
Design of SARS-CoV-2 Main Protease Inhibitors Using Artificial Intelligence and Molecular Dynamic Simulations

 , , , , and
, , , , and
Abstract
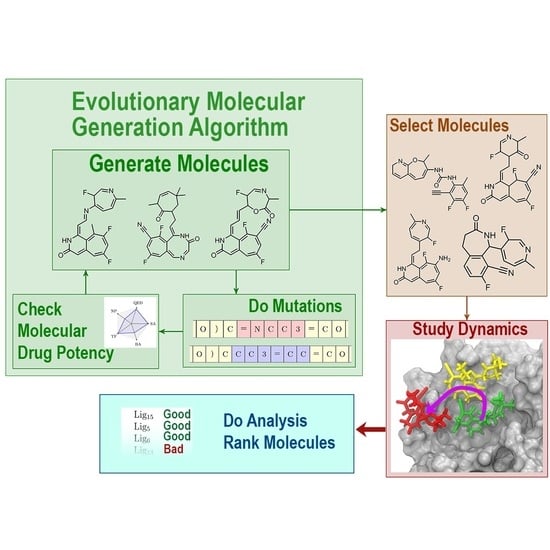
:
1. Introduction
2. Methods
2.1. Molecule Design Metrics
2.1.1. Binding Affinity (BA)
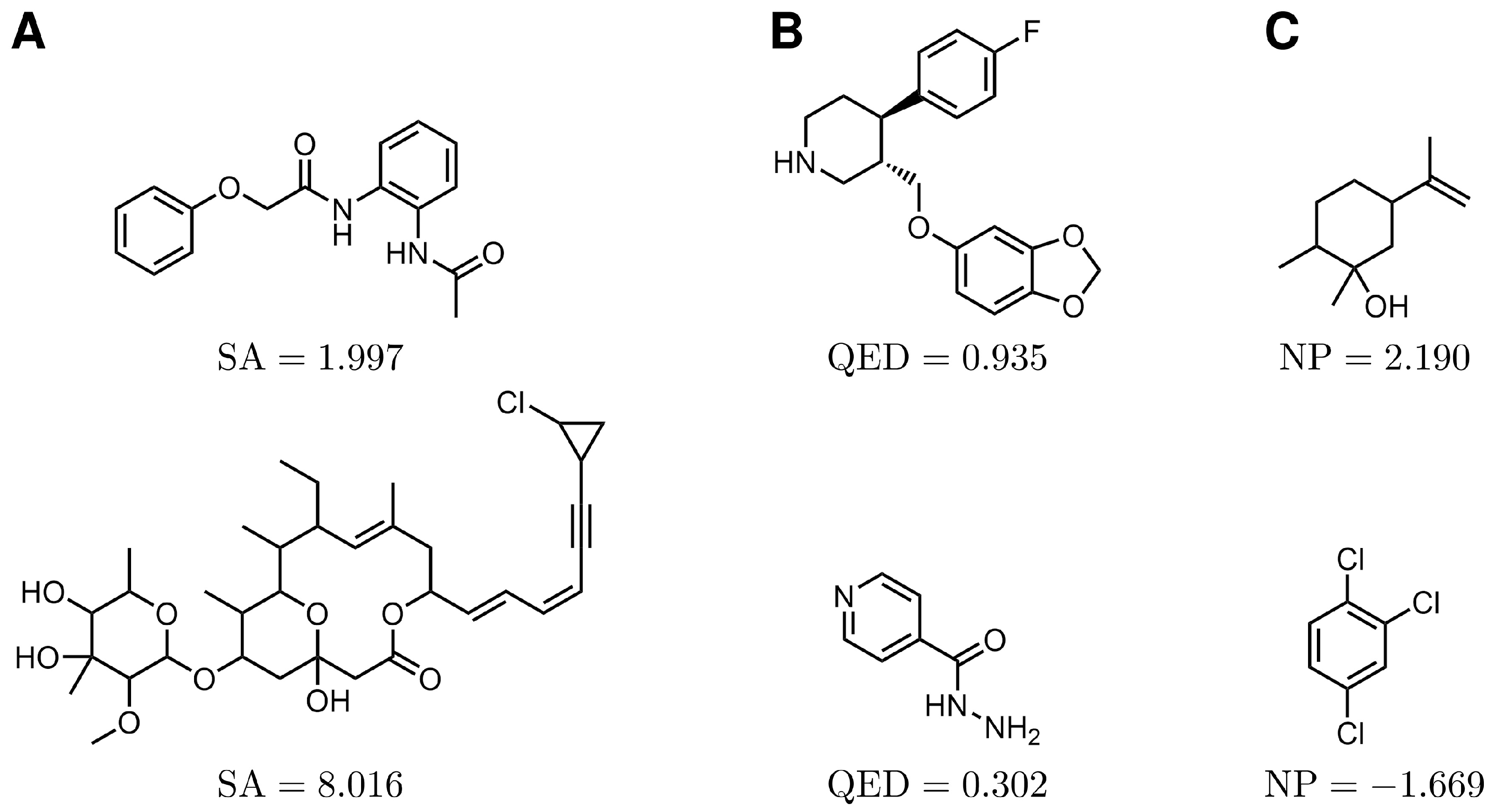
2.1.2. Synthetic Accessibility (SA)
2.1.3. Quantitative Estimate of Drug-Likeness (QED)
2.1.4. Natural Product-Likeness (NP)
2.1.5. Toxicity Filter (TF)
2.2. Fitness Evaluation
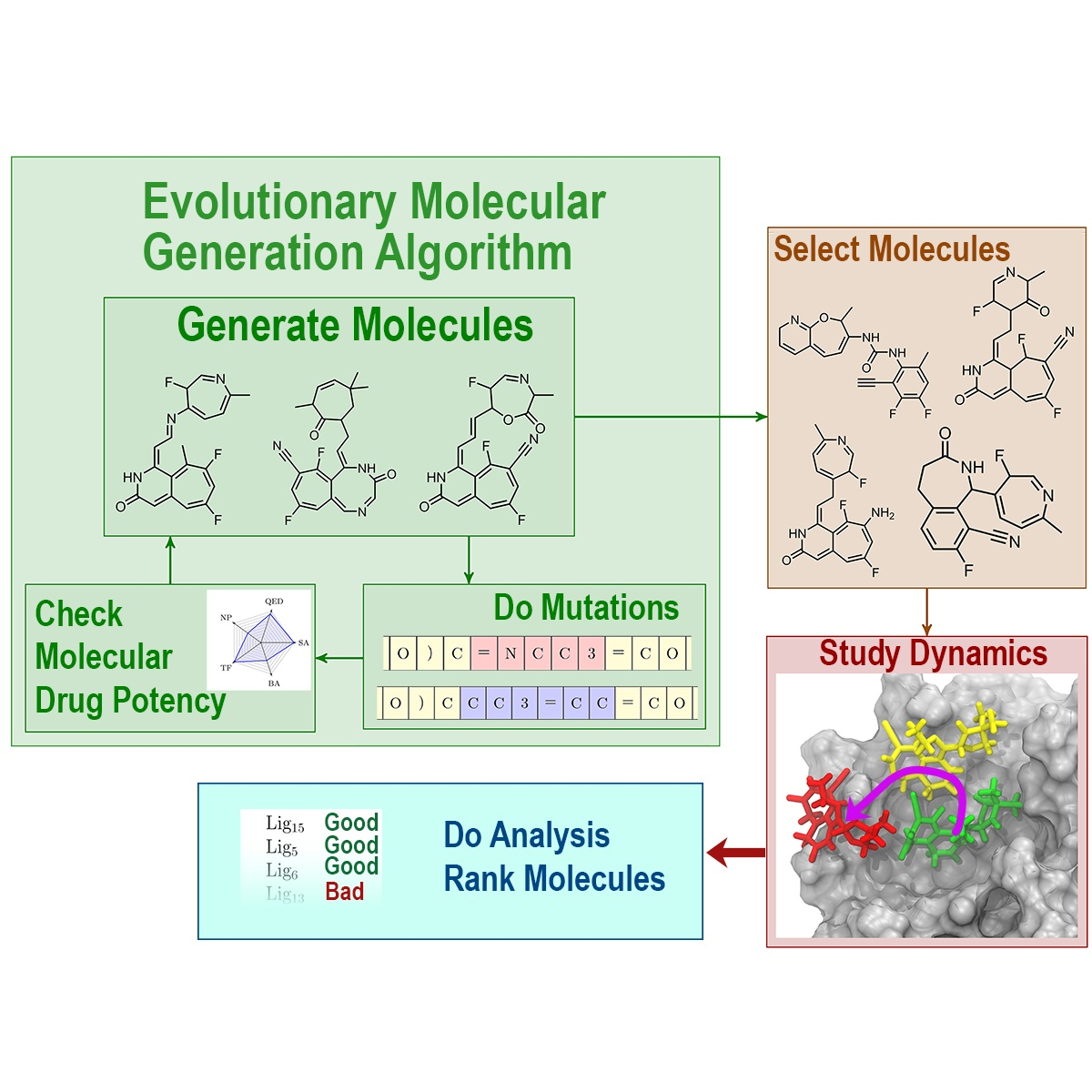
2.3. Evolutionary Molecular Generation Algorithm
2.3.1. SMILES Representation
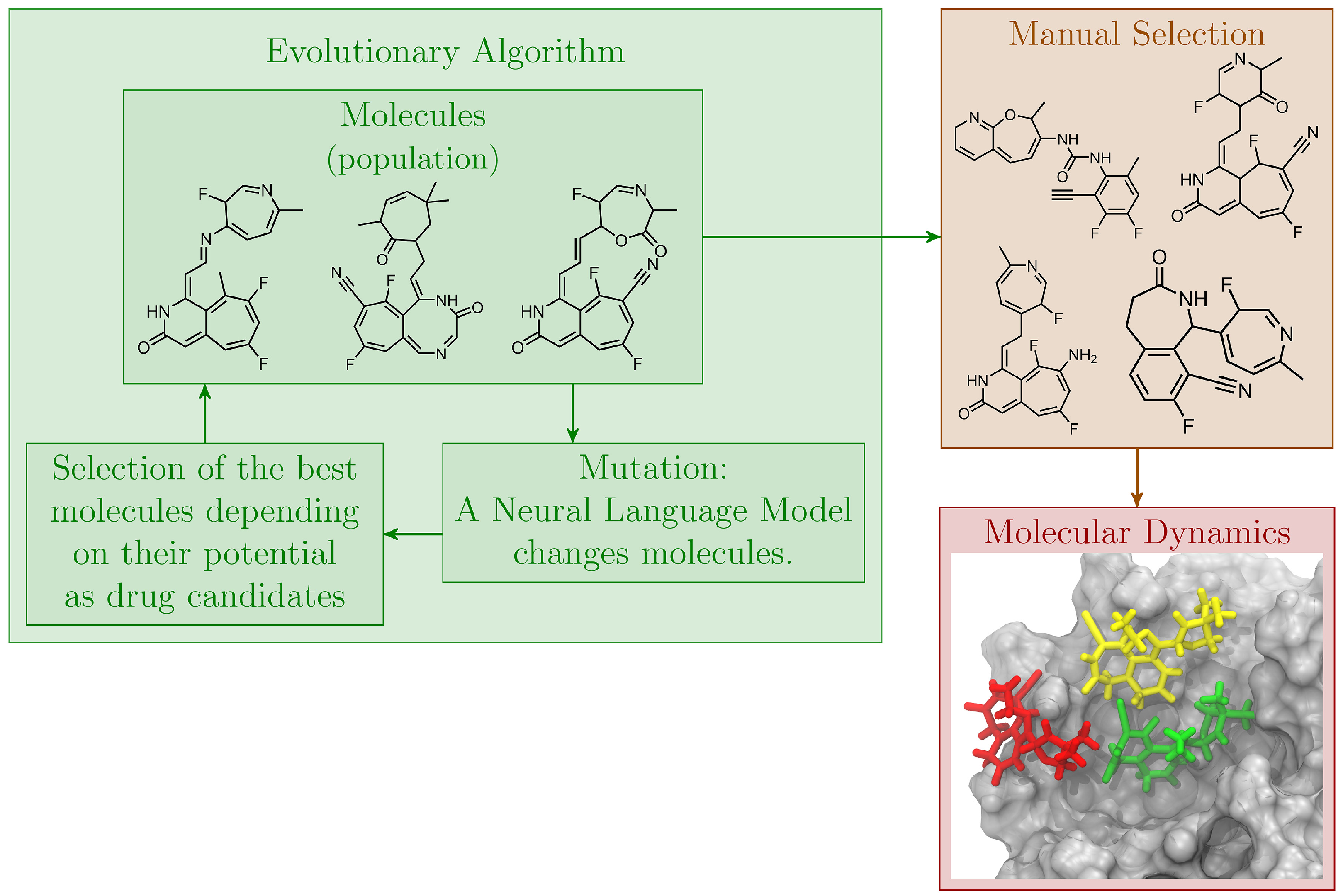
2.3.2. Evolutionary Algorithm
2.3.3. Neural Language Model
2.3.4. Evolutionary Algorithm with Language Model
2.4. Molecular Dynamics
3. Results and Discussion
3.1. Evolutionary Design of Inhibitors
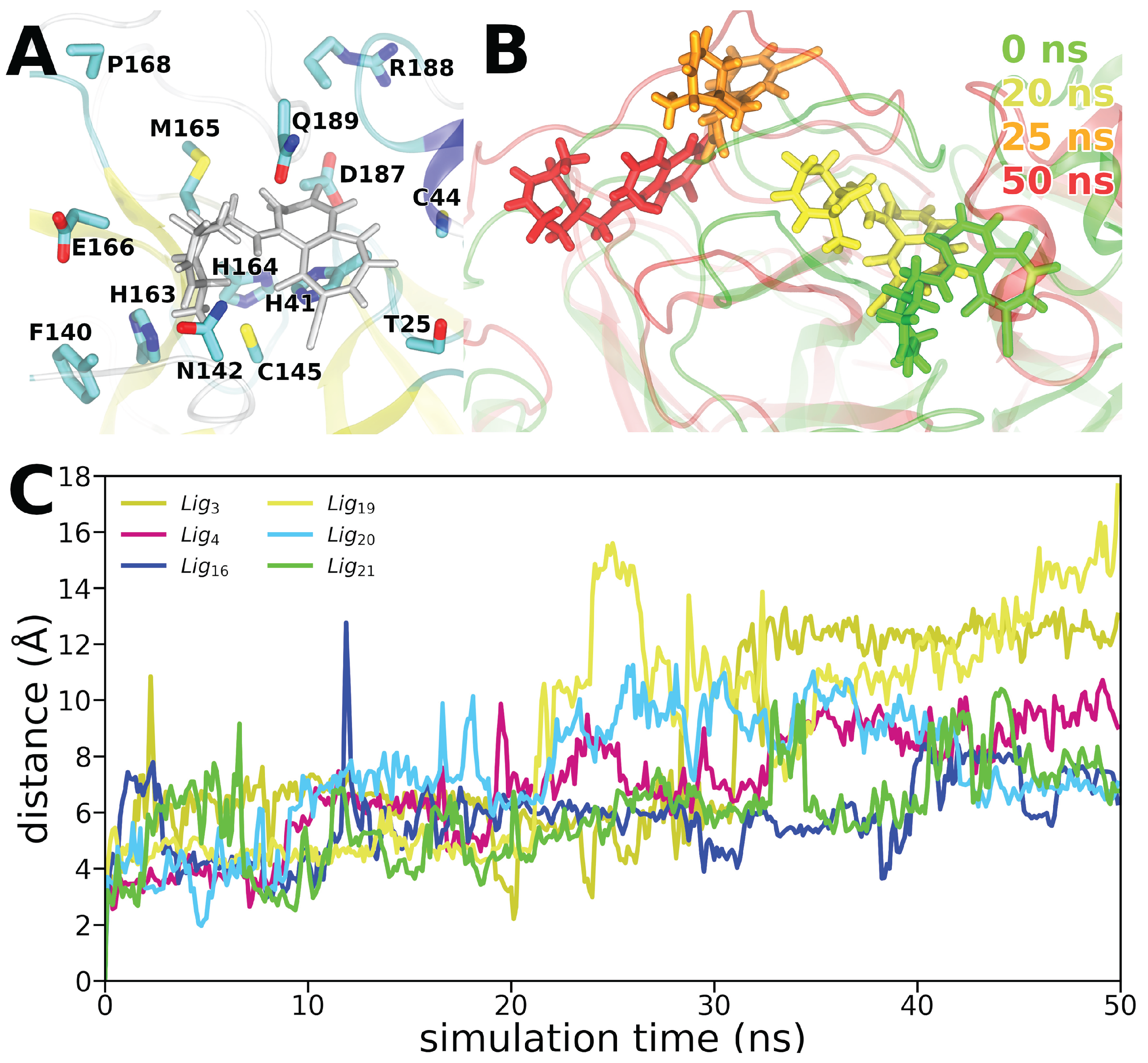
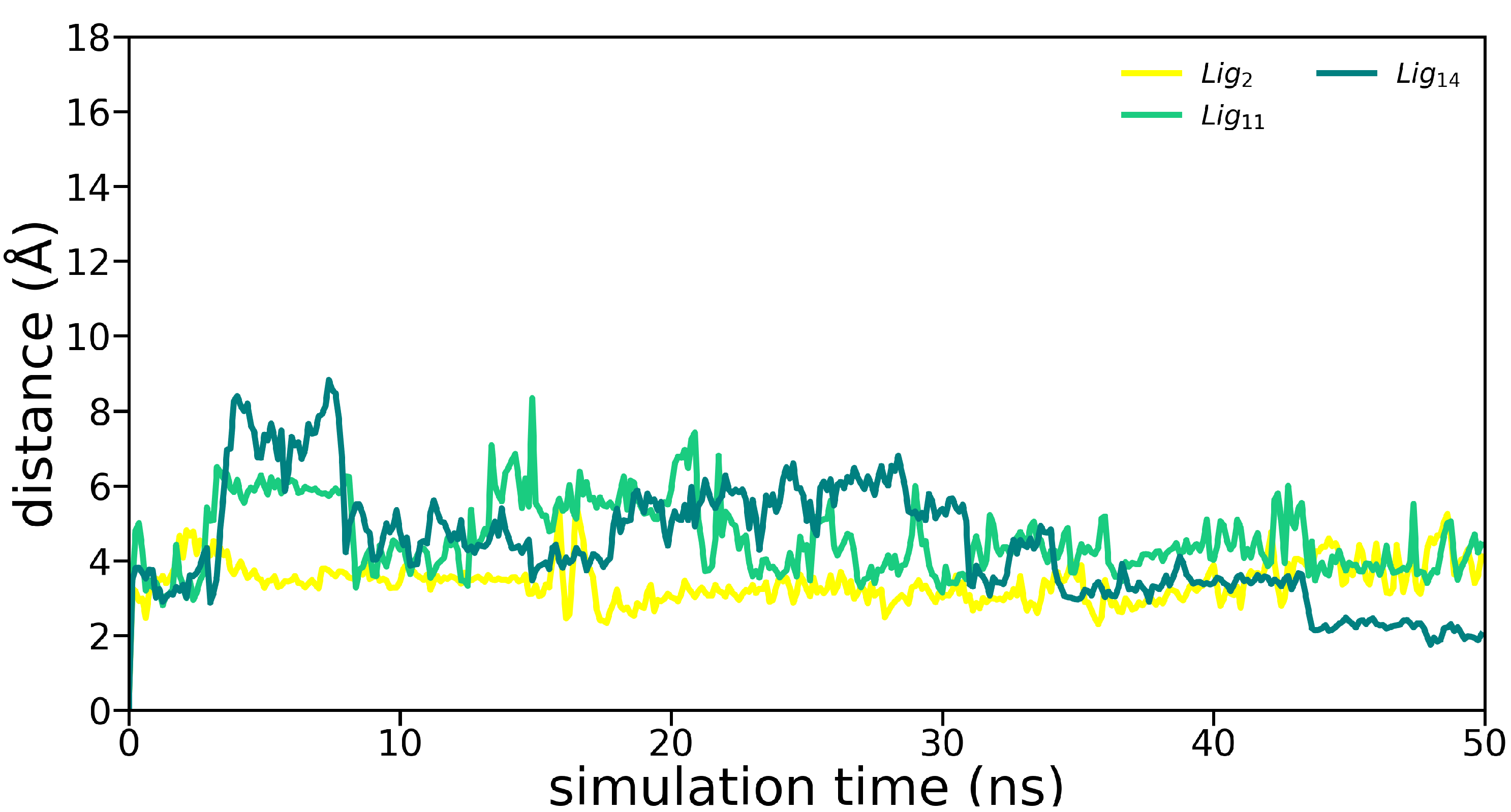
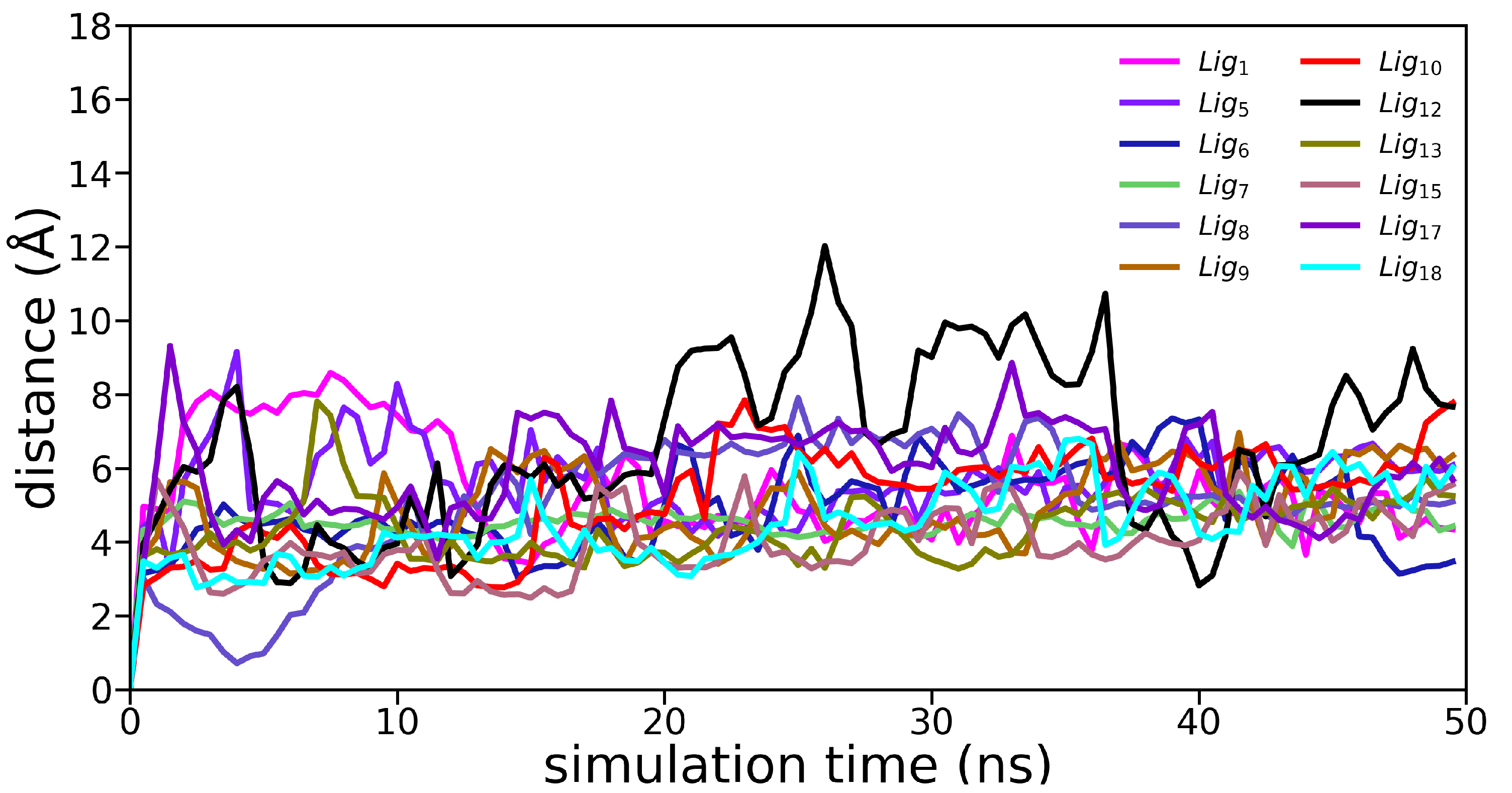
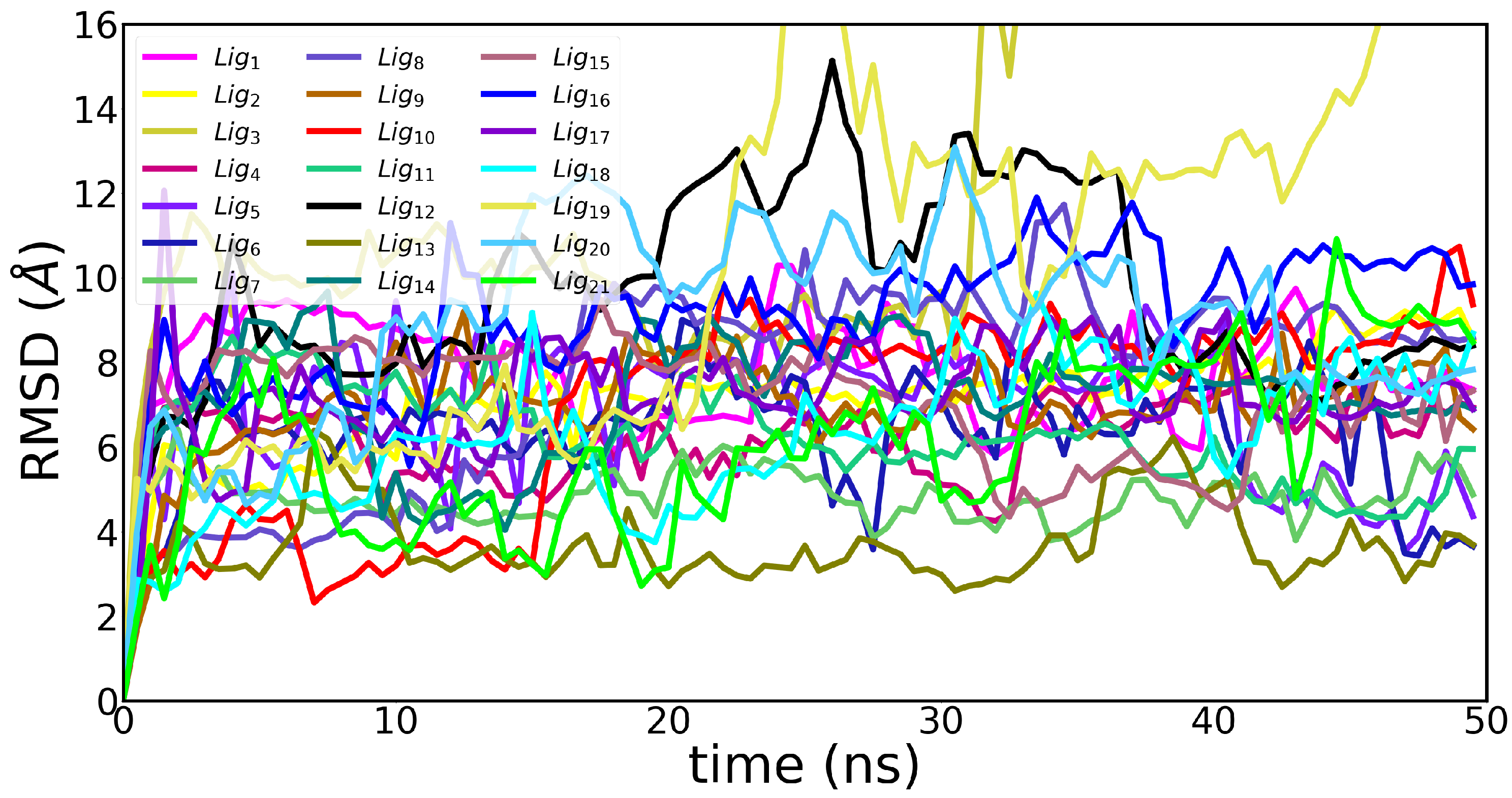
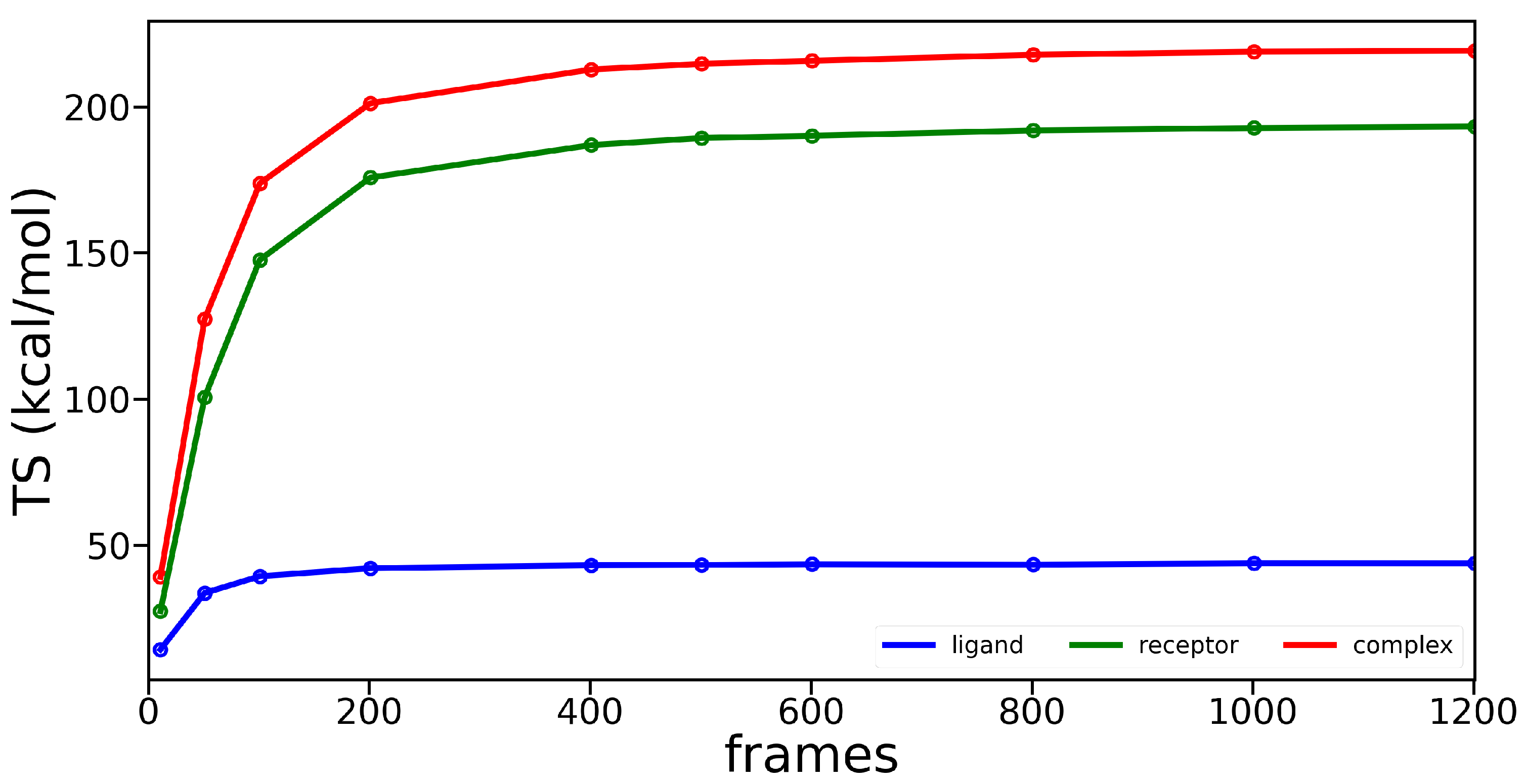
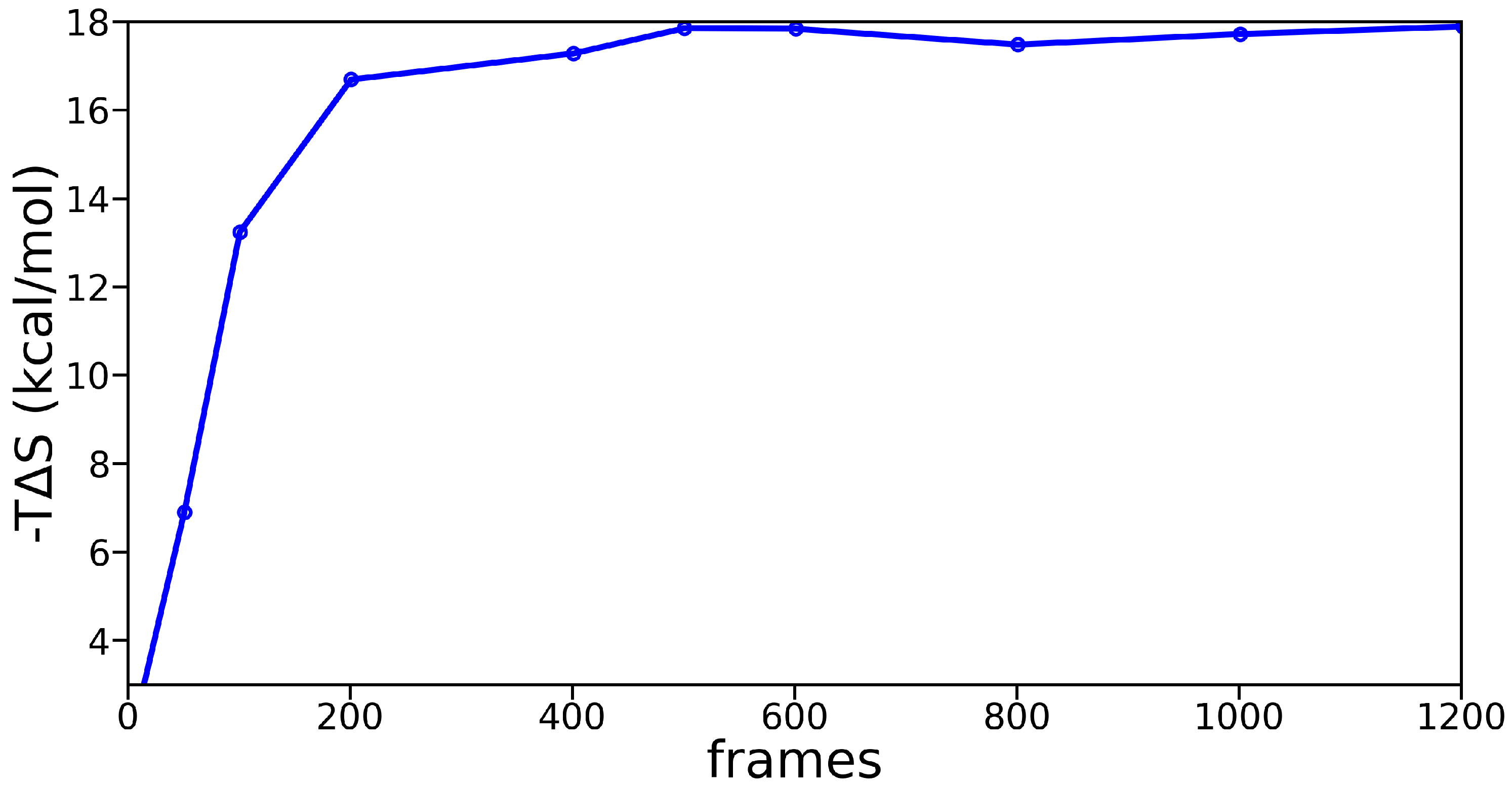
3.2. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3CLpro | 3C-like protease |
| AI | artificial intelligence |
| BA | binding affinity |
| CADD | computer-aided drug design |
| EA | evolutionary algorithm |
| EMGA | evolutionary molecular generation algorithm |
| LSTM | long short-term memory |
| MOSES | molecular sets (benchmarking plattform) |
| MD | molecular dynamics |
| Mpro | main protease |
| NP | natural product-likeness |
| QED | quantitative estimate of drug-likeness |
| RMSD | root mean square displacement |
| SA | synthetic accessibility |
| SMILES | simplified molecular input line entry system |
| TF | toxicity filter |
| vdW | van der Waals |
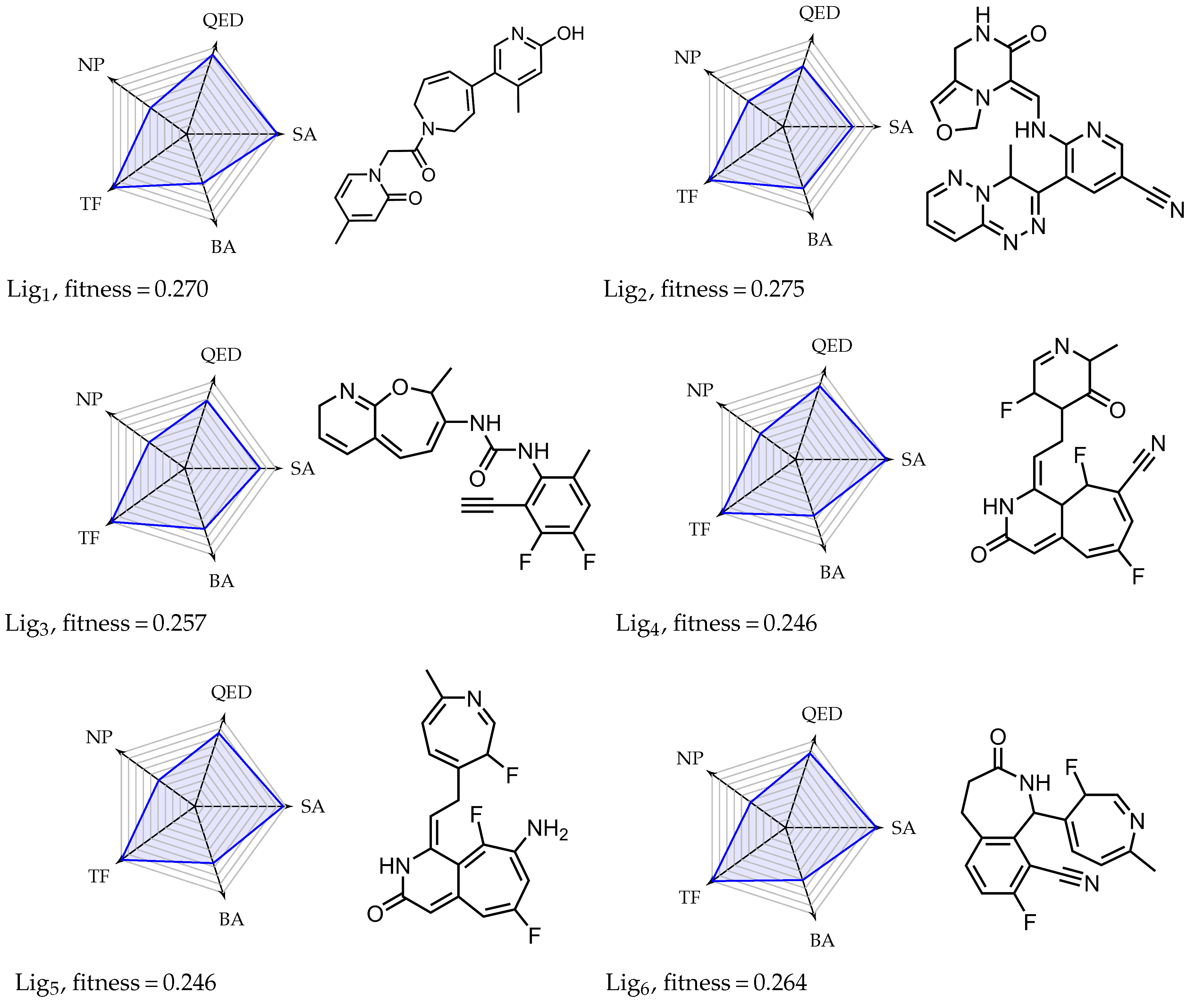
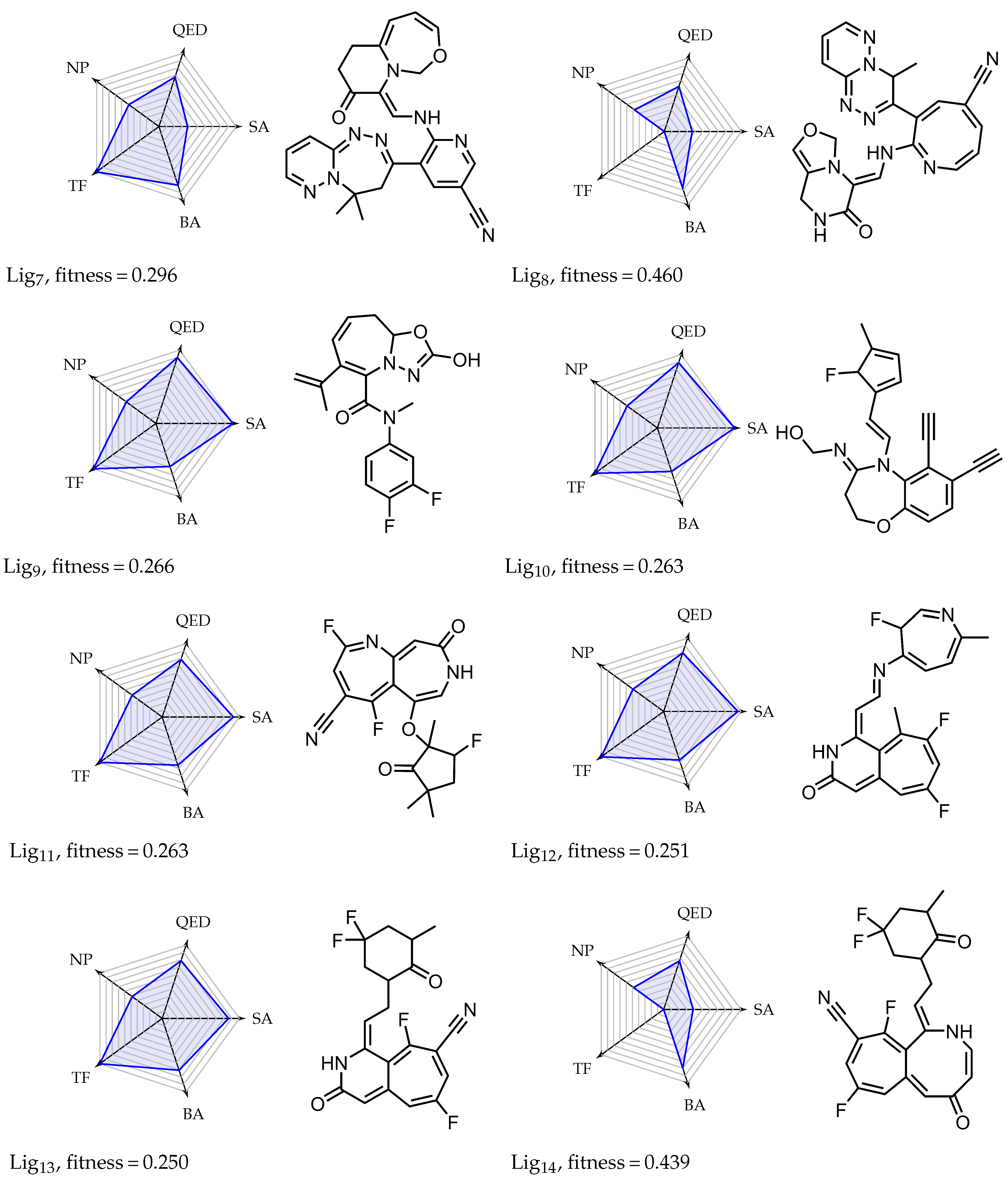
