
Analytical Methods for the Determination of 90Sr and 239,240Pu in Environmental Samples


Department of Radiochemistry, China Institute of Atomic Energy, Beijing 102413, China
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(6), 1912; https://doi.org/10.3390/molecules27061912
Submission received: 31 January 2022
/
Revised: 15 February 2022
/
Accepted: 22 February 2022
/
Published: 15 March 2022
(This article belongs to the Special Issue Actinoids in Biologic Systems and Catalysis)
Abstract
:Artificial long-lived radionuclides such as 90Sr and 239,240Pu have been long released into the environment by human nuclear activities, which have a profound impact on the ecological environment. It is of great significance to monitor the concentration of these radionuclides for environmental safety. This paper summarizes and critically discusses the separation and measurement methods for ultra-trace determination of 90Sr, 239Pu, and 240Pu in the environment. After selecting the measurement method, it is necessary to consider the decontamination of the interference from matrix elements and the key elements, and this involves the choice of the separation method. Measurement methods include both radiometric methods and non-radiometric methods. Radiometric methods, including alpha spectroscopy, liquid scintillation spectrometry, etc., are commonly used methods for measuring 239+240Pu and 90Sr. Mass spectrometry, as the representative of non-radiometric measurement methods, has been regarded as the most promising analytical method due to its high absolute sensitivity, low detection limit, and relatively short sample-analysis time. Through the comparison of various measurement methods, the future development trend of radionuclide measurement is prospected in this review. The fully automatic and rapid analysis method is a highlight. The new mass spectrometer with ultra-high sensitivity shows strong analytical capabilities for extremely low concentrations of 90Sr, 239Pu, and 240Pu, and it is expected to develop determination methods with higher sensitivity and lower detection limit.
1. Introduction
With the progress of nuclear energy technology, environmental safety, especially nuclear environmental safety, has become one of the hot issues of common concern for the academic community and the public. Due to human activities, such as nuclear tests, reprocessing of spent nuclear fuel, and nuclear accidents, different artificial radionuclides have been released into the environment. 90Sr, as a representative of typical artificial radionuclides with high fission yield and long half-life, is easy to be concentrated in the bones and teeth after ingestion and inhalation by the human body, leading to bone cancer, leukemia, etc. [1]. As an extremely toxic transuranic element with a long half-life of thousands of years, Pu is not only harmful to the environment but also will be enriched in organs after entering the human body [2,3]. As shown in Table 1, the 239,240Pu released by the nuclear weapon tests exceeded 1016 Bq, and the 90Sr was close to 1018 Bq, whose radiation pollution increasingly attracts the public’s attention. These radionuclides in the environment can be transferred to the biosphere by dispersion, sedimentation, migration, and accumulation in living organisms through the food chain. It is important for radioactive pollution assessment to monitor the concentration levels of these radionuclides in environmental samples.
The uptake of radionuclides from soil to plants is generally quantified by the ratio of activity concentrations in plant and soil, namely the transfer factor (TF). In addition to radionuclide concentration, one of the factors influencing the TF is the chemical form of the radionuclides, including oxidation state and formation of complexes [5]. The study of the distribution and migration behavior of 90Sr and 239,240Pu in the environment is important work for radioecology. The migration ability of 90Sr is very strong. The TF of Sr from soil to vegetables varied with species, higher than that of other heavy metals. Among them, the value in cabbage was as high as 0.33 [6,7]. The concentration factor (CF) indicates the ability of a biological organism to concentrate radionuclides and is defined as the ratio of the radionuclide content in the organism to the environmental medium. The CF of 90Sr by marine organisms was between 0.1 and 10, and it was easy to deposit in shellfish shells, fish scales, and bones [8]. The solubility and mobility of Pu (IV) are relatively low in the pH ranges of 4–10 relevant to the environment ([Pu (IV)aq] = 10−10 − 10−8 mol·L−1) [9], while some processes and mechanism, such as the adsorption or complexation of Pu, can increase the mobility. The Pu transport indices for Brassica juncea and Helianthus annuus increased by addition of diethylenetriaminepentaacetic (DTPA) concentration to prepare Pu-DTPA in the nutrient solution [10]. The solution-to-plant TF for the potato tubers was 0.03–0.80, and the Pu uptake from solution greatly increased by addition of the complexing agent EDTA [11]. Generally, each type of nuclear emission has its own unique isotopic composition, that is, “fingerprint” information (Table 2). Pu in environmental samples is usually derived from global fallout with the value of 240Pu/239Pu about 0.18. For example, the isotope ratio of 240Pu/239Pu obtained by Kenna was 0.161684 ± 0.001084 [12]. Understanding the concentration and isotopic composition of 239Pu and 240Pu in the environment can provide valuable information about nuclear activities in the affected areas. Therefore, accurate measurement of these radionuclides is very important for environmental risk assessment.
The complex matrix of environmental samples with the diverse and unknown existing forms of radionuclides and the trace or ultra-trace concentration level makes accurate measurement quite difficult. Researchers have tried and improved many measurement methods [19,20,21,22,23]. The purpose of this article is to review the separation methods of 90Sr, 239Pu, and 240Pu that have been released in the environment, and discuss commonly used radiometric and non-radiometric measurement methods. By comparing the capabilities of various mass spectrometry technologies, focusing on ICP–MS, the future development of 90Sr, 239Pu, and 240Pu measurements could be deduced.
2. Sample Preparation
In radiometric technology of samples, the analysis method can be divided into three steps: sample pretreatment, radiochemical separation (concentration, separation, and purification), and source preparation and measurement. The process is shown in Figure 1. Ultra-trace amounts of the target nuclides 90Sr, 239Pu, and 240Pu in environmental samples need to be separated from a large amount of matrix and interfering nuclides, and this is a great challenge for sample preparation and measurement methods.
2.1. Sample Pretreatment
The pretreatment is the most critical step when measuring the contents of 90Sr, 239Pu, and 240Pu in environmental samples. The selection of pretreatment method depends on the purpose of analysis, the method of measurement, and the type of sample. The purpose of pretreatment is to destroy organic matter and convert most of the target nuclides into solution phase, so as to facilitate the subsequent separation operation. Interfering substances should be effectively removed without cross-contamination. Choosing the appropriate pretreatment method and shortening the pretreatment time directly affect the precision and accuracy of the analysis results. At present, the commonly used pretreatment methods mainly contain dry-ashing, digestion, melting, etc.
The traditional dry-ashing method usually uses electric heating plates and muffle furnaces [24,25,26] to dry, carbonize, and ash solid samples (soil, plants, etc.). In order to shorten the ashing time, ashing aids such as HNO3, H2SO4, and H2O2 can be added. For animal samples that are more difficult to ash and milk samples that contain more water, the microwave-ashing method has more advantages [27]. The powdery sample has a bulky texture and is not easy to ash. The rising of dense smoke during carbonization and the wind of the fume hood will cause partial loss of the components. Adding a small amount of ultra-pure water to the powder sample can effectively improve this phenomenon and enhance the accuracy and reliability of the results [28]. Considering the volatilization loss of nuclides, GB14883-2016 gives recommendations on the maximum ashing temperature of food (Table 3) [29].
The wet-digestion method has been widely used for the analysis of 90Sr, 239Pu, and 240Pu in various environmental samples (Table 4). In the early days, a single acid reagent was often used. Now, mixed acids or acid reagents with H2O2 are often used to treat samples, such as HNO3 + HCl (+ HClO4, H2O2). The conventional wet digestion method has the advantages of rapid oxidation speed, convenient operation, and low loss rate of the nuclides, with a digestion temperature below 300 °C. However, its large amount of acid will increase reagent blanks and is not suitable for processing a large quantity of samples. The microwave-digestion method adopts the pressure principle for digestion, with the advantages of less acid consumption, short time, little loss, low blank value, and easy realization of automatic monitoring. It has been widely used and is suitable for the digestion of biological and geological samples. However, this method has less processing capacity, and it is also not suitable for processing a large number of samples, nor for samples with poor microwave penetration. At present, the microwave digestion–atomic absorption method is widely used to measure trace elements in environmental samples.
In addition, the melting method is sometimes used in the sample pretreatment process [30], such as processing the insoluble residues in the wet-digestion method, accelerating the isotope exchange balance between the tracer and the target nuclide, etc. The melting method can treat kilogram samples, and the treatment effect depends on flux, temperature, and so on.
The same batch of black-tea samples was selected as the research object to investigate the characteristics between different pretreatment methods [31]. The measurement results of different metals by ICP–MS found that the microwave digestion method was significantly better than the dry-ashing method and the wet-digestion method. At present, in order to increase the sample processing capacity, the dry-ashing method is combined with the microwave-digestion method, shortening the processing time and reducing the amount of acid. In order to monitor recovery, a carrier or tracer can be added during digestion.
