
New Anticancer Theobromine Derivative Targeting EGFRWT and EGFRT790M: Design, Semi-Synthesis, In Silico, and In Vitro Anticancer Studies
 , ,
, ,  , , ,
, , ,  and
and 
Abstract
:1. Introduction
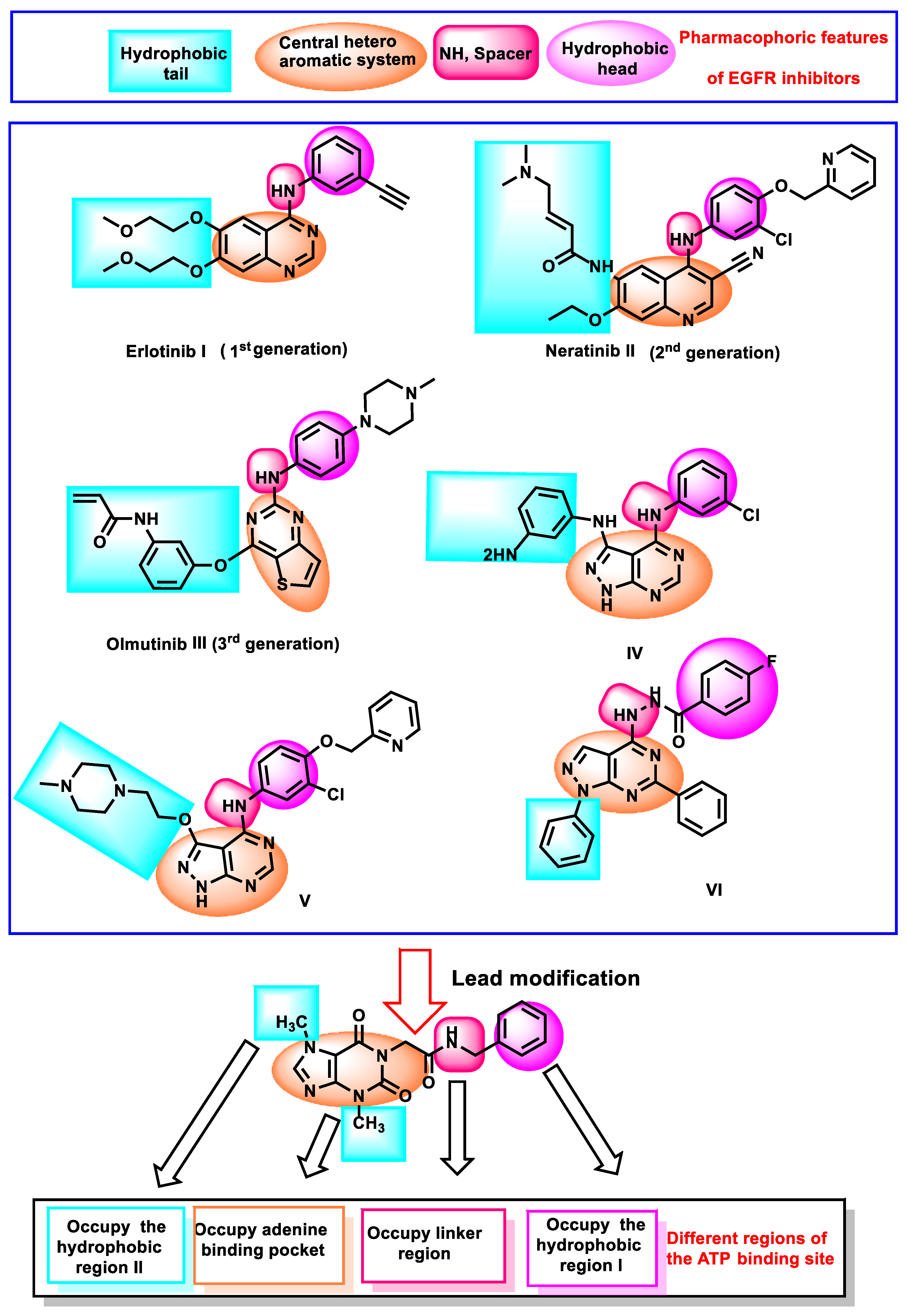
Rationale
2. Results and Discussion
2.1. In Silico Studies
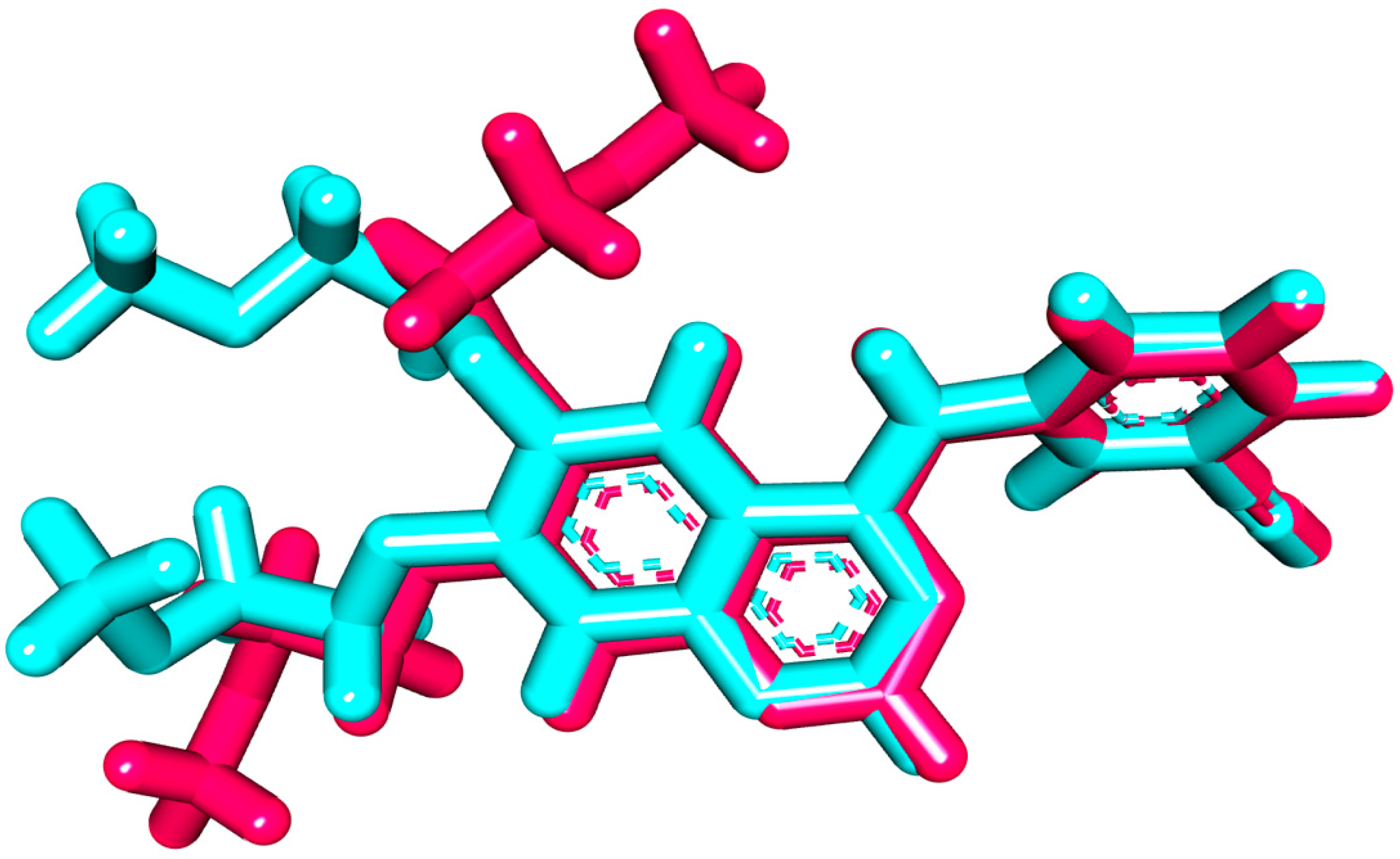
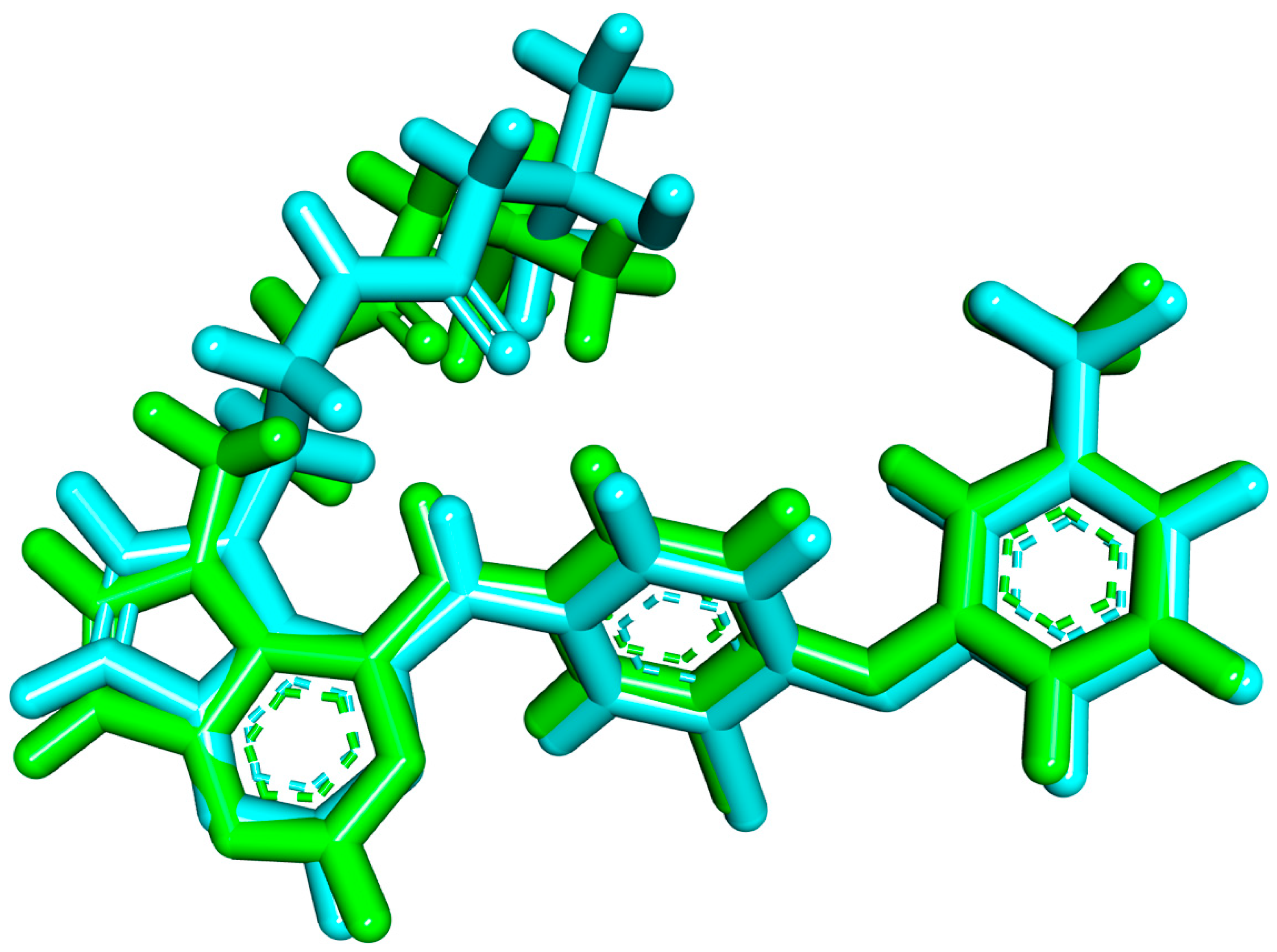
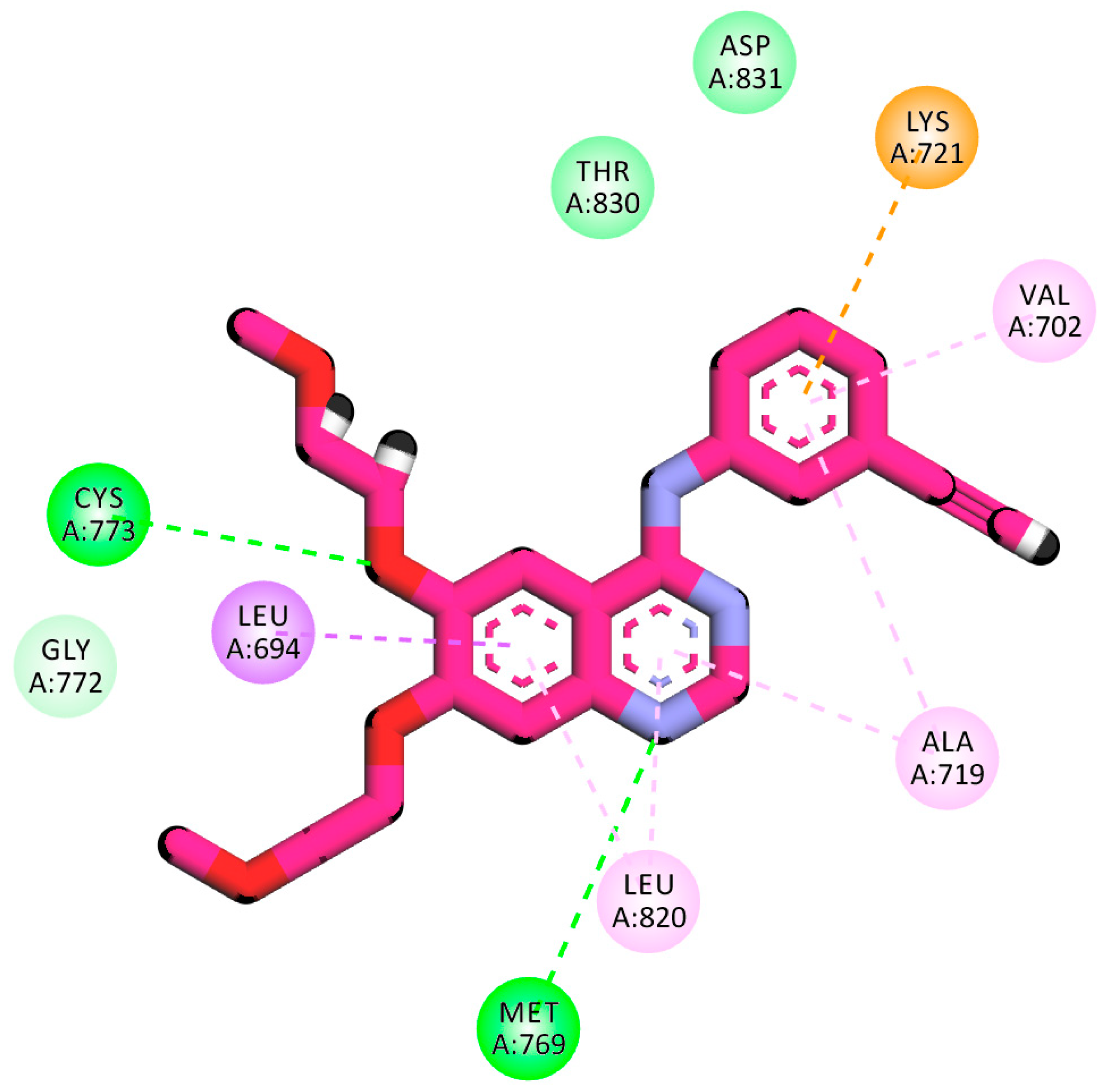