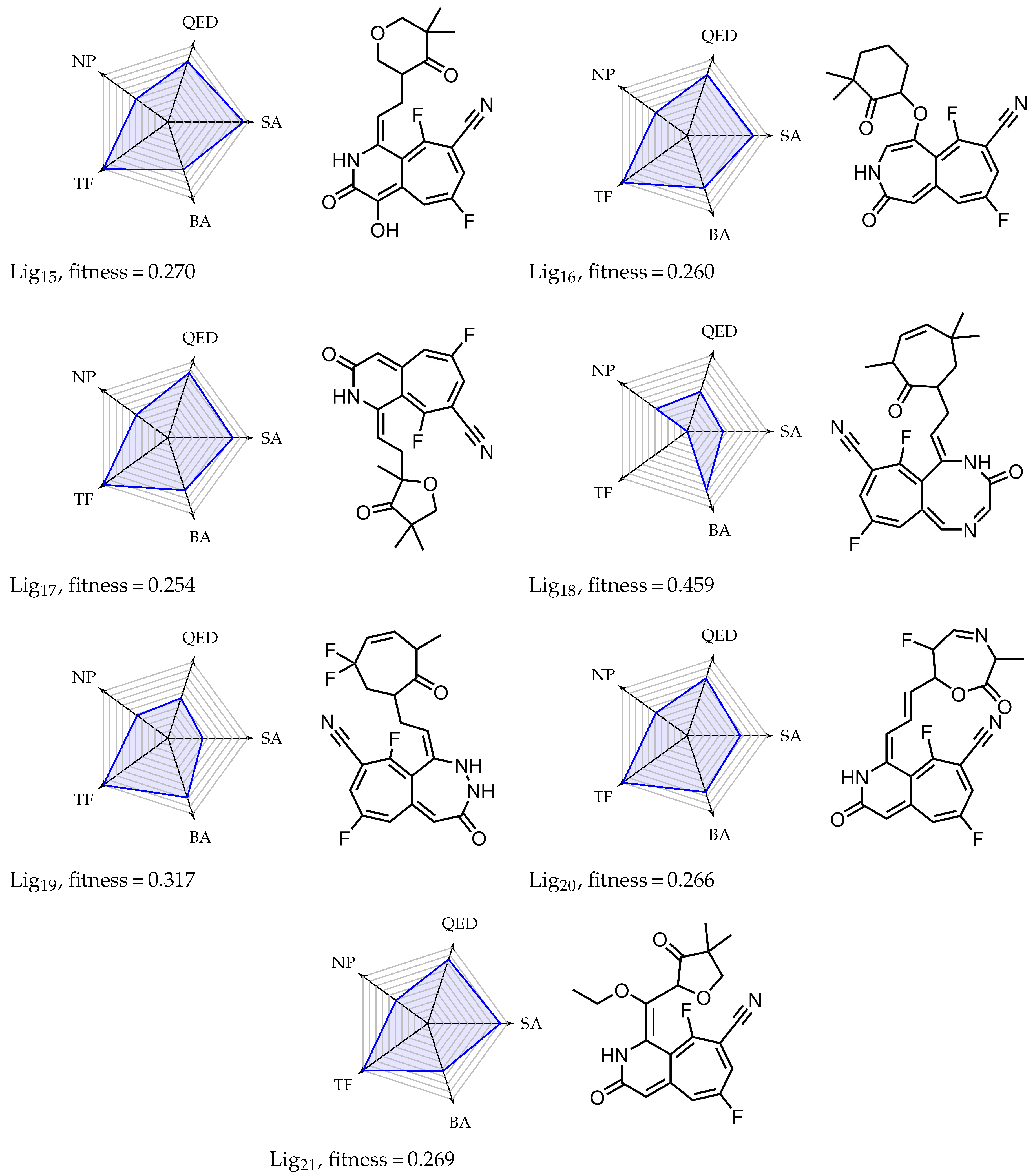
Appendix A. Overview of Simulated Ligands




{kind=link}
{kind=link}
{kind=link}
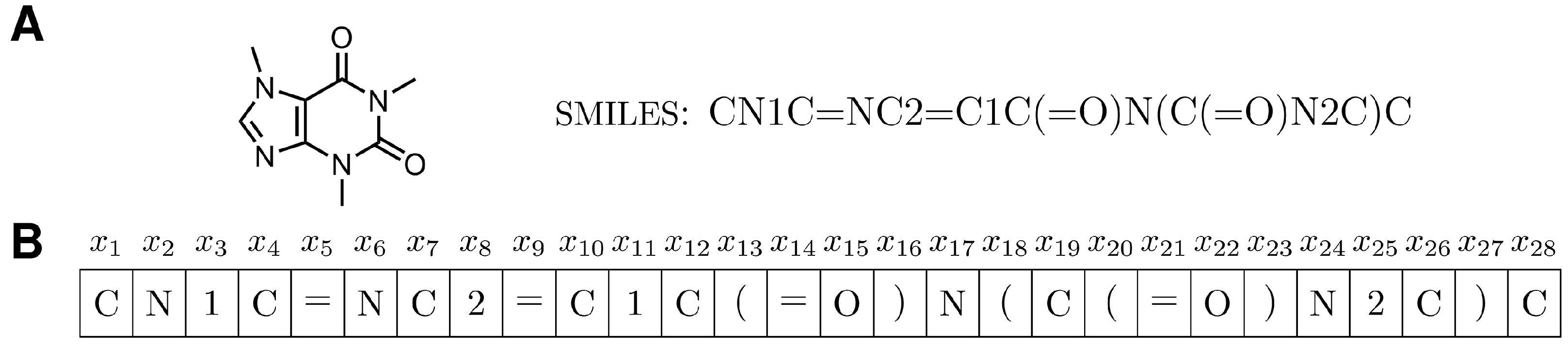{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | SMILES |
|---|---|
| Lig1 | Cc1ccn(CC(=O)N2CC=CC(c3cnc(O)cc3C)=CC2)c(=O)c1 |
| Lig2 | CC1C(c2cc(C#N)cnc2NC=C2C(=O)NCC3=COCN32)=NN=C2C=CC=NN21 |
| Lig3 | C#Cc1c(F)c(F)cc(C)c1NC(=O)NC1=CC=C2C=CCN=C2OC1C |
| Lig4 | CC1N=CC(F)C(CC=c2[nH]c(=O)cc3c2=C(F)C(C#N)=CC(F)=C3)C1=O |
| Lig5 | CC1=CC=C(CC=c2[nH]c(=O)cc3c2=C(F)C(N)=CC(F)=C3)C(F)C=N1 |
| Lig6 | CC1=CC=C(C2NC(=O)CCc3ccc(F)c(C#N)c32)C(F)C=N1 |
| Lig7 | CC1(C)CC(c2cc(C#N)cnc2NC=C2C(=O)CCC3=CC=COCN32)=NN=C2C=CC=NN21 |
| Lig8 | CC1C(C2=CC(C#N)=CC=CN=C2NC=C2C(=O)NCC3=COCN32)=NN=C2C=CC=NN21 |
| Lig9 | C=C(C)C1=C(C(=O)N(C)c2ccc(F)c(F)c2)N2N=C(O)OC2CC=C1 |
| Lig10 | CC1=CC=C(C=CN2C(=NCO)CCOc3ccc(C#N)c(C#N)c32)C1F |
| Lig11 | CC1(C)CC(F)C(C)(OC2=CNC(=O)C=C3N=C(F)C=C(C#N)C(F)=C32)C1=O |
| Lig12 | CC1=CC=C(N=CC=c2[nH]c(=O)cc3c2=C(C)C(F)=CC(F)=C3)C(F)C=N1 |
| Lig13 | CC1CC(F)(F)CC(CC=c2[nH]c(=O)cc3c2=C(F)C(C#N)=CC(F)=C3)C1=O |
| Lig14 | CC1CC(F)(F)CC(CC=C2NC=CC(=O)C=C3C=C(F)C=C(C#N)C(F)=C32)C1=O |
| Lig15 | CC1(C)COCC(CC=c2[nH]c(=O)c(O)c3c2=C(F)C(C#N)=CC(F)=C3)C1=O |
| Lig16 | CC1(C)CCCC(OC2=CNC(=O)C=C3C=C(F)C=C(C#N)C(F)=C32)C1=O |
| Lig17 | CC1(C)COC(C)(CC=c2[nH]c(=O)cc3c2=C(F)C(C#N)=CC(F)=C3)C1=O |
| Lig18 | CC1C=CC(F)(F)CC(CC=C2NC(=O)C=NC=C3C=C(F)C=C(C#N)C(F)=C32)C1=O |
| Lig19 | CC1C=CC(F)(F)CC(CC=C2NNC(=O)C=C3C=C(F)C=C(C#N)C(F)=C32)C1=O |
| Lig20 | CC1N=CC(F)C(C=CC=c2[nH]c(=O)cc3c2=C(F)C(C#N)=CC(F)=C3)OC1=O |
| Lig21 | CCOC(=c1[nH]c(=O)cc2c1=C(F)C(C#N)=CC(F)=C2)C1OCC(C)(C)C1=O |
| Ligand | BA | SA | QED | NP | TF |
|---|---|---|---|---|---|
| Lig1 | 1.019 | 0.918 | 1 | ||
| Lig2 | 3.119 | 0.699 | 1 | ||
| Lig3 | 2.543 | 0.783 | 1 | ||
| Lig4 | 1.000 | 0.850 | 1 | ||
| Lig5 | 1.254 | 0.848 | 1 | ||