2.2. Chemical Separation
The solution after sample pretreatment contains a large amount of matrix elements, which would interfere with the subsequent accurate measurement. It is necessary to use chemical methods for separation and purification to remove matrix elements and interfering nuclides. There are many chemical separation methods suitable for Sr and Pu, including precipitation, solvent extraction, ion-exchange chromatography, extraction chromatography, or a combination of these methods [36,39,40,41,42,43,44,45,46]. We mainly focus on the ability of various methods to remove elements that interfere with the analysis of 239Pu, 240Pu, and 90Sr.
2.2.1. Separation Methods of 90Sr in Environmental Samples
In the sample solution obtained by pretreatment, Sr is usually pre-concentrated by precipitation or co-precipitation via the formation of insoluble nitrates, carbonates, and oxalates. The classical method of strontium precipitation by fuming nitric acid was cumbersome and time-consuming [47]. The precipitate formed by the reaction of alkaline earth metal and carbonate was decomposed with mineral acid in order to separate Sr [48]. Rao et al. [49] used different co-precipitation methods to analyze 90Sr in environmental and dietary samples by Ĉherenkov counting of 90Y.
Alkaline earth metals, such as Sr, can be well separated from other elements via the ion-exchange method. Dowex-50 is one of the commonly used cation exchangers. Calcium, strontium, and barium were separated by cation-exchange resin, using EDTA with appropriate concentration and acidity as eluent, measuring the 90Sr in biological samples successfully [50]. The solvent extraction method is simple and convenient. The extraction rate of strontium by thenoyltrifluoroacetone (TTA) can be as high as 95%, and Bis (2-ethylhexyl) phosphate (HDEHP) can be used to separate 90Sr from its daughter 90Y. By using dicyclohexyl-18-crown-6 in chloroform solution extraction, a small amount of Sr2+ can be separated from a large quantity of Ca2+ [51].
A new analytical method was developed for the separation of 90Sr from 90Y by using DGA resin, in which the extractant system was N,N,N′,N′-tetra-n-octyldiglycolamide (Normal) or N,N,N′,N′-tetrakis-2-ethylhexyldiglycolamide (Branched) [52,53]. The working capacity of the two DGA resins is 7.23 mg Sr and 11 mg yttrium per mL of resin. Sr resin is a special resin for separation and enrichment of Sr, with strong selectivity of Sr, and can efficiently separate Sr from Ca and other elements. Sr resin can replace cation exchange resins for low-consumption (significant reduction in resin consumption and elution amount) and high-efficiency chemical separation, with the extractant of crown ether (4,4′(5′)-di-tbutylcyclohexano-18-crown-6) dissolved in octanol. The adsorption capacity of Sr enhances with the increasing of concentration of nitric acid. The study on the adsorption behavior of elements on Sr resin found that the quantitative recovery of Sr can be basically achieved by loading the column with 3 mol·L−1 nitric acid medium and desorbing with 0.05 mol·L−1 nitric acid [54]. Željko et al. [32] compared the separation effect of anion-exchange resin, Sr resin, and the combination of the two methods on Sr in environmental samples (soil, vegetation, water, and animal bones) and emphasized their influence on the accuracy of 90Sr determination, finding that the efficiency of Sr separation depended on the sample type and separation method.
The interfering nuclide 90Zr needs to be decontaminated when using mass spectrometry to measure 90Sr. Considering the high concentration of Zr in environmental samples, Sr resin was used for separation, with a 20 v/v% HNO3 system for sample loading and washing to remove 86% of Zr. Meanwhile, the outflow of Sr was about 1% [23]. With deionized water, more than 98% of Sr and about 0.2% of Zr could be eluted from the resin, which could basically separate Sr from the main interfering Zr. Two-stage extraction separation by using Sr resin could further purify Sr, reducing the Zr concentration in solution to below 5 ng·mL−1 [55].
2.2.2. Separation Methods of Pu in Environmental Samples
It is necessary to eliminate the interference of 214Am, 210Po, 224Ra, 229Th, 231Pa, 232U, and 243Am when using alpha spectroscopy to determine 238Pu and 239+240Pu. Moreover, the 238UH+ and 238 peaks produced by natural uranium interfere with the measurement of 239Pu and 240Pu by mass spectrometry. Therefore, after selecting the measurement method, it is advisable to adopt an appropriate separation method to decontaminate the corresponding interfering elements.
Over the past few decades, extraction chromatography has attracted much attention for radiochemical separation and purification of Pu due to its short sample-processing time, small acid reagent dosage, and high selectivity and recovery. Pu in the sample solution presents multiple valence states, namely Pu3+, Pu4+, , and . Pu (IV) and or Cl− form stable anionic complexes easily, which is the basis for separation of Pu on anion exchange resins or extraction chromatography. Therefore, it is necessary to adjust Pu to Pu (IV) before loading the column. The most common method is to use oxidants (such as NaNO2, , etc.) for one-step value adjustment. It is worth noting that, due to the ultra-trace amount of Pu in environmental samples, Pu (VI) may not be completely reduced to Pu (IV) under conventional experimental conditions, and the unconverted Pu (VI) fraction will be lost during the separation process. Therefore, the most appropriate adjustment method is to reduce Pu to Pu (III) with a reducing agent (such as I− and Fe2+), and then use an oxidizing agent to oxidize the generated Pu (III) to Pu (IV) [56].
A variety of extraction resins have been developed at home and abroad, such as TEVA, UTEVA, etc., with large loading capacity and good mass transfer performance, and they are widely used in the sample separation process. TEVA resin [57] is a quaternary ammonium salt, and the stationary phase is trialkylmethylammonium chloride, with a structural formula of CH3N(CnH2n+1)3Cl (n = 8–10), generally used for the separation of Np and Pu, with the best adsorption system of 2~3 mol·L−1 nitric acid [58]. Using TEVA resin to pre-concentrate Pu in a vacuum box, the decontamination factor (DF) for uranium could reach over 1 × 104, and the DF for thorium and americium was more than 1 × 103 [59]. The Pu solution separated from TEVA resin was further purified by DGA resin with the DF for uranium of 1 × 106, eliminating the interference of the uranium hydrogen peak by measuring 239Pu with ICP–MS, and the short-life isotope 238Pu was successfully measured by alpha energy spectroscopy [60].
Anion-exchange resins have become attractive methods due to the low purchase cost, and they are widely applied to the separation of Pu for the strong tolerance to matrix elements compared with extraction resins [61]. The commonly used anion-exchange resins are AG-1 × 8, Dowex-1 × 8, etc., with the best adsorption system of 8 mol·L−1 nitric acid. Yamato et al. [39] established a combined separation method of Pu and Am in biological samples. The Pu was adjusted to Pu (IV) to make it adsorb on the anion-exchange column, and Am flowed out with the effluent. Hayashi et al. [62] improved it and established an analysis method for 239/240Pu in biological samples with a recovery rate of 81.6–91.8%.
The extraction behavior of amine extractants is very similar to that of anion exchangers, with the advantages of rapid extraction speed, high sorption capacity, and high selectivity to Pu (IV) in the nitric acid system. The exchange reaction between tri-n-octylamine (TOA) resin and Pu (IV) is very rapid, and TOA extraction chromatography is a standard method for the determination of 239Pu and 240Pu in food by alpha spectroscopy [63]. Ji et al. [64] established a process for the separation of Pu by TOA resin and compared it with the anion-exchange resin (Dowex-1 × 8), which had a slightly poorer decontamination effect.
3. Measurement Methods of 239Pu, 240Pu, and 90Sr
Radionuclide measurement methods are divided into radioactive measurement methods (such as alpha spectrometry) and non-radioactive measurement methods (such as mass spectrometry). The former is to perform qualitative and quantitative analysis of the target nuclide by measuring the characteristic rays (α, β, or Ƴ rays) emitted by the decay process of radionuclides. Mass spectrometry is the main non-radioactive measurement method, with improved technology, high sensitivity, good precision, and higher equipment popularity than radioactive measurement methods. The various radioactivity measurement methods of 239Pu, 240Pu, and 90Sr are reviewed in this part and compared with mass spectrometry, focusing on mass-spectrometry measurement methods.
3.1. Radioactivity Measurement Methods
Different instruments are used to measure different kinds of rays [65]. When a certain nuclide emits different kinds of rays at the same time, the radioactivity obtained by measuring different rays is consistent. The main radiometric methods of nuclides in the environment are shown in Table 5. The radioactive analysis method is the most widely used analysis method for the determination of 90Sr, 239Pu, and 240Pu [21,45]. 90Sr is a beta emitter, while 238Pu, 239Pu, and 240Pu are alpha emitters (Table 6).