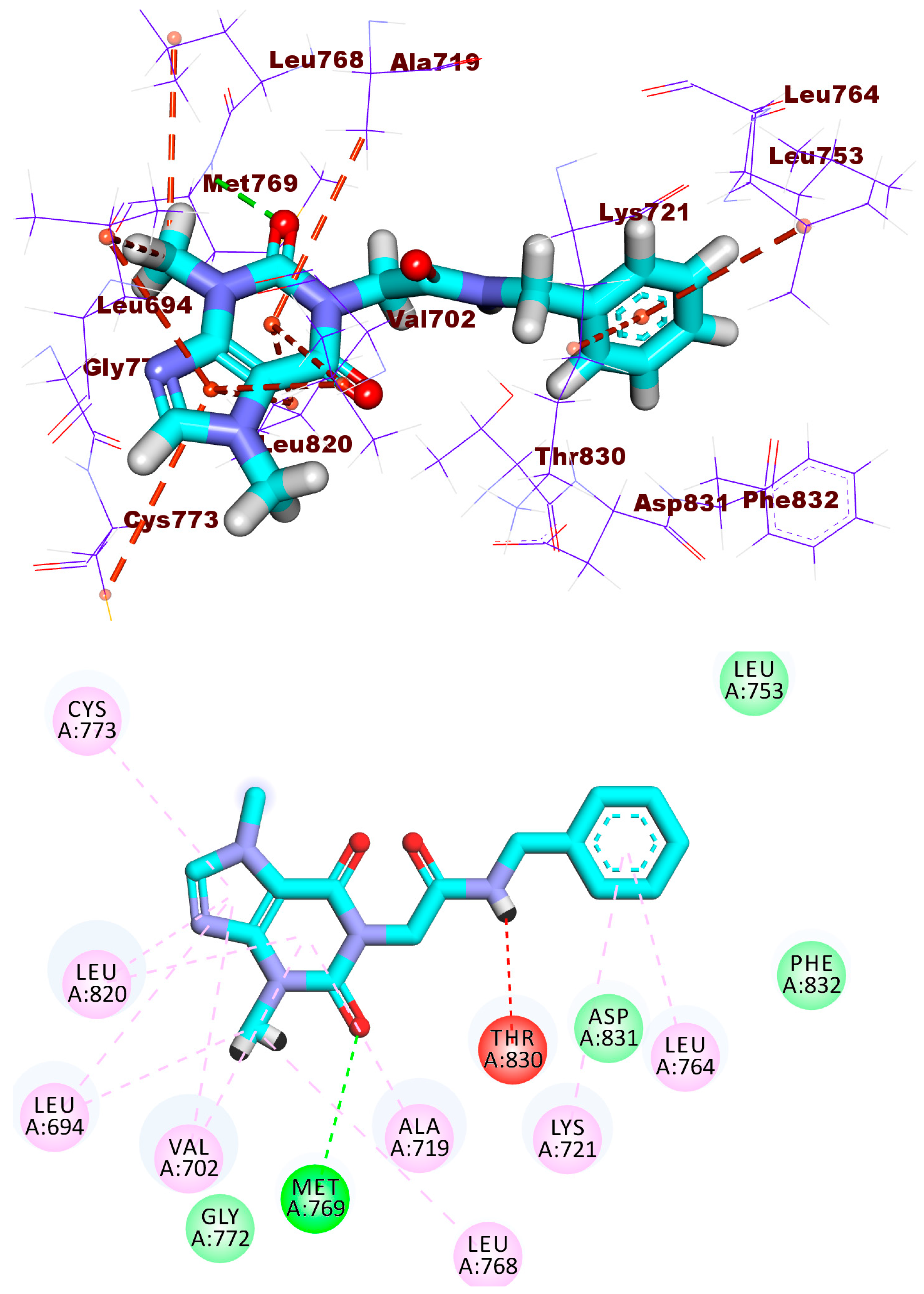
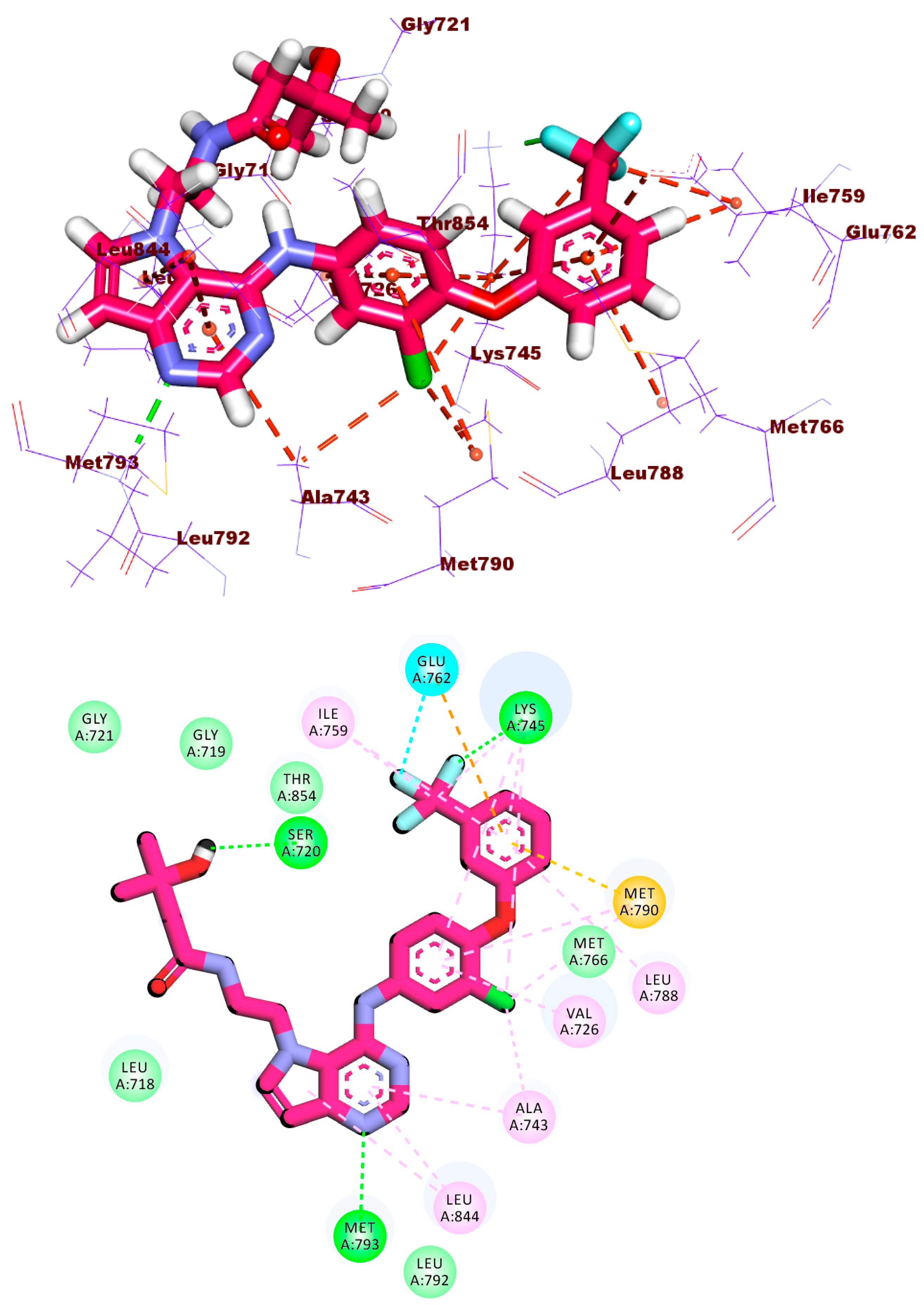
2.1.1. Molecular Docking against Wild and Mutant EGFR
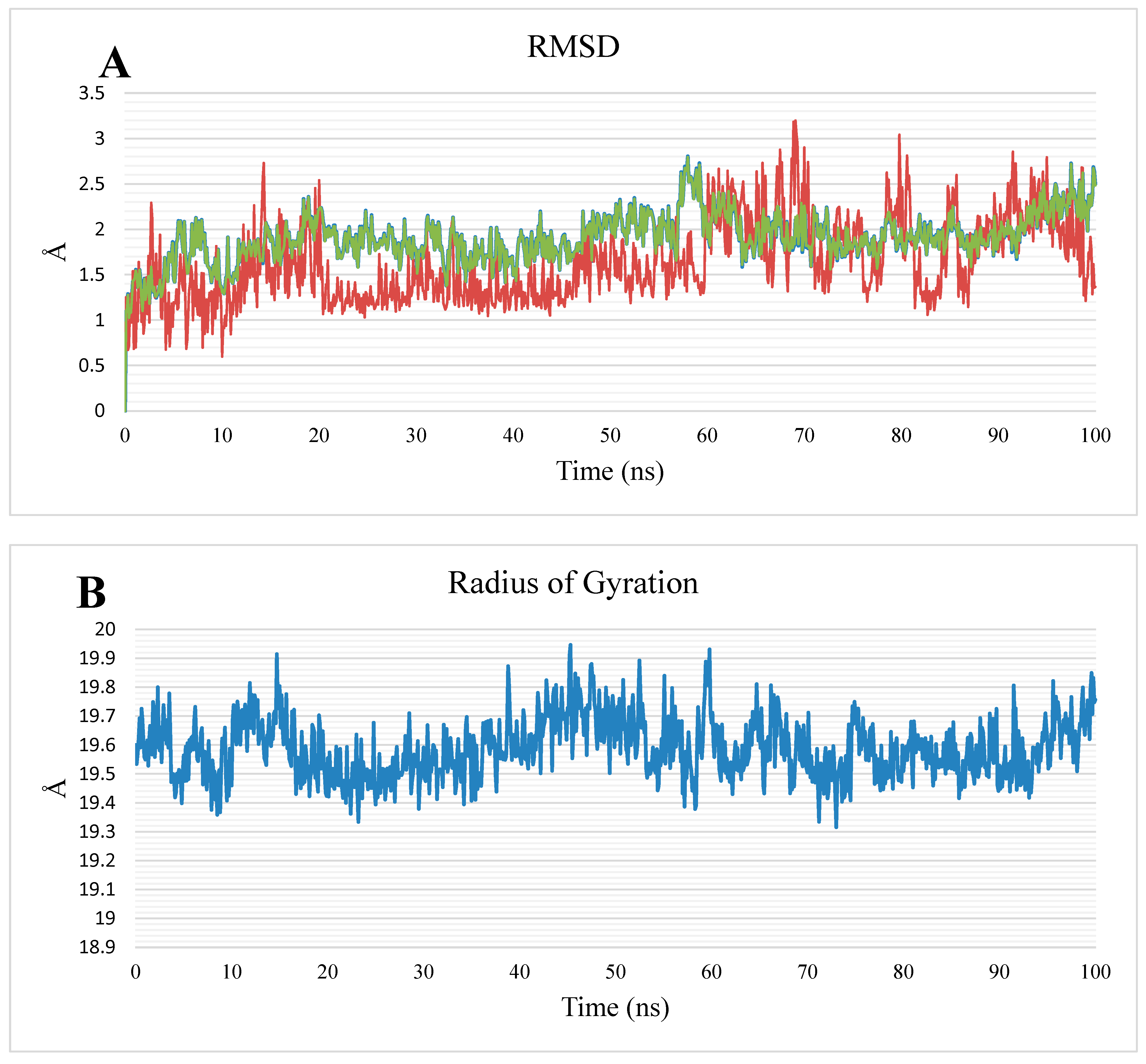