| Lig6 | 1.000 | 0.861 | 1 | ||
| Lig7 | 6.664 | 0.676 | 1 | ||
| Lig8 | 6.745 | 0.615 | 0 | ||
| Lig9 | 1.000 | 0.898 | 1 | ||
| Lig10 | 1.000 | 0.890 | 1 | ||
| Lig11 | 1.695 | 0.783 | 1 | ||
| Lig12 | 1.342 | 0.799 | 1 | ||
| Lig13 | 2.252 | 0.787 | 1 | ||
| Lig14 | 6.567 | 0.662 | 0 | ||
| Lig15 | 1.477 | 0.796 | 1 | ||
| Lig16 | 2.586 | 0.803 | 1 | ||
| Lig17 | 2.692 | 0.858 | 1 | ||
| Lig18 | 5.985 | 0.523 | 0 | ||
| Lig19 | 6.102 | 0.527 | 1 | ||
| Lig20 | 4.036 | 0.756 | 1 | ||
| Lig21 | 1.813 | 0.844 | 1 |
Appendix B. Molecular Dynamics Analysis





| Ligand | avg. COM (Å) | avg. RMSD (Å) | avg. RMSF (Å) |
|---|---|---|---|
| Lig1 | 4.82 | 7.66 | 1.37 |
| Lig2 | 3.83 | 7.80 | 1.05 |
| Lig3 | 12.43 | 14.05 | 0.99 |
| Lig4 | 9.08 | 6.40 | 1.03 |
| Lig5 | 6.15 | 6.90 | 1.15 |
| Lig6 | 4.70 | 6.46 | 0.64 |
| Lig7 | 4.75 | 4.85 | 0.51 |
| Lig8 | 5.00 | 9.13 | 1.07 |
| Lig9 | 5.87 | 7.17 | 0.39 |
| Lig10 | 6.20 | 8.59 | 0.63 |
| Lig11 | 4.23 | 5.76 | 1.32 |
| Lig12 | 6.82 | 10.44 | 1.34 |
| Lig13 | 5.04 | 3.68 | 0.85 |
| Lig14 | 2.62 | 7.67 | 1.18 |
| Lig15 | 4.85 | 6.57 | 1.10 |
| Lig16 | 7.33 | 9.95 | 0.83 |
| Lig17 | 5.24 | 7.56 | 0.45 |
| Lig18 | 5.55 | 7.19 | 1.11 |
| Lig19 | 13.28 | 13.42 | 1.15 |
| Lig20 | 7.27 | 9.50 | 1.54 |
| Lig21 | 7.93 | 7.15 | 0.71 |

References
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A.V. Advances in Docking. Curr. Med. Chem. 2019, 26, 7555–7580. [Google Scholar] [CrossRef]
- Davis, R.L. Mechanism of Action and Target Identification: A Matter of Timing in Drug Discovery. iScience 2020, 23, 101487. [Google Scholar] [CrossRef]
- Poduri, R.; Jagadeesh, G. The Concept of Receptor and Molecule Interaction in Drug Discovery and Development. In Drug Discovery and Development: From Targets and Molecules to Medicines; Poduri, R., Ed.; Springer: Singapore, 2021; pp. 67–102. [Google Scholar] [CrossRef]