Common analytical methods for the determination of 90Sr in environmental samples are liquid scintillation counting (LSC) and proportional counter [44,67]. Solatie et al. [68] used low-background LSC to measure the concentration of 90Sr in soil samples near nuclear facilities, with the activity range of 6.2–96.5 Bq·kg−1. Asgharizadeh et al. [69] measured the activity of 90Sr in soil and sediment samples from the southern coast of Iran, with the results in the range of 0.40–3.01 Bq·kg−1. At the 95% confidence level, the minimum detectable activity (MDA) of the LSC method within a 5-h counting period was 0.33 Bq·kg−1. Lee et al. [69,70] measured the activity concentration of 90Sr in the volcanic soil of Jeju Island (19.9 ± 2.1 Bq·kg−1) and determined that it was higher than that of the Korean Peninsula (8.3 ± 1.7 Bq·kg−1) by using low-level LSC. Maxwell [70] reported that the concentration of 90Sr in the soil samples from the Fukushima Nuclear Power Plant was 1.35 mBq·g−1 by a gas proportional counter. Jabbar et al. [71] used LSC to determine that the average concentration of 90Sr in soil samples and plant samples from Islamabad was 4.3 ± 0.0005 Bq·kg−1 and 1.05 ± 0.91 Bq·kg−1, respectively.
There are many methods for measuring Pu in the environment. After chemical separation, Pu is usually measured by using liquid scintillation counting or alpha spectrometry. However, the separation process of these two methods is more complicated and time-consuming. It usually takes about 1 week to complete a measurement period, and sometimes even more than 10 days. At the same time, the alpha-energy values of 239Pu (5.16, 5.14 MeV) and 240Pu (5.17, 5.12 MeV) are very close, and this is a great challenge to the energy resolution (at least 25 keV) of the current ion-implanted passivated planar silicon detector [72], leading to the fact that only the total activity of 239+240Pu can be measured. Beata Varga et al. [73] used alpha spectroscopy to measure the activity of 239+240Pu of grass samples in the hilly area of Budapest as 0.024 ± 0.01 Bq·kg−1 (fresh samples), with the MDA of 0.02 Bq·kg−1. Compared with the soil surface, the transfer factor (TF) absorbed by roots was 0.032 ± 0.014 (Bq·kg−1 fresh grass mass)/(Bq·kg−1 dry soil mass), and the TF of forage grass in the literature was in the range of 5 × 10−5–0.7. Sherrod et al. [30] determined the activity of 239Pu in rice samples as 11.8 Bq·kg−1. Lee et al. [74] measured Pu and Am isotopes in the volcanic soil of Jeju Island and the Korean Peninsula. The concentrations of 239Pu and 240Pu were 2.0 ± 0.3 Bq·kg−1 (the error is 1σ) and 0.67 ± 0.1 Bq·kg−1, respectively. Liu et al. [58] established a method to quickly determine the content of 239+240Pu in environmental water samples by using low-background LSC, and the detection limit of 5 L water samples was 6 × 10−3 Bq·L−1 within a 60-min counting time. Dagmara et al. [75] studied the distribution of Pu in the organs and tissues of seabirds and found that the highest Pu concentration was in the digestive organs and feathers, while the smallest was in the skin and muscles. The Pu content was lower in fish-eating seabirds, while it was much higher in herbivorous seabirds. It was analyzed that the main source of Pu in seabirds was global atmospheric sedimentation.
In conclusion, the commonly used methods for measuring ultra-trace 90Sr and 239,240Pu in environmental samples are liquid scintillation counting and alpha spectroscopy. The detection limit of these two methods is low, but the measurement time is long, which is not suitable for emergency monitoring. As alpha spectroscopy can only measure the total activity of 239+240Pu, the ratio of 240Pu/239Pu is unable to be given to analyze the source of Pu.
3.2. Mass Spectrometry Methods
Mass spectrometry technology is widely used in the determination of radionuclides with long life and large mass numbers, such as 235U, 239Pu, and 240Pu. For short-lived radionuclides, radioactivity-measurement methods are more commonly used. In recent years, newly developed mass spectrometry techniques with high resolution, high sensitivity, and advanced interference cancellation functions have also been used to measure radionuclides with a shorter life, such as 90Sr. This section reviews the mass spectrometry methods, focusing on ICP–MS, to determine 90Sr, 239Pu, and 240Pu in order to better understand the analytical capabilities and potential applications of mass spectrometry.
3.2.1. Measurement Method of 90Sr
The natural Sr concentration in environmental samples is about nine orders of magnitude higher than that of 90Sr. 90Sr is interfered by isobaric 90Zr interference and the stable peak trailing interference of 88Sr (82.58% abundance) when determined by mass spectrometry. The separation of 90Sr and 90Zr requires a mass resolution greater than 30,000, which exceeds the resolution capability of mass spectrometry. Researchers have tried many methods. Arslan et al. [76] made the first attempt to determine 90Sr by using accelerator mass spectrometry (AMS) after removing 90Zr by a chemical procedure. Betti et al. [77] measured the concentration of 90Sr in soil, sediment, and grass by glow-discharge mass spectrometry (GD-MS). Wendt et al. [78] reported the concentrations of 89Sr and 90Sr in soil, grass, milk, and urine after chemical separation by resonance ionization mass spectrometry (RIMS). Bushaw et al. [79] used RIMS to determine 90Sr in environmental samples with a detection limit as low as 0.8 fg. AMS, especially RIMS, has high element selectivity. However, they are not usually used in the environmental application, due to less accessibility, and complicated operation and maintenance. ICP–MS, with high sensitivity, can perform rapid and relatively cost-effective isotope analysis at ultra-trace levels [80]. High-resolution inductively coupled plasma–mass spectrometry (HR-ICP–MS) has ultra-high sensitivity and strong interference elimination ability. The ionization energy of Sr (5.70 eV) is 1.14 eV lower than that of Zr (6.84 eV). Operating under cold plasma can greatly reduce the ionization efficiency of Zr by HR-ICP–MS [33]. The isotope ratio of 90Sr/86Sr in soil samples near the nuclear facility was 6.02 × 10−9, basically reaching the sensitivity of radiochemical method for the determination of 90Sr [20].
Quadrupole inductively coupled plasma–mass spectrometry (ICP-QMS) [1,23,56] and sector-magnetic-field inductively coupled plasma–mass spectrometry (SF-ICP–MS) [19] are also used for the measurement of 90Sr. The interference signal is suppressed by improving the sample introduction method and optimizing the operating system conditions, effectively reducing the detection limit. The concentration of 90Sr was measured by tunable bandpass dynamic reaction cell–ICP–MS (DRC–ICP–MS) with relatively high abundance sensitivity [55]. O2 was used as the reaction gas to form ZrO+ with Zr in the reaction cell, effectively suppressing the interference of Zr. The concentration of 90Sr in sediments, plants and water samples was consistent with the activity value determined by Ĉherenkov count and the reference value. The detection limits of the three samples were 0.1 pg·g−1 (0.5 Bq·g−1), 0.04 pg·g−1 (0.5 Bq·g−1), and 3 pg·L−1 (5 Bq·L−1), respectively. Feuerstein et al. [1] used ICP–DRC–MS to quickly determine the concentration of 90Sr in contaminated soil samples near the Chernobyl nuclear power plant and compared with the results obtained by the previous radiometry (Table 7). The measurement results were basically consistent, while the accuracy of ICP–DCR–MS was better. Although the MDA of ICP–DRC–MS was inferior to the radiometric methods, it represented a time-saving and economical alternative technology for rapid monitoring of high levels of 90Sr contamination in environmental samples.
The interference signals can also be suppressed effectively by using dual mass spectrometry for mass discrimination. 90Sr in the reference materials was measured by triple quadrupole collision/reaction cell–CP–MS/MS in single mode and MS/MS mode with relative accuracy and precision of 0.7–3.6% and 3.5–8.1% [34]. The established method was used to measure the artificial radionuclide 90Sr in soil samples collected from the whole state of Qatar. The average concentration was 0.606 fg·g−1 (3.364 Bq·kg−1), with the detection limit of less than 0.04 pg.
3.2.2. Mass Spectrometry Measurement Methods of 239Pu and 240Pu
As mentioned above, the use of mass spectrometry to measure 239Pu and 240Pu is interfered by the uranium–hydrogen peak. It is necessary to separate 238U before measurement and absolutely eliminate polyatomic chlorides and oxides. Methods such as improving the sample introduction method, increasing the temperature of the plasma, and changing the flow rate of the atomizer can reduce the formation of hydride ions [83]. Among them, a sample introduction method, such as membrane desolvation, is a very important factor that affects hydride [84]. For hydride generation, Kim et al. [85] compared five injection methods in terms of sensitivity, specific sensitivity, and precision. A high-efficiency sample-introduction system Apex-Q combined with the sector field ICP–MS could achieve high instrumental sensitivity and reduce the formation of 238UH+/238U [86].
