur. J. Pharm. Sci. 2000, 12, 31–40. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Prates Ramalho, J.P. Fluorescent membrane probes’ behavior in lipid bilayers: Insights from molecular dynamics simulations. Biophys. Rev. 2009, 1, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Loura, L.M.S.; Ramalho, J.P.P. Recent developments in molecular dynamics simulations of fluorescent membrane probes. Molecules 2011, 16, 5437–5452. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Moreno, M.J.; Loura, L.M.S. The Secret Lives of Fluorescent Membrane Probes as Revealed by Molecular Dynamics Simulations. Molecules 2020, 25, 3424. [Google Scholar] [CrossRef]
- Duvvuri, M.; Gong, Y.; Chatterji, D.; Krise, J.P. Weak base permeability characteristics influence the intracellular sequestration site in the multidrug-resistant human leukemic cell line HL-60. J. Biol. Chem. 2004, 279, 32367–32372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramette, R.W.; Sandell, E.B. Rhodamine b equilibria. J. Am. Chem. Soc. 1956, 78, 4872–4878. [Google Scholar] [CrossRef]
- Arbeloa, I.L.; Ojeda, P.R. Molecular forms of rhodamine B. Chem. Phys. Lett. 1981, 79, 347–350. [Google Scholar] [CrossRef]
- Wang, P.; Cheng, M.; Zhang, Z. On different photodecomposition behaviors of rhodamine B on laponite and montmorillonite clay under visible light irradiation. J. Saudi Chem. Soc. 2014, 18, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Maurya, N.S.; Mittal, A.K.; Cornel, P.; Rother, E. Biosorption of dyes using dead macro fungi: Effect of dye structure, ionic strength and pH. Bioresour. Technol. 2006, 97, 512–521. [Google Scholar] [CrossRef]
- Hinckley, D.A.; Seybold, P.G. A spectroscopic/thermodynamic study of the rhodamine B lactone ⇌ zwitterion equilibrium. Spectrochim. Acta Part A Mol. Spectrosc. 1988, 44, 1053–1059. [Google Scholar] [CrossRef]
- Drexhage, K.H. Fluorescence efficiency of laser dyes. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1976, 80 A, 421–428. [Google Scholar] [CrossRef]
- Santos, H.A.F.; Vila-Viçosa, D.; Teixeira, V.H.; Baptista, A.M.; Machuqueiro, M. Constant-pH MD Simulations of DMPA/DMPC Lipid Bilayers. J. Chem. Theory Comput. 2015, 11, 5973–5979. [Google Scholar] [CrossRef]
- Radak, B.K.; Chipot, C.; Suh, D.; Jo, S.; Jiang, W.; Phillips, J.C.; Schulten, K.; Roux, B. Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. J. Chem. Theory Comput. 2017, 13, 5933–5944. [Google Scholar] [CrossRef]
- Piggot, T.J.; Piñeiro, Á.; Khalid, S. Molecular dynamics simulations of phosphatidylcholine membranes: A comparative force field study. J. Chem. Theory Comput. 2012, 8, 4593–4609. [Google Scholar] [CrossRef]
- Poger, D.; Caron, B.; Mark, A.E. Validating lipid force fields against experimental data: Progress, challenges and perspectives. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, L.S.; De Groot, B.L.; Réat, V.; Milon, A.; Czaplicki, J. Acyl chain order parameter profiles in phospholipid bilayers: Computation from molecular dynamics simulations and comparison with 2H NMR experiments. Eur. Biophys. J. 2007, 36, 919–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neale, C.; Bennett, W.F.D.; Tieleman, D.P.; Pomès, R. Statistical Convergence of Equilibrium Properties in Simulations of Molecular Solutes Embedded in Lipid Bilayers. J. Chem. Theory Comput. 2011, 7, 4175–4188. [Google Scholar] [CrossRef]
- Paloncýová, M.; Berka, K.; Otyepka, M. Convergence of free energy profile of coumarin in lipid bilayer. J. Chem. Theory Comput. 2012, 8, 1200–1211. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Moreno, M.J.; Róg, T.; Vattulainen, I.; Loura, L.M.S. How to tackle the issues in free energy simulations of long amphiphiles interacting with lipid membranes: Convergence and local membrane deformations. J. Phys. Chem. B 2014, 118, 3572–3581. [Google Scholar] [CrossRef] [PubMed]
- Neale, C.; Pomès, R. Sampling errors in free energy simulations of small molecules in lipid bilayers. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.L.; Kuczera, K. Simulating the free energy of passive membrane permeation for small molecules. Mol. Simul. 2018, 44, 1147–1157. [Google Scholar] [CrossRef]
- MacCallum, J.L.; Tieleman, D.P. Computer Simulation of the Distribution of Hexane in a Lipid Bilayer: Spatially Resolved Free Energy, Entropy, and Enthalpy Profiles. J. Am. Chem. Soc. 2005, 128, 125–130. [Google Scholar] [CrossRef]
- Paloncýová, M.; Devane, R.; Murch, B.; Berka, K.; Otyepka, M. Amphiphilic drug-like molecules accumulate in a membrane below the head group region. J. Phys. Chem. B 2014, 118, 1030–1039. [Google Scholar] [CrossRef]
- Kiametis, A.S.; Stock, L.; Cirqueira, L.; Treptow, W. Atomistic Model for Simulations of the Sedative Hypnotic Drug 2,2,2-Trichloroethanol. ACS Omega 2018, 3, 15916–15923. [Google Scholar] [CrossRef]