- Bharatam, P.V. Computer-Aided Drug Design. In Drug Discovery and Development: From Targets and Molecules to Medicines; Poduri, R., Ed.; Springer: Singapore, 2021; pp. 137–210. [Google Scholar] [CrossRef]
- Schneider, G. Automating Drug Discovery. Nat. Rev. Drug Discov. 2018, 17, 97–113. [Google Scholar] [CrossRef]
- Reymond, J.L.; van Deursen, R.; Blum, L.C.; Ruddigkeit, L. Chemical Space as a Source for New Drugs. Med. Chem. Commun. 2010, 1, 30–38. [Google Scholar] [CrossRef]
- Devi, R.V.; Sathya, S.S.; Coumar, M.S. Evolutionary Algorithms for de Novo Drug Design—A Survey. Appl. Soft Comput. 2015, 27, 543–552. [Google Scholar] [CrossRef]
- Brown, N.; Fiscato, M.; Segler, M.H.; Vaucher, A.C. GuacaMol: Benchmarking Models for de Novo Molecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef]
- Douguet, D.; Thoreau, E.; Grassy, G. A Genetic Algorithm for the Automated Generation of Small Organic Molecules: Drug Design Using an Evolutionary Algorithm. J. Comput. Aided Mol. Des. 2000, 14, 449–466. [Google Scholar] [CrossRef]
- Nigam, A.; Friederich, P.; Krenn, M.; Aspuru-Guzik, A. Augmenting Genetic Algorithms with Deep Neural Networks for Exploring the Chemical Space. In Proceedings of the International Conference on Learning Representations, New Orleans, LA, USA, 6–9 May 2019. [Google Scholar]
- Pegg, S.C.H.; Haresco, J.J.; Kuntz, I.D. A Genetic Algorithm for Structure-Based de Novo Design. J. Comput. Aided Mol. Des. 2001, 15, 911–933. [Google Scholar] [CrossRef]
- Yuan, Y.; Pei, J.; Lai, L. LigBuilder V3: A Multi-Target de Novo Drug Design Approach. Front. Chem. 2020, 8, 142. [Google Scholar] [CrossRef] [Green Version]