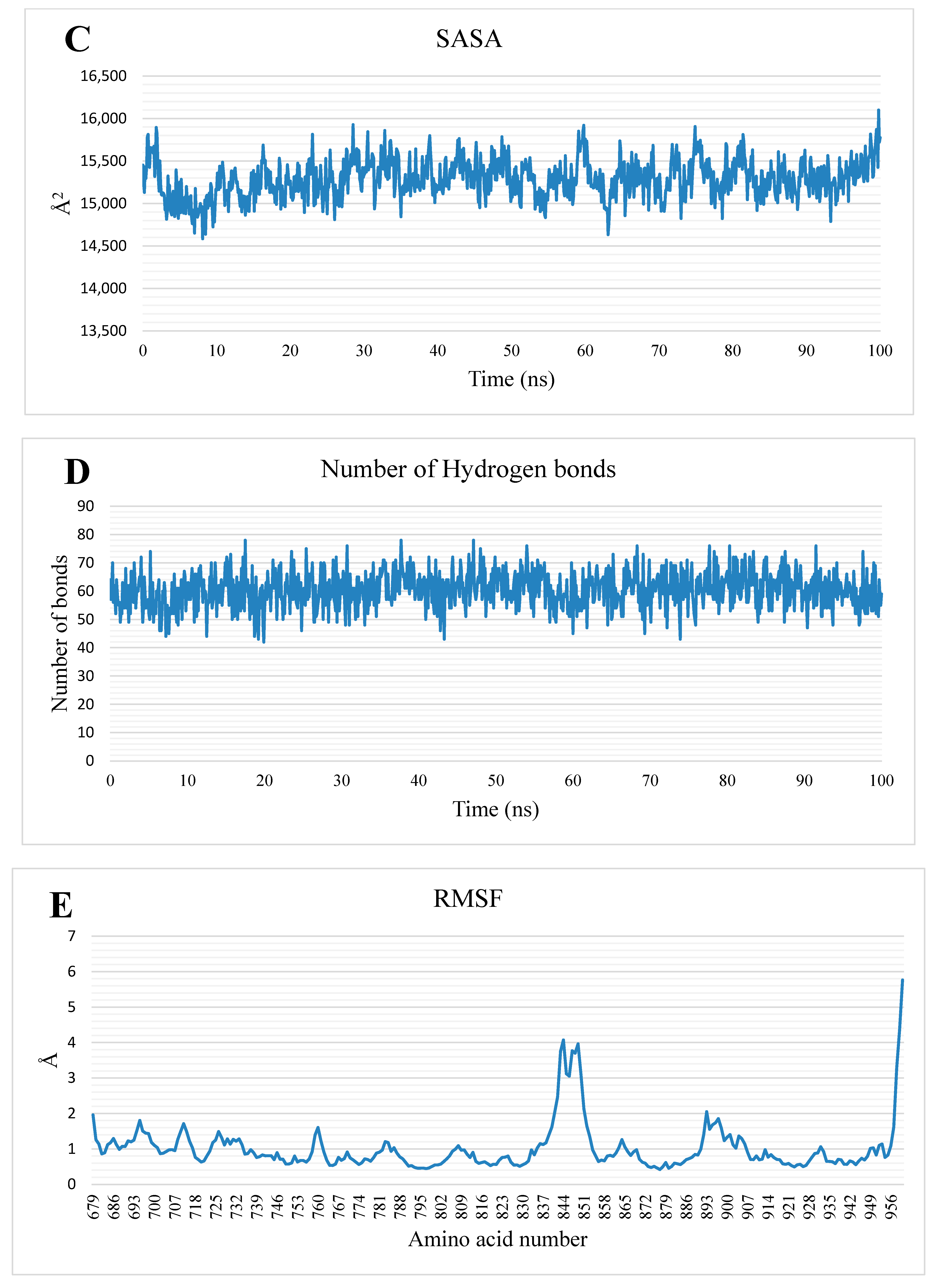
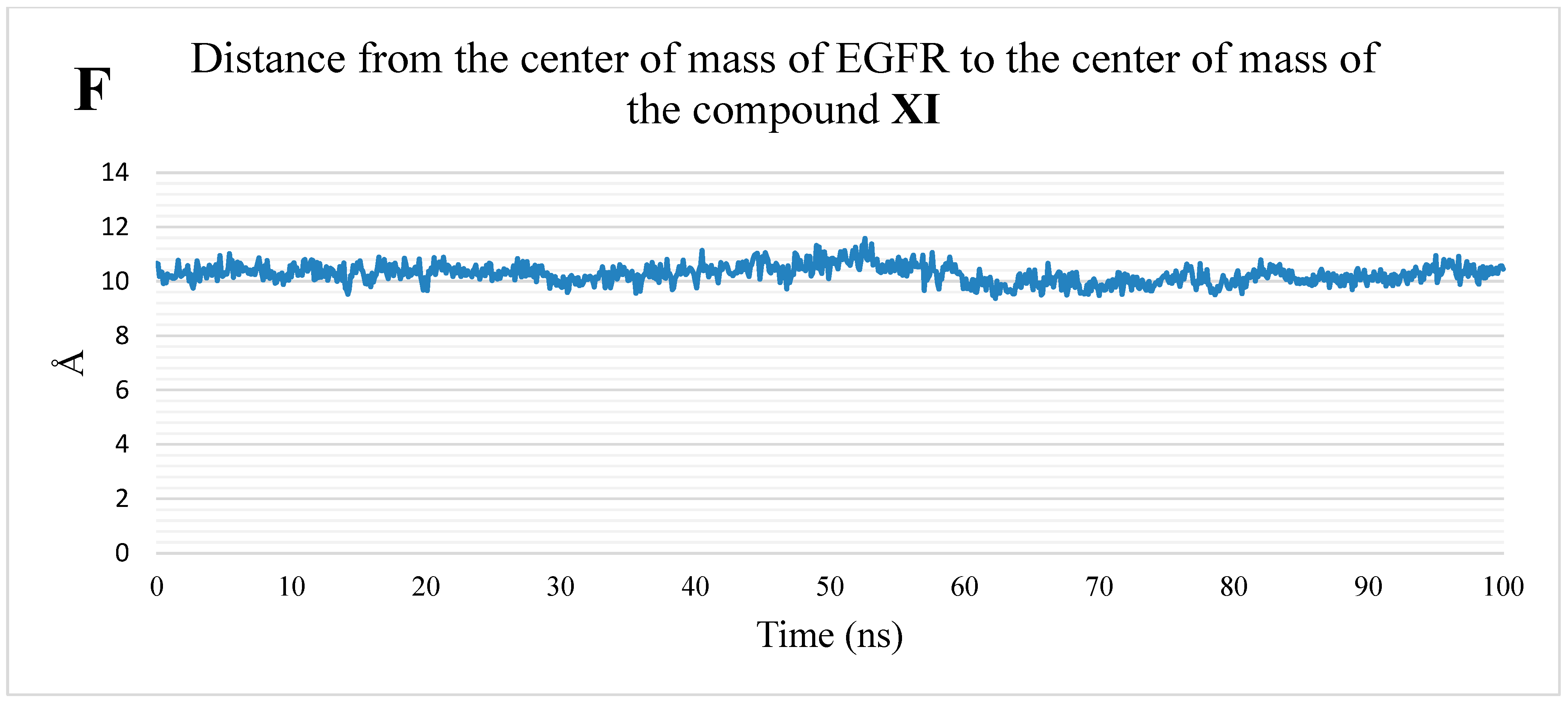
2.1.2. MD Simulations
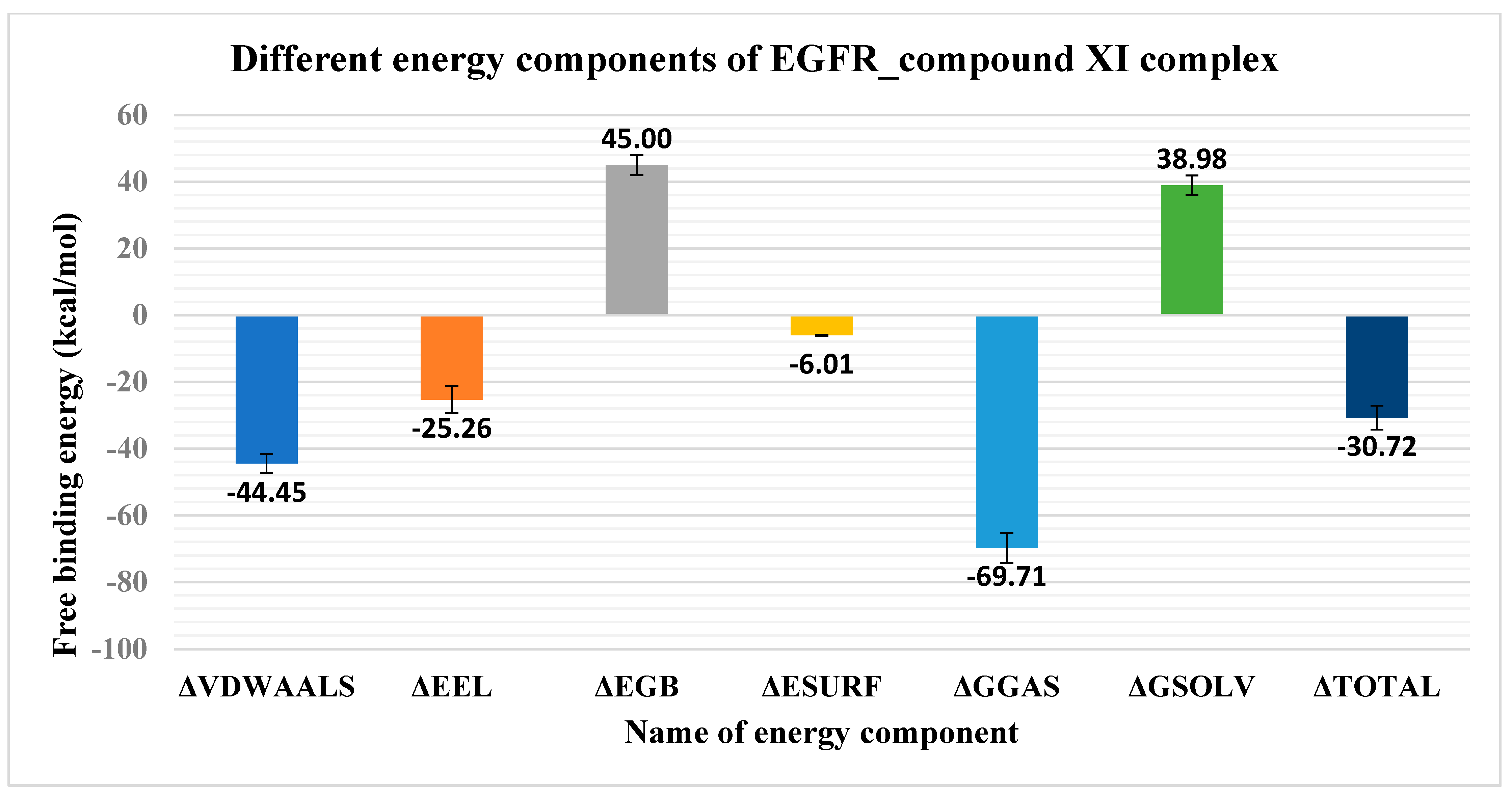
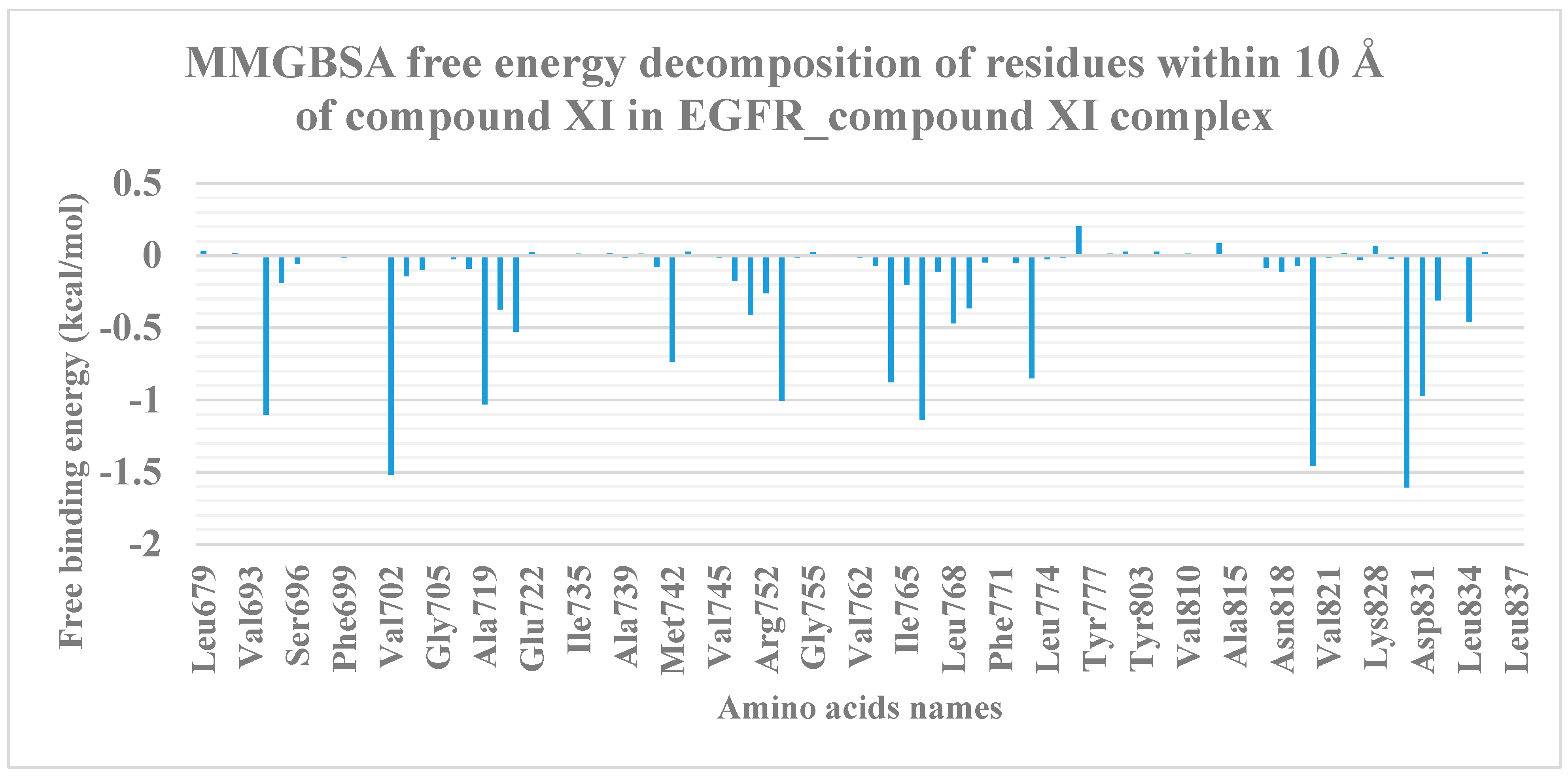
2.1.3. MM-GBSA