Thermal ionization mass spectrometry (TIMS) is one of the main methods to determine isotope composition and ratio. The accuracy of isotope measurement is less than 5 × 10−4, and the abundance sensitivity is better than 5 × 10−8. McCarthy et al. [87] measured the isotopic ratios of 240/239Pu and 241/239Pu in Oxfordshire soil to be 0.184 (RSD < 1%) and 0.0049 (RSD < 5%). The average concentration of 239+240Pu was 0.257 Bq·kg−1, close to the average value of radioactive deposition in Britain [88]. Buesseler et al. [89] found that TIMS was one order of magnitude more sensitive than the traditional alpha-counting technique when used to detect Pu in sediments, seawater, and coral samples.
TIMS has high precision and abundance sensitivity; however, with a complicated sample-preparation process (requiring pure samples), long measurement time, and a large amount of samples, it is not as simple as ICP–MS to operate. By using the sector-field ICP–MS to measure 239Pu, Ni et al. [85] investigated the transfer of global fallout Pu from paddy soil to rice grain in 12 Japanese prefectures, with the TF ranging from 4.5 × 10−6 to 1.2 × 10−4. Chiappini et al. [90] used high-sensitivity ICP–MS to measure the contribution of Pu deposited in the Mururoa and Fangataufa sediments in the French atmospheric nuclear test for the first time. The contribution was very small and could only be observed in the territorial sea boundary area 22 km away from the atoll. The concentration of Pu in the seawater was only 2 mBq·m−3 higher than that observed in Rangiroa. For standard solutions without interference, the detection limits of ICP–MS/HP/Mistral (high-performance ICP–MS associated with Mistral desolvation atomizer (HP/Mistral)) were compared with alpha spectrometry (Table 8). The measurement period of alpha spectrometry was 4000 min. It should be noted that alpha spectrometry measures the sum of the activities of 239Pu and 240Pu, while the ICP–MS measures the activities of 239Pu and 240Pu, respectively. On this basis, ICP–MS/HP and alpha spectroscopy were compared for the measured values of 239+240Pu in Sellafield sediments, Mururoa lagoon sediments, and grouper samples. These comparisons confirmed that the detection limit of ICP–MS/HP/Mistral was lower than that of alpha spectroscopy when measuring environmental samples. In addition, the introduction of collision/reaction cell (CRC) technology in ICP–MS can effectively reduce the interfering effects of the uranium-hydrogen peaks on measuring 239Pu and 240Pu. Recently, a CRC–ICP–MS/MS method reduced 238/238U16 ratio to 4.82 × 10−9, 2-to-3 orders of magnitude lower than previously reported values, and accurately measured the ultra-trace level 239Pu with the concentration ratio as low as 10−10 by 239Pu/238U [91].
HR-ICP–MS is also mostly used when measuring the concentration of 239,240Pu in the environment. Helal et al. [20] measured the isotope ratio of 240Pu/239Pu of 0.17 in the soil near nuclear facilities. Stefan et al. [92] used the ultrasonic atomization method to measure the Pu isotope activity and 240Pu/239Pu isotope ratio in environmental samples, with the measurement accuracy (RSD) of the ratio of 240Pu/239Pu at about 2%. The total plutonium activity (239+240Pu) measured was very consistent with the data obtained by the alpha spectroscopy, with the detection limits of 239Pu and 240Pu to be 5 and 1 fg·mL−1, respectively. Wang et al. [93] measured the activity of 239+240Pu in the surface sediments of the Bohai Sea and the North Yellow Sea by the isotope dilution method to be 0.001–0.288 and 0.040–0.269 Bq·kg−1, respectively. The atomic ratio of 240Pu/239Pu was 0.173–0.256 and 0.196–0.275, respectively, slightly higher than the global sedimentation value of 0.18.
In addition, Mathew et al. [94] used AMS to distinguish Pu from the FDNPP accident and Nagasaki explosion from global sediments. It was found that the isotope ratio of 241Pu/239Pu was more sensitive than that of 240Pu/239Pu when Pu was traced.
Overall, mass spectrometry has been widely used in the measurement of ultra-trace nuclides. In recent years, CRC–ICP–MS/MS is a promising method with ultra-high sensitivity, which can significantly eliminate the 90Zr and 238U interferences in the measurement of 90Sr and 239,240Pu by using O2 as the reaction gas.
4. Conclusions
Concerns have increased regarding the environmental radioactive contamination occurring in large-scale nuclear or radiological accident/incident. Carrying out relevant monitoring is of great significance, because it reflects the affected degree of biological chain to a certain extent. In this work, we reviewed and summarized the pretreatment, separation, and measurement methods for analyzing 90Sr and 239,240Pu in environmental samples with the characteristics of ultra-trace concentration and complex matrix. The pretreatment of samples usually costs most of the experimental time. Combining the dry-ashing and microwave-digestion methods greatly increases the processing capacity and shortens the processing time. The classical separation method of Sr is fuming nitric acid precipitation, which is time-consuming and cumbersome. In contrast, the solvent-extraction method is quicker and simpler. In recent years, the commonly used Sr high-efficiency specific resin extraction method has been more selective. As for the separation and purification of Pu in the environment, adjusting the valence state of Pu to Pu (IV) is the primary task. Among the commonly used methods for the separation of trace amounts of Pu, the ion-exchange method is fast and economical, while the exchange capacity of the leaching resin is high. Then we analyzed and compared the nuclear measurement techniques of 90Sr and 239,240Pu in the environment. The highly sensitive liquid scintillation spectrometry and alpha spectrometry are the general methods for measuring 90Sr and 239,240Pu, as their instrument maintenance is simpler than mass spectrometry. By improving sample introduction methods and optimizing the operating conditions of the instrument, mass spectrometry has the ability to successfully measure the concentrations of 90Sr and 239,240Pu in the environment with equal sensitivity and a lower detection limit to radiometric methods.
The pretreatment of a large number of samples and the low detection limit of the instrument are required for the complex matrix of environmental samples and the ultra-trace level of radionuclides. Here are some prospects for the radionuclide analysis of environmental samples. First, as sample preprocessing is often cumbersome and time-consuming, it is critical to establish a rapid, convenient, efficient, and clean pretreatment method in the future. Second, based on the characteristics of radionuclides in environmental samples, research on developing high-capacity, high-efficiency, and specific separation resins for major beta nuclides and actinides is the development trend. Providing rapid and ultra-sensitive analysis technology is of great significance to environmental emergency monitoring. Therefore, automatic separation and measurement technology will become a promising direction due to its fast and convenient advantages. In addition, research on mobility of 90Sr and 239,240Pu in the environment is the basis for predicting the degree of ecological environment pollution, as well as the restoration study of soil, vegetation, and water after contamination with these nuclides.