- Cofala, T.; Elend, L.; Mirbach, P.; Prellberg, J.; Teusch, T.; Kramer, O. Evolutionary Multi-objective Design of SARS-CoV-2 Protease Inhibitor Candidates. In Parallel Problem Solving from Nature – PPSN XVI; Lecture Notes in Computer Science; Bäck, T., Preuss, M., Deutz, A., Wang, H., Doerr, C., Emmerich, M., Trautmann, H., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 357–371. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome–Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from COVID-19 Virus and Discovery of Its Inhibitors. Nature 2020, 582, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, P.K.; Arul, M.N.; Patel, P.; Verma, S.K.; Luo, W.; Rubahn, H.G.; Mishra, Y.K.; Suar, M.; Ahuja, R. Structure-Based Drug Designing and Immunoinformatics Approach for SARS-CoV-2. Sci. Adv. 2020, 6, eabb8097. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Liu, Q.; Wan, J.; Wang, G. A Survey on Computational Methods in Discovering Protein Inhibitors of SARS-CoV-2. Briefings Bioinform. 2022, 23, bbab416. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.K.; Singh, R.; Das, P.; Purohit, R. Evaluation of Acridinedione Analogs as Potential SARS-CoV-2 Main Protease Inhibitors and Their Comparison with Repurposed Anti-Viral Drugs. Comput. Biol. Med. 2021, 128, 104117. [Google Scholar] [CrossRef]
- Sharma, J.; Kumar Bhardwaj, V.; Singh, R.; Rajendran, V.; Purohit, R.; Kumar, S. An In-Silico Evaluation of Different Bioactive Molecules of Tea for Their Inhibition Potency against Non Structural Protein-15 of SARS-CoV-2. Food Chem. 2021, 346, 128933. [Google Scholar] [CrossRef]
- Singh, R.; Bhardwaj, V.K.; Das, P.; Purohit, R. A Computational Approach for Rational Discovery of Inhibitors for Non-Structural Protein 1 of SARS-CoV-2. Comput. Biol. Med. 2021, 135, 104555. [Google Scholar] [CrossRef]