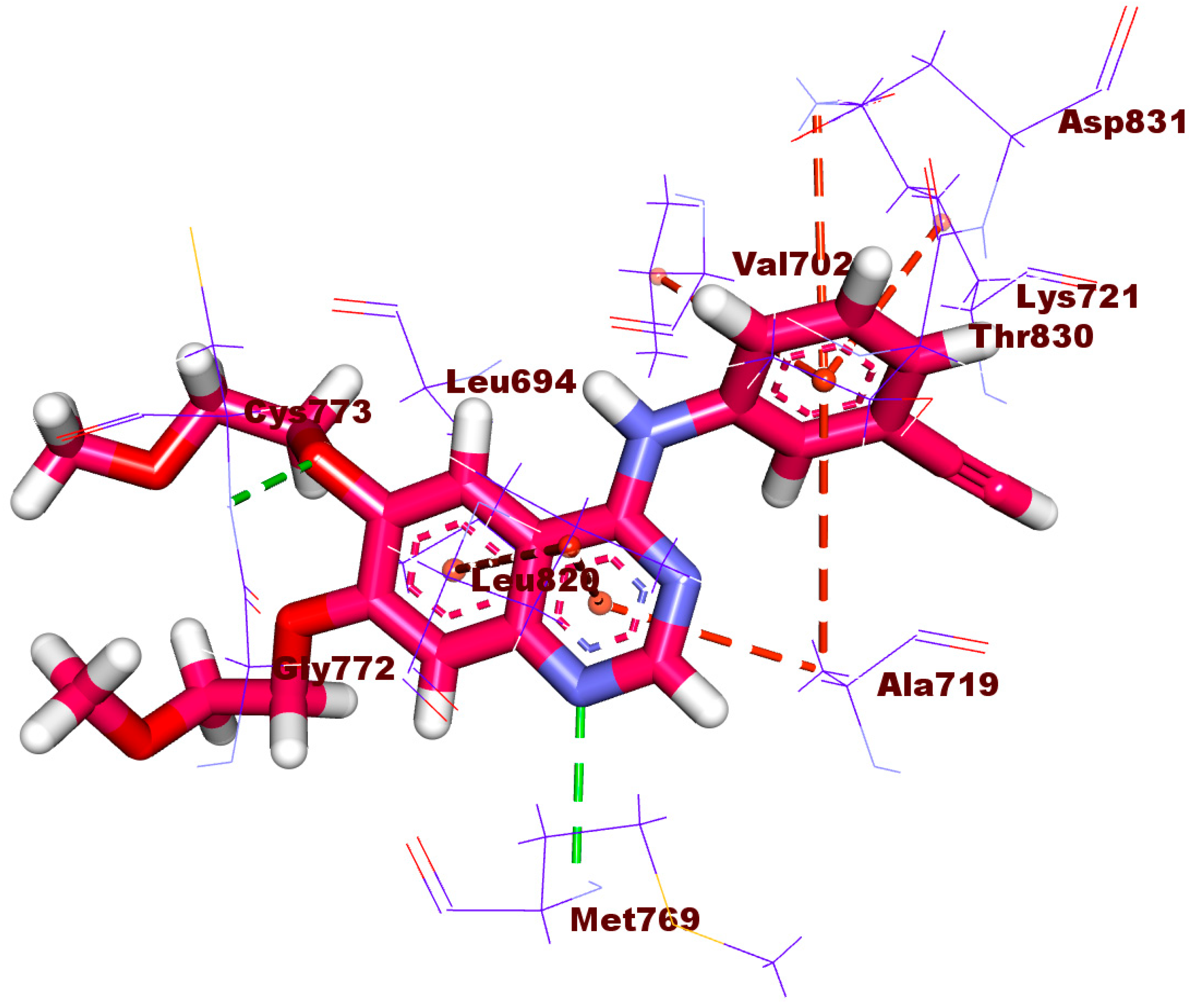
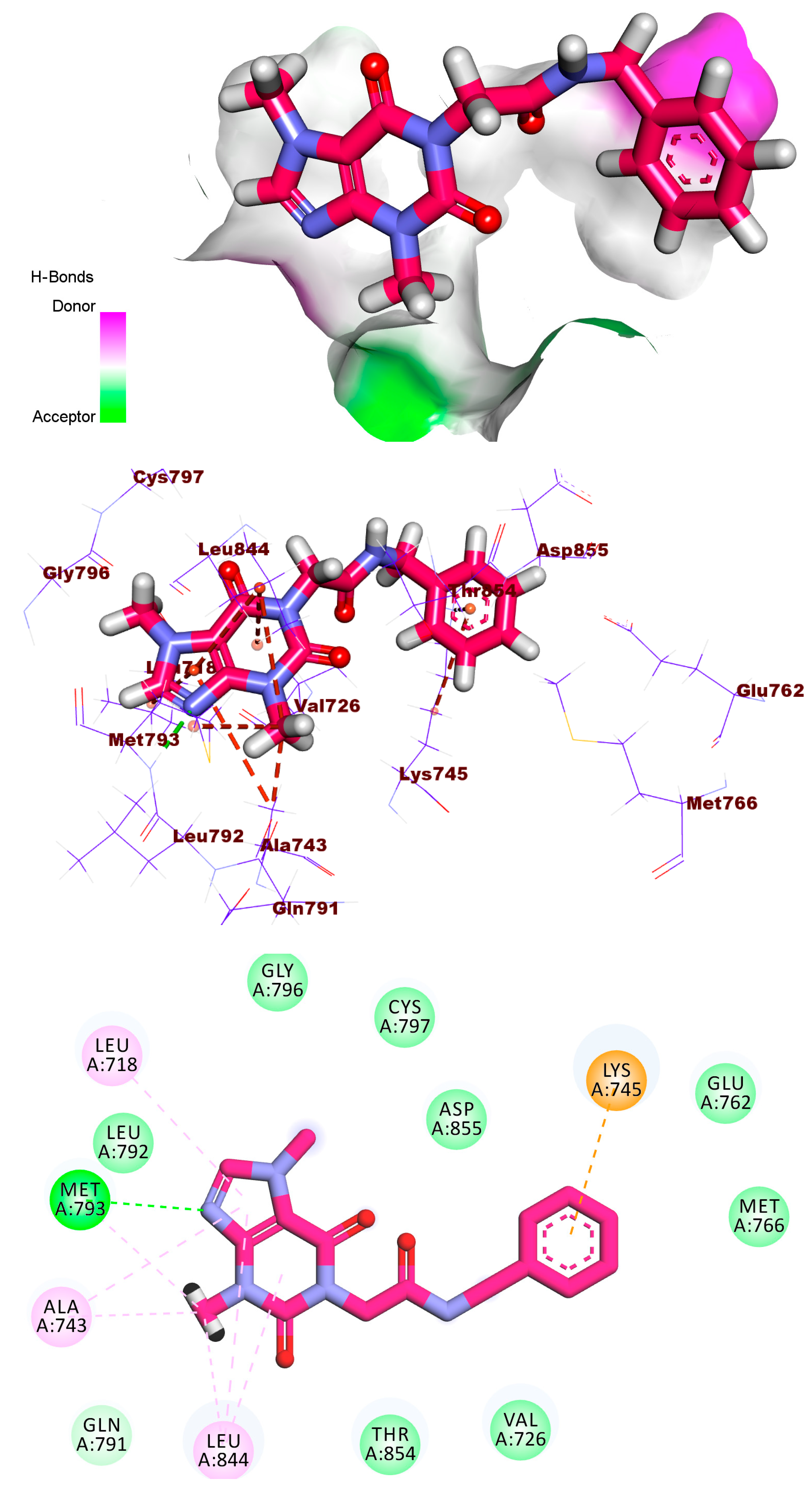
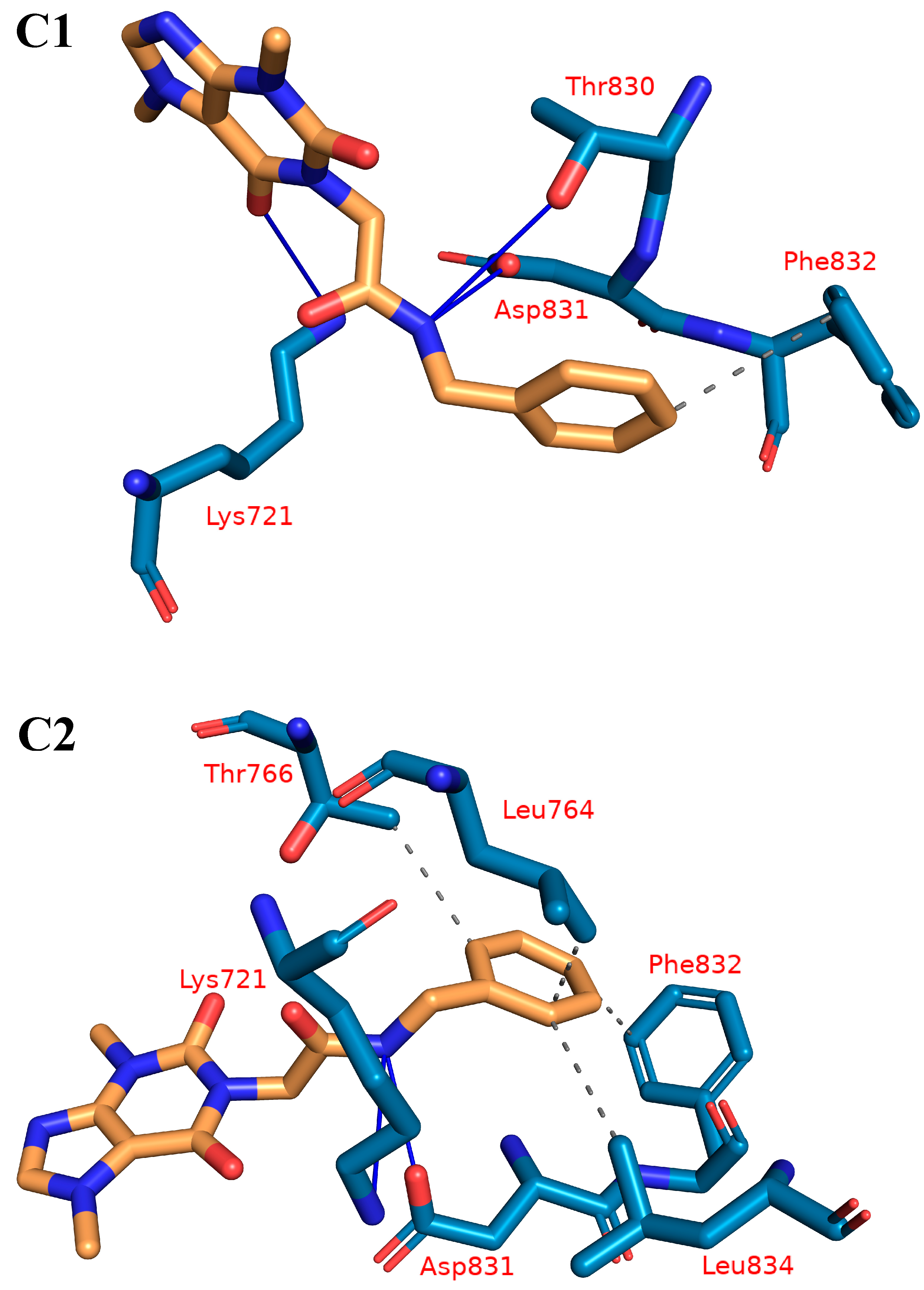
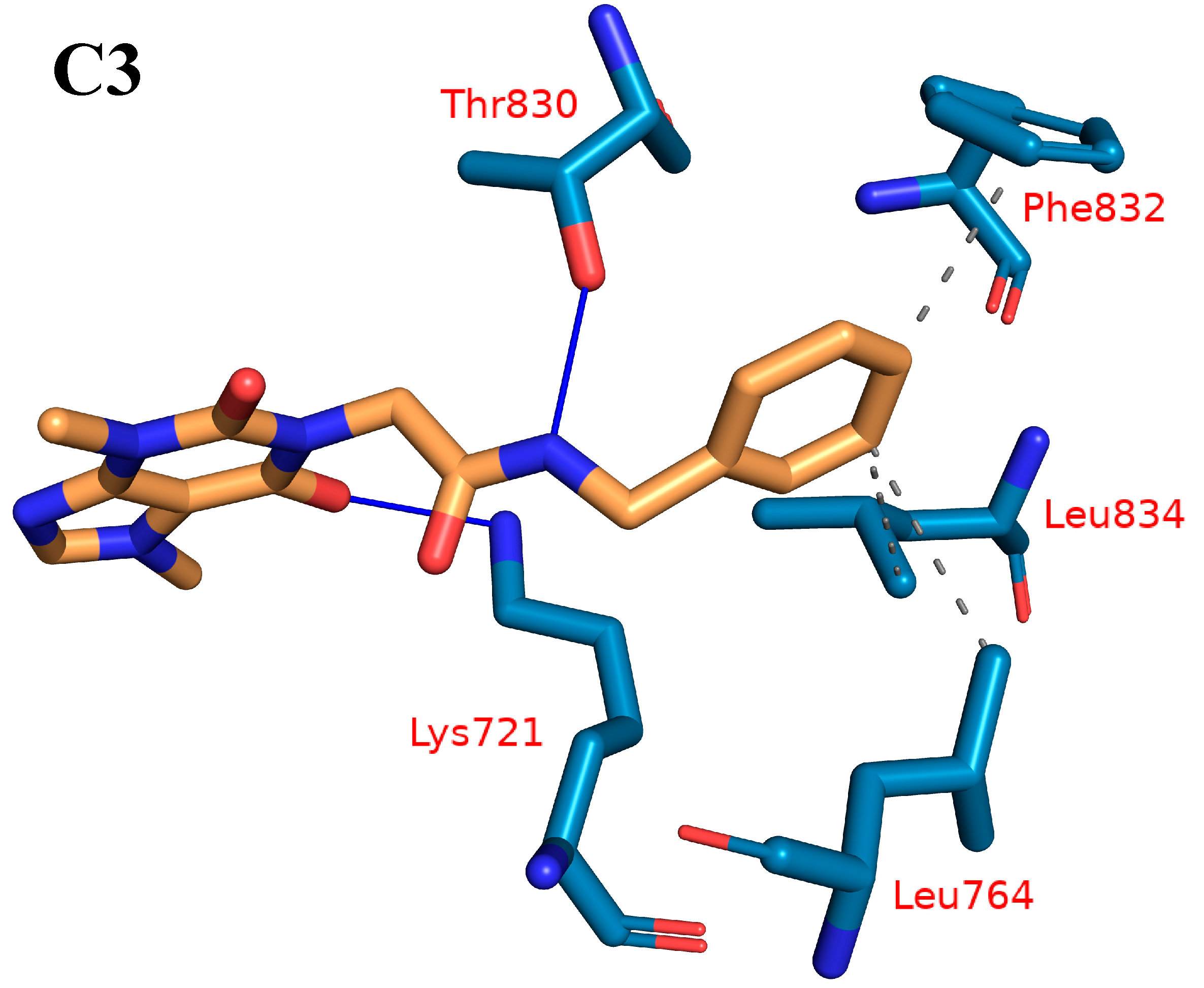
2.1.4. Protein–Ligand Interaction Profiler (PLIP) Studies