Author Contributions
Writing—original draft, N.Z.; writing—review and editing, N.Z., L.L., X.Y. and Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Key R&D Program of China, grant number 2018YFC1602500.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Feuerstein, J.; Boulyga, S.F.; Galler, P.; Stingeder, G.; Prohaska, T. Determination of 90Sr in soil samples using inductively coupled plasma mass spectrometry equipped with dynamic reaction cell (ICP-DRC-MS). J. Environ. Radioact. 2008, 99, 1764–1769. [Google Scholar] [CrossRef]
- Mole, R.H. Working with plutonium. Lancet 1975, 305, 701–702. [Google Scholar] [CrossRef]
- Langham, W.H.; Healy, J. Maximum permissible body burdens and concentrations of plutonium: Biological basis and history of development. In Uranium-Plutonium Transplutonic Elements; Springer: Berlin/Heidelberg, Germany, 1973; Volume 36, pp. 569–592. [Google Scholar]
- Aliyu, A.S.; Evangeliou, N.; Mousseau, T.A.; Wu, J.; Ramli, A.T. An overview of current knowledge concerning the health and environmental consequences of the Fukushima Daiichi Nuclear Power Plant (FDNPP) accident. Environ. Int. 2015, 85, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Runde, W. The chemical interactions of actinides in the environment. Alamos Sci. 2000, 26, 392–411. [Google Scholar]
- Li, M.F.; Luo, D.L.; Weng, H.S.; Zhang, Z.; Yu, J.Y. The transfer coefficients of Ba, Sr, and Zr in Guangdong cabbage. J. Sun Yat-sen Univ. Nat. Sci. 1997, 36, 109–112. [Google Scholar]
- Weng, C.W.; Huang, H.S.; Xu, C.X. Study on the accumulation effect of 11 heavy metals in vegetables. Food Res. Dev. 2018, 39, 194–200. [Google Scholar]
- Cai, F.L.; Chen, Y.; Huang, L.Y. Research on Mytilus viridis linnaeus as indicator organisms for 90Sr pollution in sea area. J. Oceanogr. 1990, 12, 261–264. [Google Scholar]
- Neck, V.; Altmaier, M.; Seibert, A.; Yun, J.I.; Marquardt, C.M.; Fanghanel, T. Solubility and redox reactions of Pu(IV) hydrous oxide: Evidence for the formation of PuO2+x(s, hyd). Radiochim. Acta 2007, 95, 137d9–207. [Google Scholar] [CrossRef]
- Lee, J.H.; Hossnera, L.R.; Attrep, M.; Kung, K.S. Uptake and translocation of plutonium in two plant species using hydroponics. Environ. Pollut. 2002, 117, 61–68. [Google Scholar] [CrossRef]
- Tawussi, F.; Gupta, D.; Muhr-Ebert, E.; Schneider, S.; Bister, S.; Walther, C. Uptake of Plutonium-238 into Solanum tuberosum L. (potato plants) in presence of complexing agent EDTA. J. Environ. Radioact. 2017, 178-179, 186–192. [Google Scholar] [CrossRef]
- Kenna, T.C. Determination of plutonium isotopes and neptunium-237 in environmental samples by inductively coupled plasma mass spectrometry with total sample dissolution. J. Anal. At. Spectrom. 2002, 17, 1471–1479. [Google Scholar] [CrossRef]
- Mitchell, P.I.; Vintro, L.L.; Dahlgard, H.; Gasco, C.; Sanchez-Cabeza, J.A. Perturbation in the 240Pu239Pu global fallout ratio in local sediments following the nuclear accidents at Thule (Greenland) and Palomares (Spain). Sci. Total Environ. 1997, 202, 147–153. [Google Scholar] [CrossRef]
- Erdmann, N.; Herrmann, G.; Huber, G.; Kohler, S.; Kratz, J.V.; Mansel, A.; Nunnemann, M.; Passler, G.; Trautmann, N.; Turchin, A.; et al. Resonance Ionization Mass Spectroscopy for Trace Determination of Plutonium in Environmental Samples. Fresenius’ J. Anal. Chem. 1997, 359, 378–381. [Google Scholar] [CrossRef]
- Eriksson, M.; Lindahl, P.; Roos, P.; Dahlgaard, H.; Holm, E. U, Pu, and Am nuclear signatures of the thule hydrogen bomb debris. Environ. Sci. Technol. 2008, 42, 4717–4722. [Google Scholar] [CrossRef]
- Krey, P.W.; Hardy, E.P.; Pachucki, C.; Rourke, F.; Coluzza, J.; Benson, W.K. Mass Isotopic Composition of Global Fall-Out Plutonium in Soil. In Transuranium Nuclides in the Environment, Proceedings of the Symposium on Transuranium Nuclides in the Environment, San Francisco, CA, USA, 17–21 November 1975; International Atomic Energy Agency: Vienna, Austria, 1976; pp. 671–678. [Google Scholar]
- Igarashi, J.; Zheng, J.; Zhang, Z.; Ninomiya, K.; Satou, Y.; Fukuda, M.; Ni, Y.; Aono, T.; Shinohara, A. First determination of Pu isotopes (239Pu, 240Pu and 241Pu) in radioactive particles derived from Fukushima Daiichi Nuclear Power Plant accident. Sci. Rep. 2019, 9, 11807. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, Y.; Rühm, W.; Yoshida, S.; Tagami, K.; Uchida, S.; Wirth, E. Concentrations of 239Pu and 240Pu and their isotopic ratios determined by ICP-MS in soils collected from the Chernobyl 30-km zone. Environ. Sci. Technol. 2000, 34, 2913–2917. [Google Scholar] [CrossRef]
- Vonderheide, A.P.; Zoriy, M.V.; Izmer, A.V.; Pickhardt, C.; Caruso, J.A.; Ostapczuk, P.; Hille, R.; Becker, J.S. Determination of 90Sr at ultratrace levels in urine by ICP-MS. J. Anal. At. Spectrom. 2004, 19, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Helal, A.I.; Zahran, N.F.; Amr, M.A.; El-Lateef, A.M.A.; Bashter, I.I.; Mohsen, H.T.; Abbas, Y. Ultratrace and isotope ratios analyses of some radionuclides by ICP-MS. Radiochim. Acta 2004, 92, 369–374. [Google Scholar] [CrossRef]
- Amr, M.A.; Abdel-Lateef, A.M. Comparing the capability of collision/reaction cell quadrupole and sector field inductively coupled plasma mass spectrometers for interference removal from 90Sr, 137Cs, and 226Ra. Int. J. Mass Spectrom. 2011, 299, 184–190. [Google Scholar] [CrossRef]
- Goldstein, S.; Price, A.; Hinrichs, K.; Lamont, S.; Nunn, A.; Amato, R.; Cardon, A.; Gurganus, D. High-precision measurement of U-Pu-Np-Am concentrations and isotope ratios in environmental reference materials by mass spectrometry. J. Environ. Radioact. 2021, 237, 106689. [Google Scholar] [CrossRef]
- Takagai, Y.; Furukawa, M.; Kameo, Y.; Suzuki, K. Sequential inductively coupled plasma quadrupole mass-spectrometric quantification of radioactive strontium-90 incorporating cascade separation steps for radioactive contamination rapid survey. Anal. Methods 2014, 6, 355–362. [Google Scholar] [CrossRef]
- Florou, H.; Kehagia, K.; Chaloulou, C.; Koukouliou, V.; Lykomitrou, C. Determination of radionuclides in Mytilus galloprovicialis by Alpha and Gamma-Spectroscopy. Mediterr. Mar. Sci. 2012, 5, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Li, K.X.; Qin, S.C. Pretreatment of biological samples in the background investigation of radiation environment. J. Nav. Med. 2005, 26, 201–203. [Google Scholar]
- GB/T 16145—2020; Gamma Spectrometry Method of Analysing Radionuclides in Biological Samples. National Standardization Administration: Beijing, China, 2020.
- Ma, Y.; Tang, X.Z.; Ding, T.T. The application of microwave ashing analysis in the pretreatment of oil metal content. Anal. Test. Technol. Instrum. 2010, 16, 206–208. [Google Scholar]
- Bao, M.M.; Kang, J.W.; Jiang, D.C.; Li, P.; Wang, L.Y.; Zhu, S.W. Improvement of powdery sample detection dry ashing pretreatment. Light Ind. Technol. Food Biol. 2013, 12, 4–5. [Google Scholar]
- GB 14883.1—2016; General Rules for the Inspection of Radioactive Substances in Food. National Health and Family Planning: Beijing, China, 2017.