- Arshia, A.H.; Shadravan, S.; Solhjoo, A.; Sakhteman, A.; Sami, A. De Novo Design of Novel Protease Inhibitor Candidates in the Treatment of SARS-CoV-2 Using Deep Learning, Docking, and Molecular Dynamic Simulations. Comput. Biol. Med. 2021, 139, 104967. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Strodel, B.; Olubiyi, O.; Olagunju, M.; Keutmann, M.; Loschwitz, J. High Throughput Virtual Screening to Discover Inhibitors of the Main Protease of the Coronavirus SARS-CoV-2. Molecules 2020, 25, 3193. [Google Scholar] [CrossRef]
- Alhossary, A.; Handoko, S.D.; Mu, Y.; Kwoh, C.K. Fast, Accurate, and Reliable Molecular Docking with QuickVina 2. Bioinformatics 2015, 31, 2214–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model. 2018, 58, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the Chemical Beauty of Drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural Product-likeness Score and Its Application for Prioritization of Compound Libraries. J. Chem. Inf. Model. 2008, 48, 68–74. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Front. Pharmacol. 2020, 11, 1931. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimek, M.; Perelstein, M. Neural Network-Based Approach to Phase Space Integration. SciPost Phys. 2020, 9, 053. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Lameijer, E.W.; Kok, J.N.; Bäck, T.; IJzerman, A.P. The Molecule Evoluator. An Interactive Evolutionary Algorithm for the Design of Drug-Like Molecules. J. Chem. Inf. Model. 2006, 46, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.; McKay, B.; Gilardoni, F.; Gasteiger, J. A Graph-Based Genetic Algorithm and Its Application to the Multiobjective Evolution of Median Molecules. J. Chem. Inf. Comput. Sci. 2004, 44, 1079–1087. [Google Scholar] [CrossRef]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Beyer, H.G.; Schwefel, H.P. Evolution Strategies—A Comprehensive Introduction. Nat. Comput. 2002, 1, 3–52. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Dai, Z.; Yang, Y.; Carbonell, J.; Salakhutdinov, R.R.; Le, Q.V. XLNet: Generalized Autoregressive Pretraining for Language Understanding. In Advances in Neural Information Processing Systems; Curran Associates, Inc.: Red Hook, NY, USA, 2019; Volume 32. [Google Scholar]