- Tang, P.K.; Chakraborty, K.; Hu, W.; Kang, M.; Loverde, S.M. Interaction of Camptothecin with Model Cellular Membranes. J. Chem. Theory Comput. 2020, 16, 3373–3384. [Google Scholar] [CrossRef]
- Piasentin, N.; Lian, G.; Cai, Q. Evaluation of Constrained and Restrained Molecular Dynamics Simulation Methods for Predicting Skin Lipid Permeability. ACS Omega 2021, acsomega.1c04684. [Google Scholar] [CrossRef] [PubMed]
- Eytan, G.D.; Regev, R.; Oren, G.; Assaraf, Y.G. The Role of Passive Transbilayer Drug Movement in Multidrug Resistance and Its Modulation. J. Biol. Chem. 1996, 271, 12897–12902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paketurytė, V.; Petrauskas, V.; Zubrienė, A.; Abian, O.; Bastos, M.; Chen, W.Y.; Moreno, M.J.; Krainer, G.; Linkuvienė, V.; Sedivy, A.; et al. Uncertainty in protein–Ligand binding constants: Asymmetric confidence intervals versus standard errors. Eur. Biophys. J. 2021, 50, 661–670. [Google Scholar] [CrossRef]
- Vogel, M.; Rettig, W.; Sens, R.; Drexhage, K.H. Structural relaxation of rhodamine dyes with different N-substitution patterns: A study of fluorescence decay times and quantum yields. Chem. Phys. Lett. 1988, 147, 452–460. [Google Scholar] [CrossRef]
- Zhang, X.F.; Zhang, Y.; Liu, L. Fluorescence lifetimes and quantum yields of ten rhodamine derivatives: Structural effect on emission mechanism in different solvents. J. Lumin. 2014, 145, 448–453. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density—functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1998, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Besler, B.H.; Merz, K.M.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in Electronic Structure Theory: GAMESS a Decade Later. In Theory and Applications of Computational Chemistry; Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. ISBN 9780444517197. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Poger, D.; Van Gunsteren, W.F.; Mark, A.E. A new force field for simulating phosphatidylcholine bilayers. J. Comput. Chem. 2010, 31, 1117–1125. [Google Scholar] [CrossRef]
- Poger, D.; Mark, A.E. On the validation of molecular dynamics simulations of saturated and cis-monounsaturated phosphatidylcholine lipid bilayers: A comparison with experiment. J. Chem. Theory Comput. 2010, 6, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces; Pullman, B., Ed.; Springer: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated force field Topology Builder (ATB) and repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated Topology Builder Version 3.0: Prediction of Solvation Free Enthalpies in Water and Hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Santos, L.S.; Prates Ramalho, J.P.; Moreno, M.J.; Loura, L.M.S. Behaviour of NBD-head group labelled phosphatidylethanolamines in POPC bilayers: A molecular dynamics study. Phys. Chem. Chem. Phys. 2015, 17, 20066–20079. [Google Scholar] [CrossRef] [PubMed]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Hub, J.S.; De Groot, B.L.; Van Der Spoel, D. g_wham—A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef] [Green Version]
- Samelo, J.; Mora, M.J.; Granero, G.E.; Moreno, M.J. Partition of amphiphilic molecules to lipid bilayers by ITC: Low-affinity solutes. ACS Omega 2017, 2, 6863–6869. [Google Scholar] [CrossRef] [PubMed]
- Wiener, M.C.; White, S.H. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of X-ray and neutron diffraction data. III. Complete structure. Biophys. J. 1992, 61, 434–447. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental | Rh123 | RhB | |||
|---|---|---|---|---|---|
| Experimental KP | 1.8 × 102 1 | 2.2 × 104 2 | |||
| “Normalized KP” | 1 | 1.2 × 102 | |||
| Computational | neutral | cation | lactone | zwit. | cation |
| Normalized integral 3 | 0.18 | 1.0 | 1.3 × 106 | 6.2 × 102 | 8.2 × 102 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magalhães, N.; Simões, G.M.; Ramos, C.; Samelo, J.; Oliveira, A.C.; Filipe, H.A.L.; Ramalho, J.P.P.; Moreno, M.J.; Loura, L.M.S. Interactions between Rhodamine Dyes and Model Membrane Systems—Insights from Molecular Dynamics Simulations. Molecules 2022, 27, 1420. https://doi.org/10.3390/molecules27041420
Magalhães N, Simões GM, Ramos C, Samelo J, Oliveira AC, Filipe HAL, Ramalho JPP, Moreno MJ, Loura LMS. Interactions between Rhodamine Dyes and Model Membrane Systems—Insights from Molecular Dynamics Simulations. Molecules. 2022; 27(4):1420. https://doi.org/10.3390/molecules27041420
Chicago/Turabian StyleMagalhães, Nisa, Guilherme M. Simões, Cristiana Ramos, Jaime Samelo, Alexandre C. Oliveira, Hugo A. L. Filipe, João P. Prates Ramalho, Maria João Moreno, and Luís M. S. Loura. 2022. "Interactions between Rhodamine Dyes and Model Membrane Systems—Insights from Molecular Dynamics Simulations" Molecules 27, no. 4: 1420. https://doi.org/10.3390/molecules27041420