- Maxwell, S.L.; Culligan, B.K.; Hutchison, J.B. Rapid fusion method for determination of plutonium isotopes in large rice samples. J. Radioanal. Nucl. Chem. 2013, 298, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Comparison of different digestion methods-inductively coupled plasma mass spectrometry for the determination of 15 metal elements in black tea. J. Tea 2019, 60, 151–155. [Google Scholar]
- Grahek, Z.; Dulanska, S.; Karanovic, G.; Coha, I.; Tucakovic, I.; Nodilo, M.; Matel, L. Comparison of different methodologies for the 90Sr determination in environmental samples. J. Environ. Radioact. 2018, 181, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Zheng, J.; Liu, X.; Long, K.; Hu, S.; Uchida, S. Mass spectrometry for the determination of fission products 135Cs, 137Cs and 90Sr: A review of methodology and applications. Spectrochim. Acta B At. Spectrosc. 2016, 119, 65–75. [Google Scholar] [CrossRef]
- Amr, M.; Helal, A.-F.; Al-Kinani, A.T.; Balakrishnan, P. Ultra-trace determination of 90Sr, 137Cs, 238Pu, 239Pu, and 240Pu by triple quadruple collision/reaction cell-ICP-MS/MS: Establishing a baseline for global fallout in Qatar soil and sediments. J. Environ. Radioact. 2016, 153, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Mola, M.; Avivar, J.; Nieto, A.; Peñalver, A.; Aguilar, C.; Ferrer, L.; Cerdà, V.; Borrull, F. Determination of 90Sr and 210Pb in sludge samples using a LOV-MSFIA system and liquid scintillation counting. Appl. Radiat. Isot. 2014, 86, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Bermudez, S.; Mas, J.L.; Villa-Alfageme, M. A sequential determination of 90Sr and 210Po in food samples. Food Chem. 2017, 229, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.; Elahi, S.; Lee, K.; Fairman, B. A rapid and accurate method for the determination of plutonium in food using magnetic sector ICP-MS with an ultra-sonic nebuliser and ion chromatography. J. Environ. Monit. 2003, 5, 175–179. [Google Scholar] [CrossRef]
- Holm, E.; Ballestra, S.; Fukai, R. A method for ion-exchange separation of low levels of americium in environmental materials. Talanta 1979, 26, 791–794. [Google Scholar] [CrossRef]
- Hamato, A. An anion exchange method for the determination of 241Am and plutonium in environmental and biological samples. J. Radioanal. Nucl. Chem. 1982, 75, 265. [Google Scholar] [CrossRef]
- Matishov, G.G.; Matishov, D.G.; Namjatov, A.A.; Carroll, J.; Dahle, S. Anthropogenic radionuclides in Kola and Motovsky bays of the barents Sea, Russia. J. Environ. Radioact. 1999, 43, 77–88. [Google Scholar] [CrossRef]
- Lee, Y.K.; Bakhtiar, S.N.; Akbarzadeh, M.; Lee, J.S. Sequential isotopic determination of strontium, thorium, plutonium, uranium, and americium in bioassay samples. J. Radioanal. Nucl. Chem. 2000, 243, 525. [Google Scholar] [CrossRef]
- Heldal, H.E.; Varskog, P.; Føyn, L. Distribution of selected anthropogenic radionuclides (137Cs, 238Pu, 239,240Pu and 241Am) in marine sediments with emphasis on the Spitsbergene-Bear Island Area. Sci. Total Environ. 2002, 293, 233–245. [Google Scholar] [CrossRef]
- Qiao, J.X.; Hou, X.L.; Miro, M.; Roos, P. Determination of plutonium isotopes in waters and environmental solids: A Review. Anal. Chim. Acta 2009, 652, 66–84. [Google Scholar] [CrossRef] [Green Version]
- Vajda, N.; Kim, C.K. Determination of radiostrontium isotopes: A review of analytical methodology. Appl. Radiat. Isot. 2010, 68, 2306–2326. [Google Scholar] [CrossRef]
- Marzo, G.A. Atmospheric transport and deposition of radionuclides released after the Fukushima Daichi accident and resulting effective dose. Atmos. Environ. 2014, 94, 709–722. [Google Scholar] [CrossRef]
- Tayeb, M.; Dai, X.; Sdraulig, S. Rapid and simultaneous determination of Strontium-89 and Strontium-90 in seawater. J. Environ. Radioact. 2016, 153, 214–221. [Google Scholar] [CrossRef]
- Bojanowski, R.; Knapinska-Skiba, D. Determination of low level 90Sr in environmental samples: A novel approach to the classical method. J. Radioanal. Nucl. Chem. 1990, 138, 207–218. [Google Scholar] [CrossRef]
- Chobola, R.; Mell, P.; Daroczi, L.; Vincze, A. Rapid determination of radiostrontium isotopes in samples of NPP origin. J. Radioanal. Nucl. Chem. 2006, 267, 297–304. [Google Scholar] [CrossRef]
- Rao, D.D.; Mehendarge, S.T.; Chandramouli, S.; Hegde, A.G.; Mishra, U.C. Application of Cherenkov radiation counting for determination of 90Sr in environmental samples. J. Environ. Radio. 2000, 48, 49–57. [Google Scholar] [CrossRef]
- Tera, F.; Morrison, G.H. Radiochemical separations by isotopic ion exchange. Anal. Chem. 1966, 38, 959–964. [Google Scholar] [CrossRef]
- Kimura, T.; Iwashima, K.; Ishimori, T.; Hamada, T. Separation of strontium-89 and -90 from calcium in milk with a macrocyclic ether. Anal. Chem. 1979, 51, 1113–1116. [Google Scholar] [CrossRef]
- Pawlak, D.; Parus, J.; Dziel, T.; Muklaniwicz, A.; Mikolajczak, R. Determination of 90Sr traces in medical 90Y after separation on DGA column. Talanta 2013, 114, 1–4. [Google Scholar] [CrossRef]
- Horwitz, E.P.; McAlister, D.R.; Bond, A.H.; Barrans, R.E. Novel Extraction of Chromatographic Resins Based on Tetraalkyldiglycolamides: Characterization and Potential Applications. Solvent Extr. Ion Exch. 2005, 23, 319–344. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Chiarizia, R.; Dietz, M.L. A novel strontium-selective extraction chromatographic resin. Solvent Extr. Ion Exch. 1992, 10, 313–336. [Google Scholar] [CrossRef]
- Taylor, V.F.; Evans, R.D.; Cornett, R.J. Determination of 90Sr in contaminated environmental samples by tuneable bandpass dynamic reaction cell ICP–MS. Anal. Bioanal. Chem. 2007, 387, 343–350. [Google Scholar] [CrossRef]
- Odoh, S.O.; Bylaska, E.J.; de Jong, W.A. Coordination and hydrolysis of plutonium ions in aqueous solution using Car–Parrinello molecular dynamics free energy simulations. J. Phys. Chem. A 2013, 117, 12256–12267. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Dietz, M.L.; Chiarizia, R.; Diamond, H.; Maxwell, S.L.; Nelson, M.R. Separation and preconcentration of actinides by extraction chromatography using a supported liquid anion exchanger: Application to the characterization of high-level nuclear waste solutions. Anal. Chim. Acta 1995, 310, 63–78. [Google Scholar] [CrossRef]
- Liu, B.; Shi, K.L.; Ye, G.Y.; Guo, Z.J.; Wu, W.S. Method development for plutonium analysis in environmental water samples using TEVA microextraction chromatography separation and low background liquid scintillation counter measurement. Microchem. J. 2016, 124, 824–830. [Google Scholar] [CrossRef]
- Maxwell, S.L.; Jones, V.D. Rapid determination of actinides in urine by inductively coupled plasma mass spectrometry and alpha spectrometry: A hybrid approach. Talanta 2009, 80, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, S.L.; Culligan, B.K.; Jones, V.D.; Nichols, S.T.; Bernard, M.A.; Noyes, G.W. Determination of 237Np and Pu isotopes in large soil samples by inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2010, 682, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.G.; Bu, W.T.; Zheng, J.; Pan, S.M.; Wang, Z.T.; Uchida, S.G. Plutonium determination in seawater by inductively coupled plasma mass spectrometry: A review. Talanta 2016, 151, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Ishida, J.; Yamato, A.; Iwai, M.; Kinoshita, M. Determination of 239,240Pu and 241Am in Environmental Samples. J. Radioanal. Nucl. Chem. 1987, 115, 369–376. [Google Scholar] [CrossRef]
- GB 14883.8—2016; Determination of Radioactive Substance Plutonium-239 and 240 in Food (Extraction Chromatography). National Health and Family Planning: Beijing, China, 2017.
- Ji, Y.Q. Study on Inductively Coupled Plasma Mass Spectrometry (ICP-MS) Analysis Method of Trace Uranium, Thorium, Neptunium and Plutonium in Environmental Samples. Ph.D. Thesis, China Institute of Atomic Energy, Beijing, China, September 2001. [Google Scholar]
- Ding, W.Z.; Wang, Y.; Huang, H.Q.; Wang, M. Principles of Alpha Spectrum Measurement of Natural Radionuclides. Nucl. Electron. Detect. Technol. 2010, 30, 817–820. [Google Scholar]
- Decay Radiation Search, Decay Radiation Database Version of 9 September, 2021; National Nuclear Data Center (NNDC). Available online: https://www.nndc.bnl.gov/nudat3/indx_dec.jsp (accessed on 30 January 2022).