- Landrum, G. RDKit: Open-Source Cheminformatics Software. 2016. Available online: https://github.com/rdkit/rdkit/releases/tag/Release_2016_09_4 (accessed on 19 June 2022).
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Brieg, M.; Setzler, J.; Albert, S.; Wenzel, W. Generalized Born Implicit Solvent Models for Small Molecule Hydration Free Energies. Phys. Chem. Chem. Phys. 2017, 19, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB(GB)SA studies on the protein–protein complex Ras–Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Srinivasan, J.; Trevathan, M.W.; Beroza, P.; Case, D.A. Application of a Pairwise Generalized Born Model to Proteins and Nucleic Acids: Inclusion of Salt Effects. Theor. Chem. Acc. 1999, 101, 426–434. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Case, D.A. Generalized Born Implicit Solvent Models for Biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef]
- Bernardi, R.; Bhandarkar, M.; Bhatele, A.; Bohm, E.; Brunner, R.; Buch, R.; Buelens, F.; Chen, H.; Chipot, C.; Dalke, A.; et al. NAMD 2.14 User’s Guide. Available online: https://www.ks.uiuc.edu/Research/namd/2.14/ug/ (accessed on 19 June 2022).
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring Protein Native States and Large-Scale Conformational Changes with a Modified Generalized Born Model. Proteins: Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Schlitter, J. Estimation of Absolute and Relative Entropies of Macromolecules Using the Covariance Matrix. Chem. Phys. Lett. 1993, 215, 617–621. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Aktulga, H.; Belfon, K.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T.; Cisneros, G.; Cruzeiro, V.; Darden, T.; et al. AmberTools21. Available online: https://ambermd.org/AmberTools.php (accessed on 19 June 2022).