2.1.5. DFT
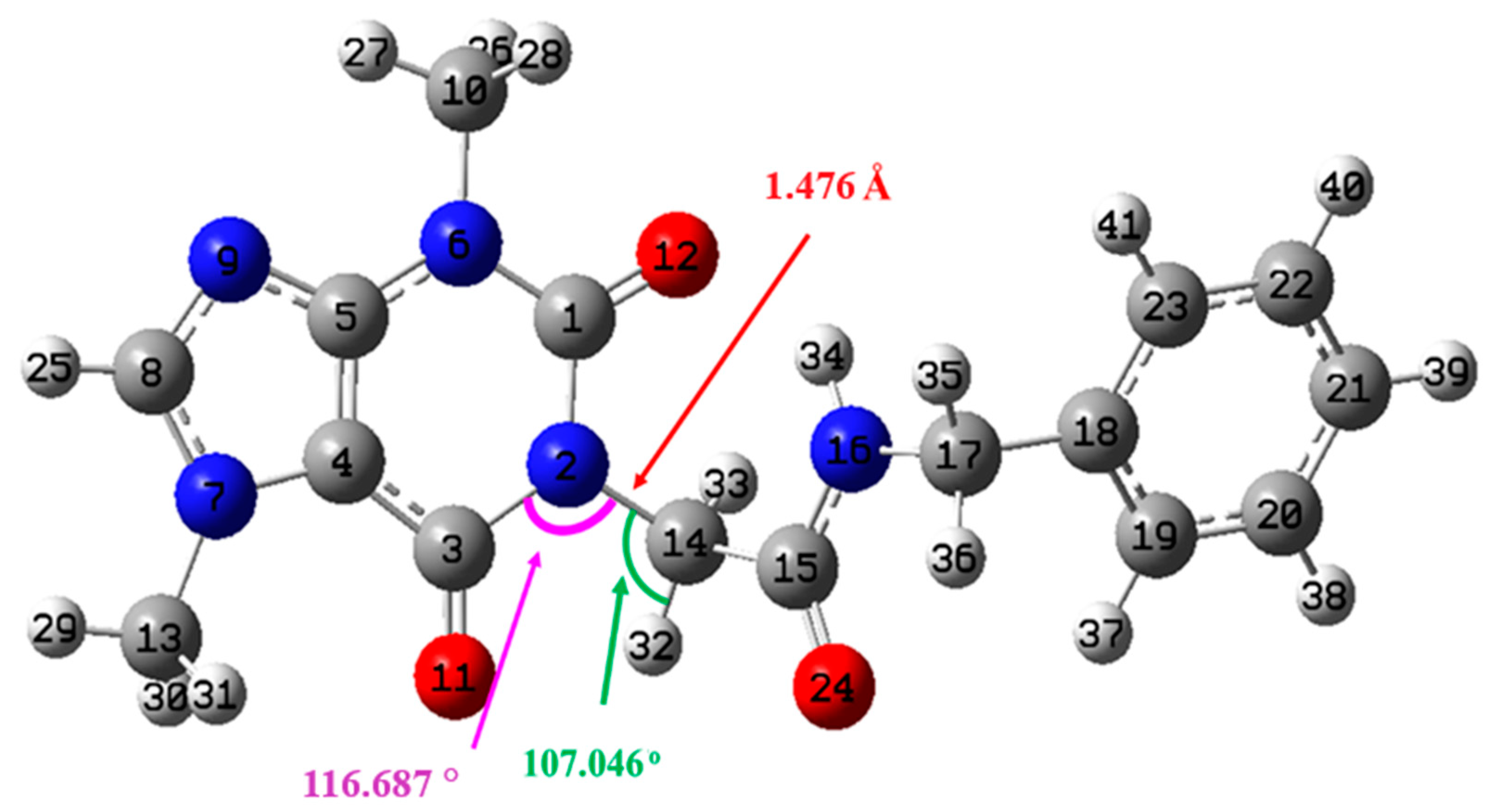
Geometry Optimization
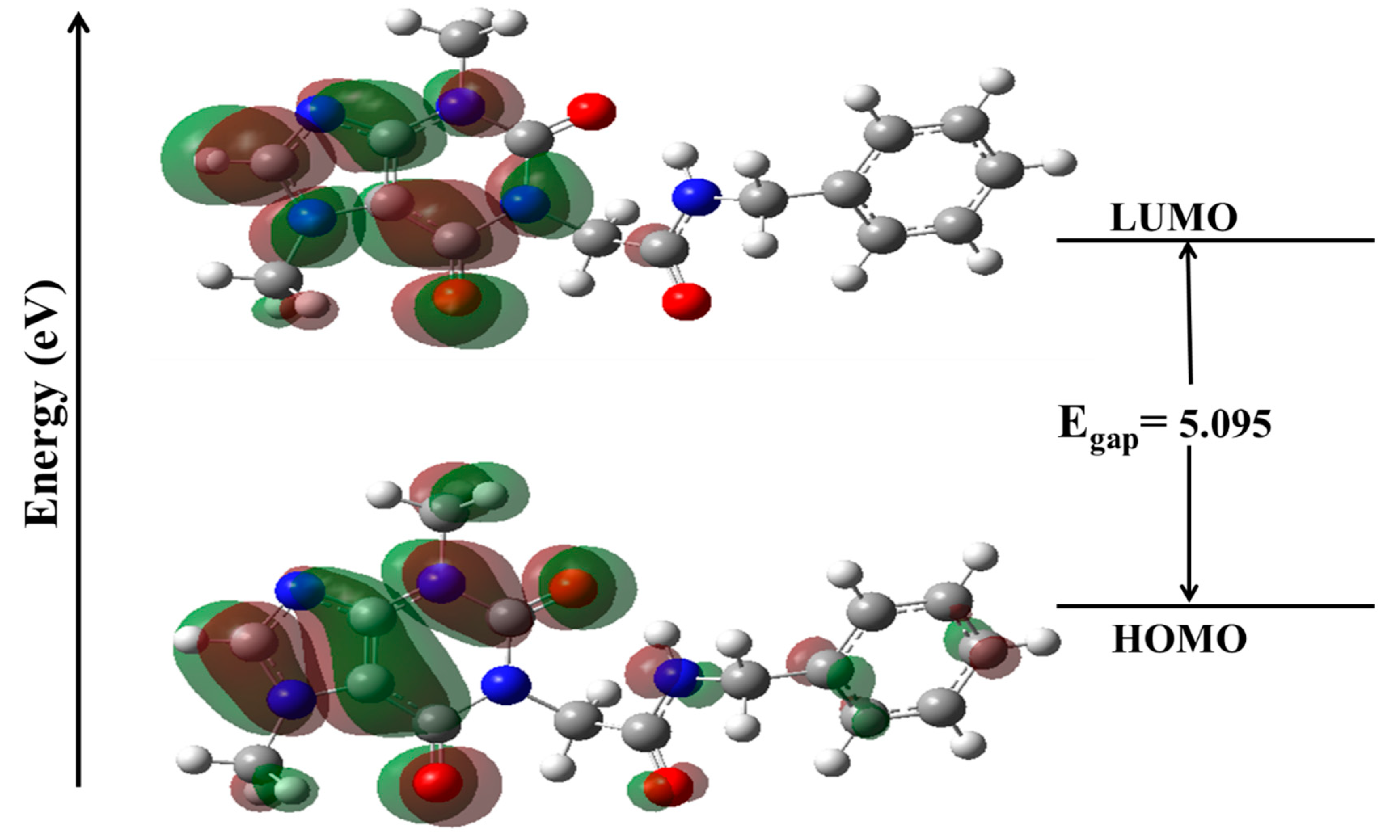
Frontier Molecular Orbital (FMO) Analysis
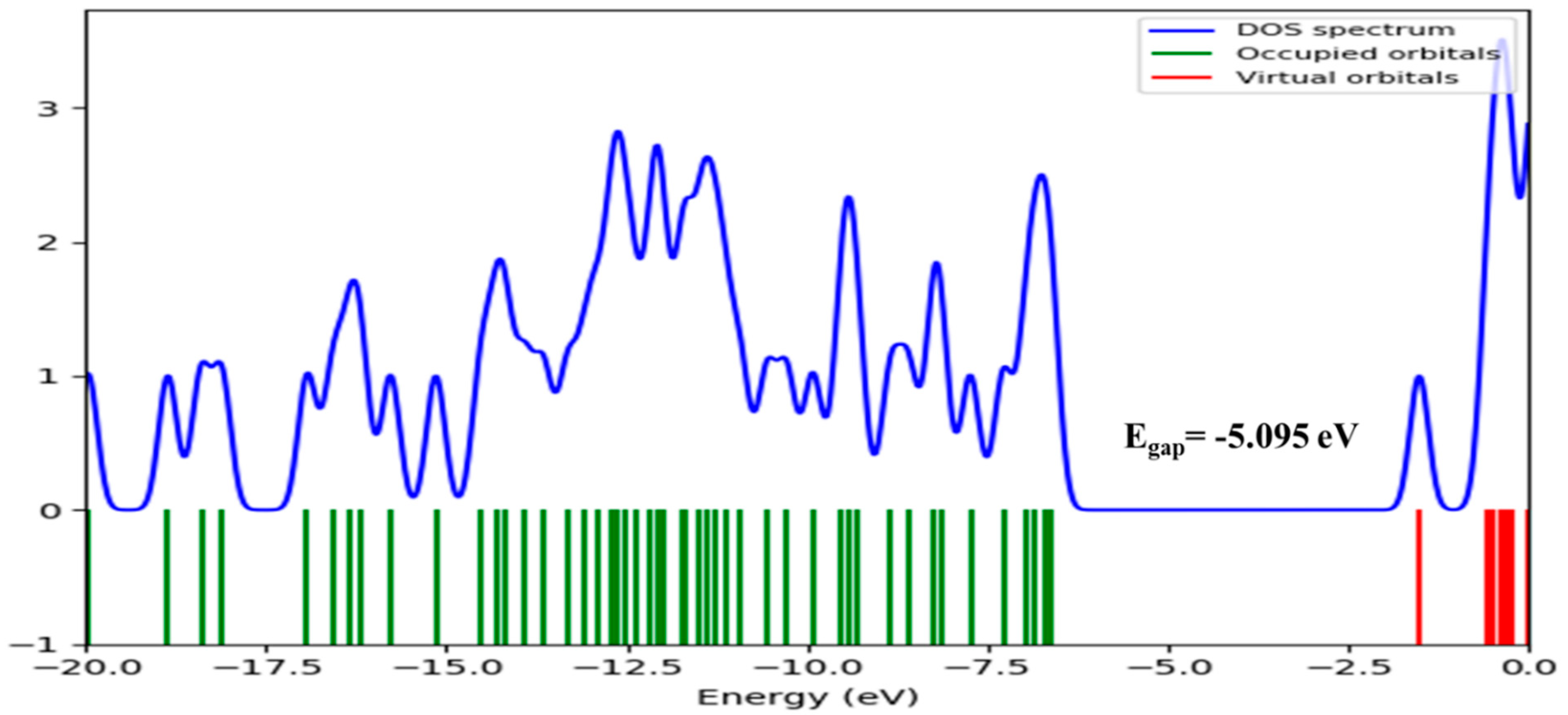