- Matthews, K.M.; Kim, C.K.; Martin, P. Determination of 210Po in environmental materials: A review of analytical methodology. Appl. Radiat. Isot. 2007, 65, 267–279. [Google Scholar] [CrossRef]
- Solatie, D.; Carbol, P.; Hrnecek, E.; Jaakkola, T.; Betti, M. Sample preparation methods for the determination of plutonium and strontium in environmental samples by low level liquid scintillation counting and α-spectrometry. Radiochim. Acta 2002, 90, 447–454. [Google Scholar] [CrossRef]
- Asgharizadeh, F.; Salimi, B.; Maragheh, M.G.; Mahani, M.K.; Aliabadi, M. Determination of 90Sr concentration in soil and sediment samples from southern shores of Iran using a Sr resin and LSC method. Adv. Liq. Scintill. Spectrom. 2009, 299–303. [Google Scholar]
- Maxwell, S.L.; Culligan, B.K.; Shaw, P.J. Rapid determination of radiostrontium in large soil samples. J. Radioanal. Nucl. Chem. 2013, 295, 965–971. [Google Scholar] [CrossRef] [Green Version]
- Jabbar, T.; Subhani, M.S.; Khan, K.; Rashid, A.; Orfi, S.D.; Khan, A.Y. Natural and fallout radionuclide concentrations in the environment of Islamabad. J. Radioanal. Nucl. Chem. 2003, 258, 143–149. [Google Scholar] [CrossRef]
- Gao, R.Q.; Hou, X.L.; Zhang, L.Y.; Zhang, W.C.; Zhang, M.T. Determination of Ultra-Low Level Plutonium Isotopes in Large Volume Environmental Water Samples. Chin. J. Anal. Chem. 2020, 48, 765–773. [Google Scholar] [CrossRef]
- Varga, B.; Tarjan, S.; Vajda, N. Plutonium isotopes in the Hungarian environment. J. Environ. Radioact. 2008, 99, 641–648. [Google Scholar] [CrossRef]
- Lee, M.H.; Ahn, H.J.; Park, J.H.; Park, Y.J.; Song, K. Rapid sequential determination of Pu, 90Sr and 241Am nuclides in environmental samples using an anion exchange and Sr-Spec resins. Appl. Radiat. Isot. 2011, 69, 295–298. [Google Scholar] [CrossRef]
- Strumińska-Parulska, D.I.; Skwarzec, B.; Fabisiak, J. Plutonium bioaccumulation in seabirds. J. Environ. Radioact. 2011, 102, 1105–1111. [Google Scholar] [CrossRef]
- Arslan, F.; Behrendt, M.; Ernst, W.; Finckh, E.; Greb, G.; Gumbmann, F.; Haller, M.; Hofmann, S.; Karschnick, R.; Klein, M.; et al. 14C and 90Sr measurements at the Erlangen AMS facility. Nucl. Instrum. Methods Phys. Res. B Beam Interact. Mater. At. 1994, 92, 39–42. [Google Scholar] [CrossRef]
- Betti, M.; Giannarelli, S.; Hiernaut, T.; Rasmussen, G.; Koch, L. Detection of trace radioisotopes in soil, sediment and vegetation by glow discharge mass spectrometry. Fresenius’ J. Anal. Chem. 1996, 355, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Wendt, K.; Kratz, J.V.; Lantzsch, J.; Müller, P.; Northershauser, W.; Seibert, A.; Trautmann, N.; Waldek, A.; Zimmer, K. Rapid ultratrace determination of 89Sr and 90Sr in environmental samples by collinear laser resonance ionization spectrometry. Kerntechnik 1997, 62, 81–84. [Google Scholar] [CrossRef]
- Bushaw, B.A.; Cannon, B.D. Diode laser based resonance ionization mass spectrometric measurement of strontium-90. Spectrochim. Acta B At. Spectrosc. 1997, 52, 1839–1854. [Google Scholar] [CrossRef]
- Becker, J.S. Inductively coupled plasma mass spectrometry (ICP-MS) and laser ablation ICP-MS for isotope analysis of long-lived radionuclides. Int. J. Mass Spectrom. 2005, 242, 183–195. [Google Scholar] [CrossRef]
- Alvarez, A.; Navarro, N. Method for actinides and Sr-90 determination in urine samples. Appl. Radiat. Isot. 1996, 47, 869–873. [Google Scholar] [CrossRef]
- Shiraishi, K.; Ko, S.; Arae, H.; Ayama, K. Rapid analysis technique for strontium, thorium, and uranium in urine samples. J. Radioanal. Nucl. Chem. 2007, 273, 307–310. [Google Scholar] [CrossRef]
- Alves, L.; Wiederin, D.; Houk, R. Reduction of Polyatomic Ion Interferences in Inductively Coupled Plasma Mass Spectrometry by Cryogenic Desolvation. Anal. Chem. 1992, 64, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- Minnich, M.G.; Houk, R.S. Comparison of cryogenic and membrane desolvation for attenuation of oxide, hydride and hydroxide ions and ions containing chlorine in inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 1998, 13, 167–174. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, C.K.; Lee, J.I.; Lee, K.J. Rapid determination of Pu isotopes and atom ratios in small amounts of environmental samples by an on-line sample pre-treatment system and isotope dilution high resolution inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 2000, 15, 247–255. [Google Scholar] [CrossRef]
- Ni, Y.; Wang, Z.; Zheng, J.; Tagami, K.; Guo, Q.; Uchida, S.; Tsukada, H. The transfer of fallout plutonium from paddy soil to rice: A field study in Japan. J. Environ. Radioact. 2019, 196, 22–28. [Google Scholar] [CrossRef]
- McCarthy, W.; Nicholls, T.M. Mass-Spectrometric Analysis of Plutonium in Soils Near Sellafield. J. Environ. Radioact. 1990, 12, 1–12. [Google Scholar] [CrossRef]
- Cawse, P.A.; Horrill, A.D. A Survey of Caesium-137 and Plutonium in British Soils in 1977; AERE-R10155; UK Atomic Energy Authority: Harwell, UK, 1986.
- Buesseler, K.O. The isotopic signature of fallout plutonium in the North Pacific. J. Environ. Radioact. 1997, 36, 69–83. [Google Scholar] [CrossRef]
- Chiappini, R.; Pointurier, F.; Millies-Lacroix, J.C.; Lepetit, G.; Hemet, P. 240Pu/239Pu isotopic ratios and 239+240Pu total measurements in surface and deep waters around Mururoa and Fangataufa atolls compared with Rangiroa atoll French Polynesiaž. Sci. Total Environ. 1999, 237–238, 269–276. [Google Scholar] [CrossRef]
- Zhang, W.C.; Lin, J.F.; Fang, S.; Li, C.; Yi, X.; Hou, X.; Chen, N.; Zhang, H.; Xu, Y.; Dang, H.; et al. Determination of ultra-trace level plutonium isotopes in soil samples by triple-quadrupole inductively coupled plasma-mass spectrometry with mass-shift mode combined with UTEVA chromatographic separation. Talanta 2021, 234, 122652. [Google Scholar] [CrossRef]
- Sturup, S.; Dahlgaard, H.; Nielsen, S. High resolution inductively coupled plasma mass spectrometry for the trace determination of plutonium isotopes and isotope ratios in environmental samples. J. Anal. At. Spectrom. 1998, 13, 1321–1326. [Google Scholar] [CrossRef]
- Wang, J.L.; Du, J.Z.; Qu, J.G.; Bi, Q.Q. Distribution of Pu isotopes and 210Pb in the Bohai Sea and Yellow Sea: Implications for provenance and transportation. Chemosphere 2021, 263, 127896. [Google Scholar] [CrossRef]
- Mathew, P.J.; Donovan, A.; David, C.; Michael, A.C.H.; Hirofumi, T.; Kei, O.; Thomas, G.H. Differentiating Fukushima and Nagasaki plutonium from global fallout using 241Pu/239Pu atom ratios: Pu vs. Cs uptake and dose to biota. Sci. Total Environ. 2021, 754, 141890. [Google Scholar]
Figure 1.
Separation process of radionuclides in environmental samples.