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Brünger, A.T. X-PLOR: Version 3.1: A System for X-ray Crystallography and NMR; Yale University Press: New Haven, CT, USA, 1992. [Google Scholar]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef]
- Sharma, M.; Prasher, P.; Mehta, M.; Zacconi, F.C.; Singh, Y.; Kapoor, D.N.; Dureja, H.; Pardhi, D.M.; Tambuwala, M.M.; Gupta, G.; et al. Probing 3CL Protease: Rationally Designed Chemical Moieties for COVID-19. Drug Dev. Res. 2020, 81, 911–918. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Liu, F.; Li, C.; Li, Y.; et al. Structure-Based Design of Antiviral Drug Candidates Targeting the SARS-CoV-2 Main Protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]








| BA [kcal/mol] | SA | QED | NP | TF | |
|---|---|---|---|---|---|
| score range | |||||
| optimum | 1 | 1 | 5 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elend, L.; Jacobsen, L.; Cofala, T.; Prellberg, J.; Teusch, T.; Kramer, O.; Solov’yov, I.A. Design of SARS-CoV-2 Main Protease Inhibitors Using Artificial Intelligence and Molecular Dynamic Simulations. Molecules 2022, 27, 4020. https://doi.org/10.3390/molecules27134020
Elend L, Jacobsen L, Cofala T, Prellberg J, Teusch T, Kramer O, Solov’yov IA. Design of SARS-CoV-2 Main Protease Inhibitors Using Artificial Intelligence and Molecular Dynamic Simulations. Molecules. 2022; 27(13):4020. https://doi.org/10.3390/molecules27134020
Chicago/Turabian StyleElend, Lars, Luise Jacobsen, Tim Cofala, Jonas Prellberg, Thomas Teusch, Oliver Kramer, and Ilia A. Solov’yov. 2022. "Design of SARS-CoV-2 Main Protease Inhibitors Using Artificial Intelligence and Molecular Dynamic Simulations" Molecules 27, no. 13: 4020. https://doi.org/10.3390/molecules27134020