Global Reactive Indices and Total Density of State (TDOS)
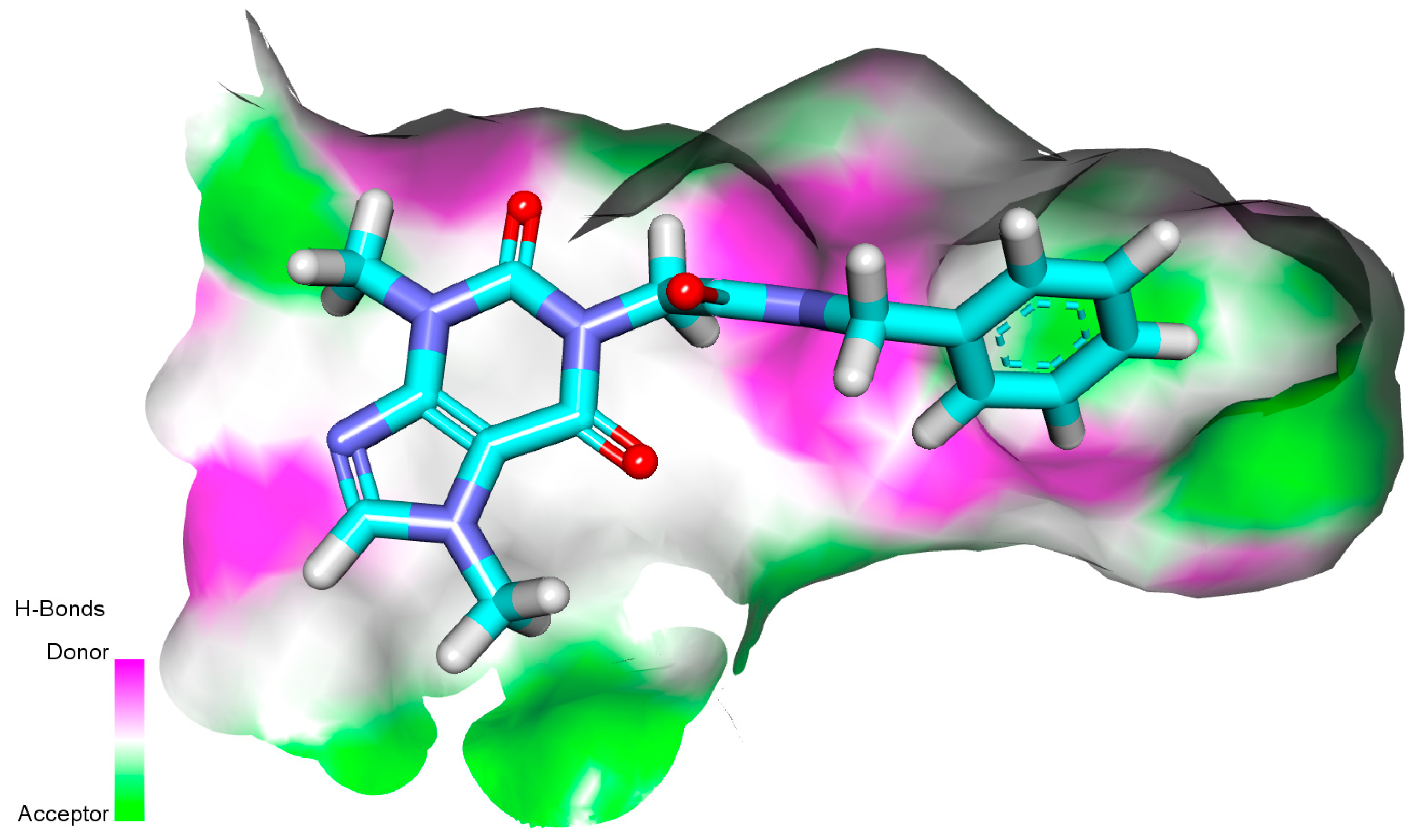
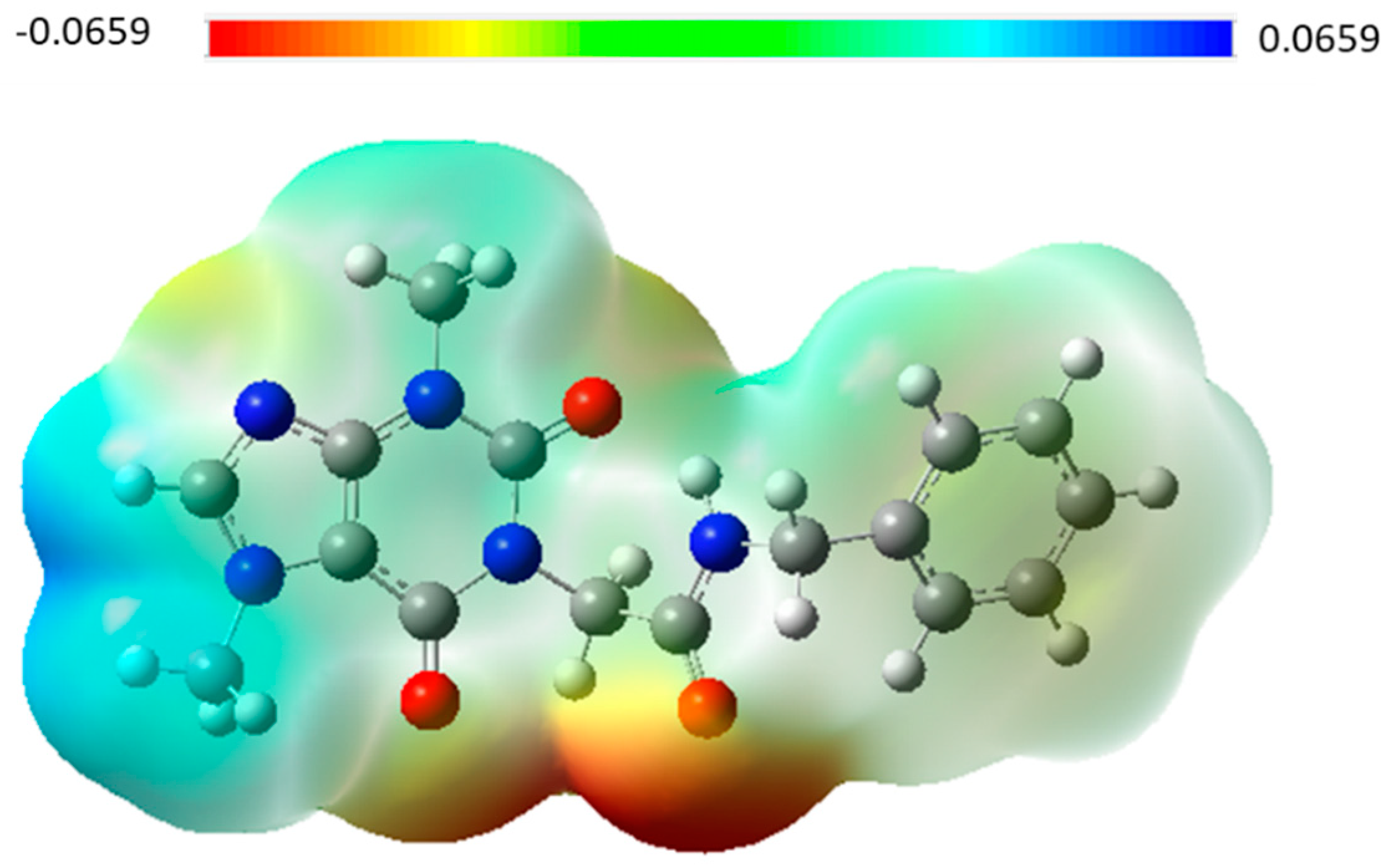
Surface Potential Mapping
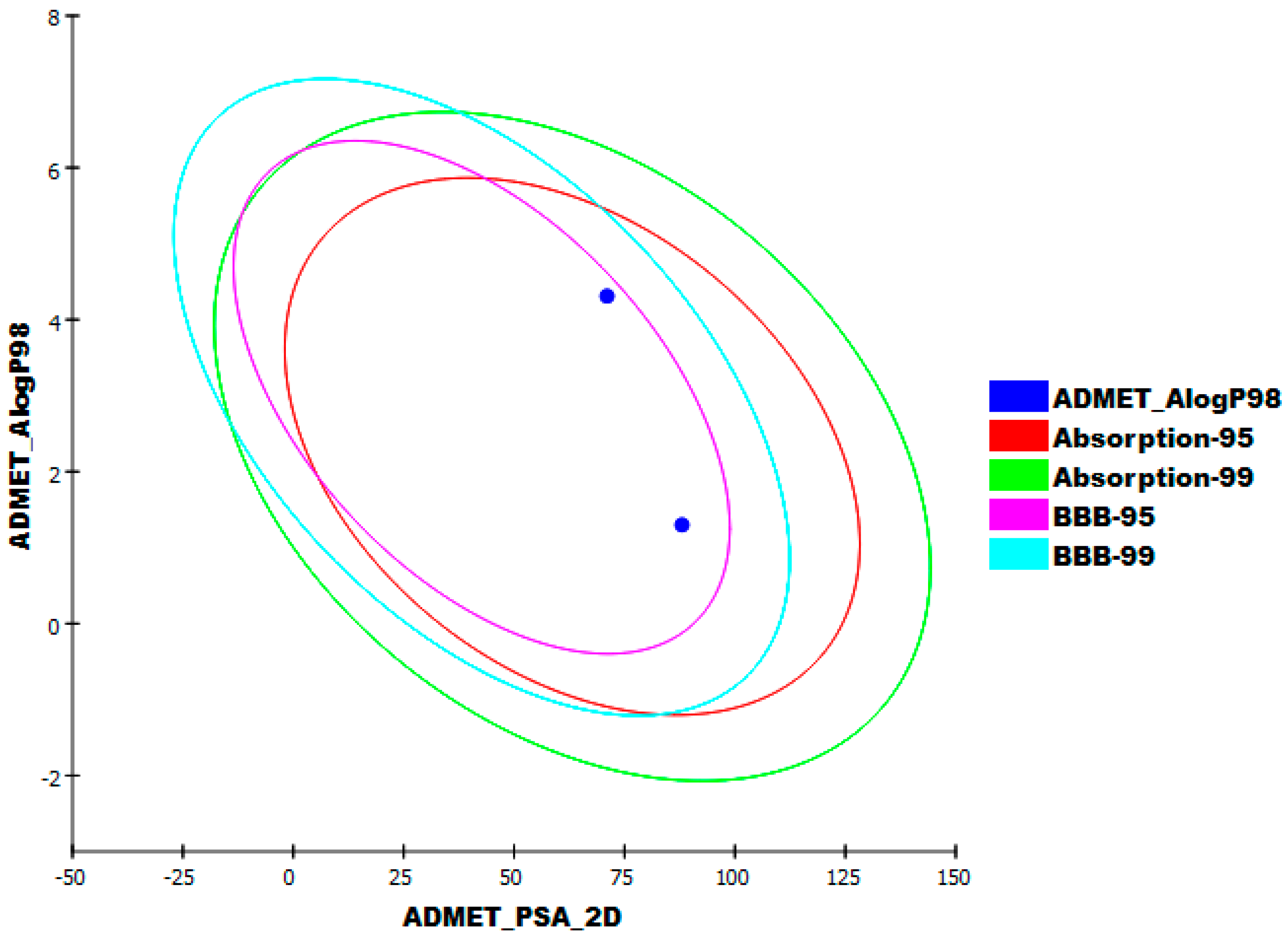