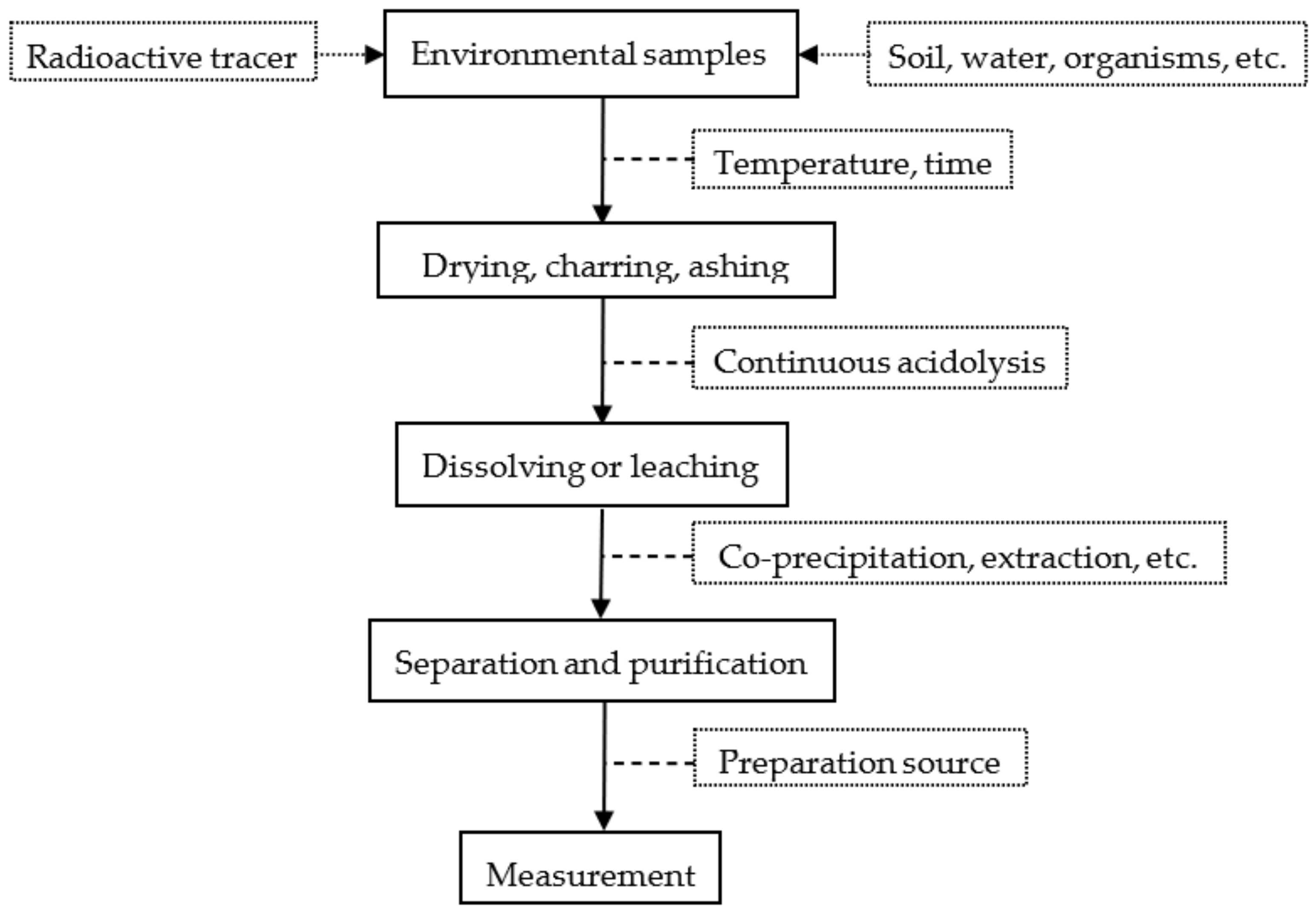{kind=link}
Table 1.
Source and total release of artificial radionuclides (PBq). Adapted with permission from ref. [4].
Table 1.
Source and total release of artificial radionuclides (PBq). Adapted with permission from ref. [4].
| Radionuclide | Weapon Tests | Chernobyl Accident | Fukushima Accident | |||
|---|---|---|---|---|---|---|
| Atmosphere | Ocean | Atmosphere | Ocean | Atmosphere | Ocean | |
| 131I | - | - | 1760 | - | 150–160 | - |
| 137Cs | 950 | 600 | 85 | 16 | 12–20 | 4–27 |
| 90Sr | 600 | 380 | 1 | - | 0.01–0.14 | 0.1–2.2 |
| 239,240Pu | 10.87 | 6.6 | 0.087 | - | (1–2.4) × 10−6 | - |
Table 2.
Pu isotope ratios in pollution from different nuclear events.
| 240Pu/239Pu | Ratio | References |
|---|---|---|
| Spent fuel reprocessing | 0.02–0.06 | [13,14] |
| Weapons-grade | <0.06 | [15] |
| Global fallout | 0.176 ± 0.014 | [16] |
| Fukushima | 0.330–0.415 | [17] |
| Chernobyl | 0.4 | [18] |
Table 3.
Maximum ashing temperature of food samples specified in GB14883-2016.
| Analysis Project | 89Sr, 90Sr | 137Cs | 147Pm | 226Ra | Natural Thorium | Natural Uranium | 239Pu |
|---|---|---|---|---|---|---|---|
| Temperature (°C) | 550 | 450 | 450 | 550 | 550 | 550 | 450 |
Table 4.
Digestion methods applied to the analysis of 90Sr, 239Pu, and 240Pu in the environment.
| Research Object | Acid Reagent | Digestion Equipment | References |
|---|---|---|---|
| 10 or 20 g soil sample | 1 mL Concentrated HNO3, 150 mL Deionized water | [32] | |
| 5 g plant sample ash | 100 mL 5 mol·L−1 HNO3 | Infrared Lamp | [32] |
| 1 g soil sample | 4.8 mL HF, 1.2 mL HClO4 | Microwave Digestion Apparatus | [33] |
| 50 g reference material + 500 g Qatari soil | 500 mL Concentrated HNO3, 250 mL HCl | Electric heating plate | [34] |
| 0.5 g sludge ash sample from sewage treatment plant | 12 mL HNO3:HCl (3:1) mixture | Microwave Digestion Apparatus | [35] |
| 5 g dried food | 1:1 HNO3, H2O2 | Electric heating plate | [36] |
| 0.5 g food sample | Concentrated HNO3 | Microwave Digestion Apparatus | [37] |
| 1 g soil sample | 4.8 mL HF, 1.2 mL HClO4 | Microwave Digestion Apparatus | [38] |
Table 5.
Main radioactive detection methods of radionuclides in environmental samples.
| Analytical Method | Ray Type | Main Detection Nuclides | Advantage | Disadvantage |
|---|---|---|---|---|
| Alpha spectrometry | α | 238Pu, 239Pu, 210Po, 241Am | Low detection limit and high sensitivity | Complicated process and time-consuming |
| Beta spectrometry | β | 3H, 89Sr, 90Sr, 226Ra, 137Cs | Low background and high detection efficiency | Tedious preprocessing and significant spectrum interference |
| Gamma spectrometry | γ | 55Fe, 60Co, 65Zn, 95Zr, 110Ag, 131I, 134Cs, 137Cs | Simple preprocessing, high detection efficiency, and strong energy resolution | High cost and many influencing factors |
| Liquid scintillation | α, low energy β | 3H, 14C, 89Sr, 90Sr, 239Pu | High detection efficiency, high sensitivity, and high precision | Complicated separation and time-consuming |
Table 6.
Half-life, decay mode, and strongest decay energy of 90Sr, 238Pu, 239Pu, and 240Pu [66].
Table 6.
Half-life, decay mode, and strongest decay energy of 90Sr, 238Pu, 239Pu, and 240Pu [66].
| Nuclides | Half-Life | Main Alpha Particles | Main Gamma Rays | Main Beta Particles | |||
|---|---|---|---|---|---|---|---|
| Energy/keV | Intensity/% | Energy/keV | Intensity/% | Energy/keV | Intensity/% | ||
| 90Sr | 28.9 a* | 545.9 | 100.0 | ||||
| 238Pu | 87.7 a | 5499.03 | 70.91 | 43.498 | 0.0392 | ||
| 239Pu | 24,110 a | 5156.59 | 70.77 | 51.624 | 0.0272 | ||
| 240Pu | 6561 a | 5168.17 | 72.8 | 45.244 | 0.0447 | ||
* ‘a’ represents the unit of years.
Table 7.
Determination of 90Sr activity in soil samples by ICP–DRC–MS and radiometric method [81,82].
| Sample | ICP–DCR–MS | Radiometric Method | ||
|---|---|---|---|---|
| (pg·g−1) | (Bq·g−1) | (Bq·g−1) 1996 | (Bq·g−1) 2007 | |
| soil 1 | 4.66 ± 0.27 | 23.7 ± 1.3 | 45 ± 9 | 35 ± 7 |
| soil 2 | 13.48 ± 0.68 | 68.6 ± 3.5 | 82 ± 16 | 63 ± 13 |
| soil 3 | 12.9 ± 1.5 | 65.6 ± 7.8 | 99 ± 18 | 76 ± 15 |
Table 8.
Instrument detection limits of ICP–MS/HP/Mistral and alpha spectroscopy [90].
Table 8.
Instrument detection limits of ICP–MS/HP/Mistral and alpha spectroscopy [90].
| Isotope | ICP–MS/HP/Mistral | Alpha Spectrometry | |
|---|---|---|---|
| Mass/g | Activity/Bq | Activity/Bq | |
| 239Pu | 1.2 × 10−15 | 2.8 × 10−6 | 1 × 10−4 |
| 240Pu | 1.2 × 10−15 | 1.0 × 10−5 | 1 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhong, N.; Li, L.; Yang, X.; Zhao, Y. Analytical Methods for the Determination of 90Sr and 239,240Pu in Environmental Samples. Molecules 2022, 27, 1912. https://doi.org/10.3390/molecules27061912
AMA Style
Zhong N, Li L, Yang X, Zhao Y. Analytical Methods for the Determination of 90Sr and 239,240Pu in Environmental Samples. Molecules. 2022; 27(6):1912. https://doi.org/10.3390/molecules27061912
Chicago/Turabian StyleZhong, Ningjie, Lili Li, Xiaofan Yang, and Yonggang Zhao. 2022. "Analytical Methods for the Determination of 90Sr and 239,240Pu in Environmental Samples" Molecules 27, no. 6: 1912. https://doi.org/10.3390/molecules27061912