2.1.6. Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Profiling Study
2.1.7. In Silico Toxicity Studies
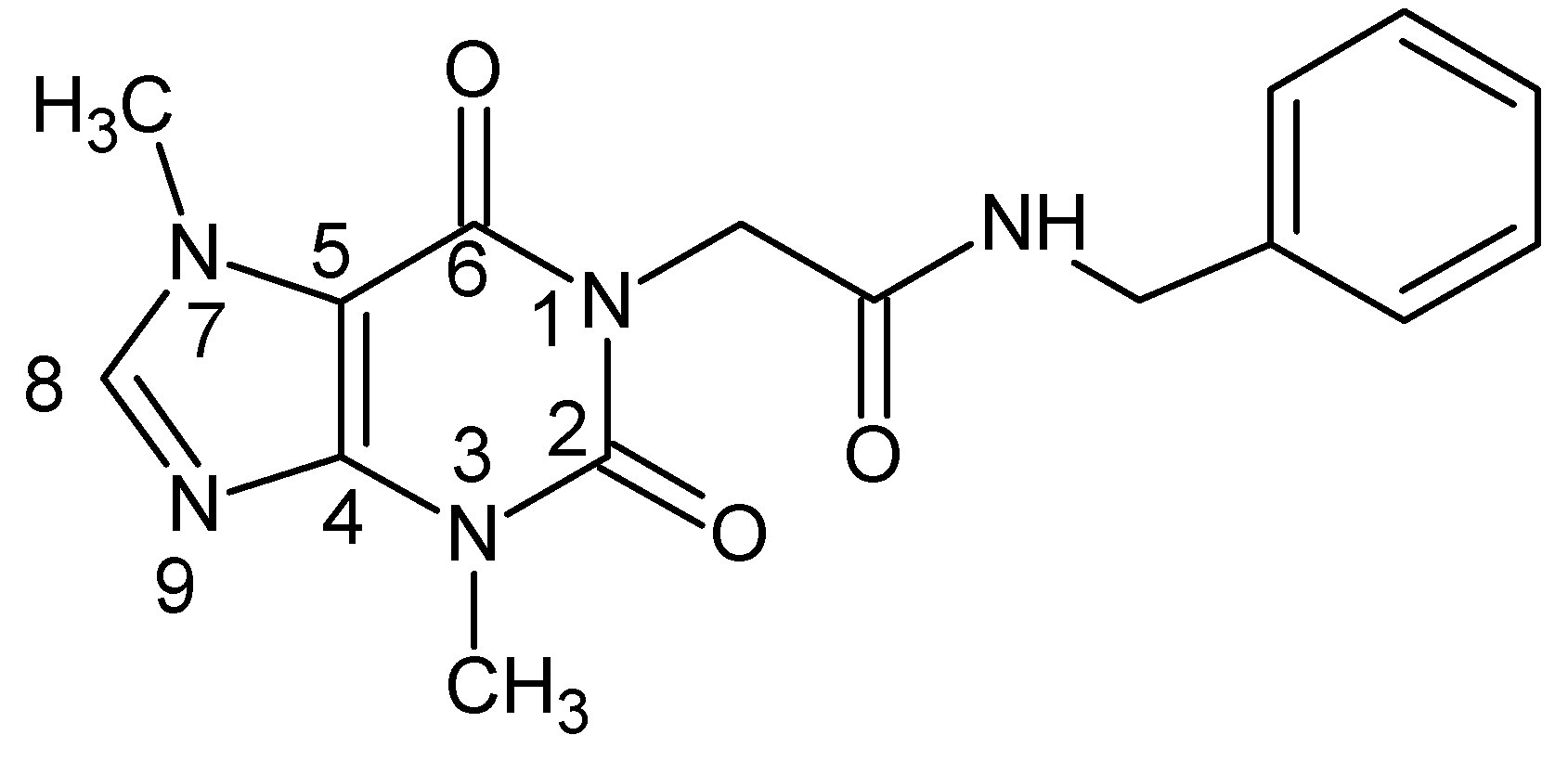
2.2. Chemistry
2.3. Biological Evaluation
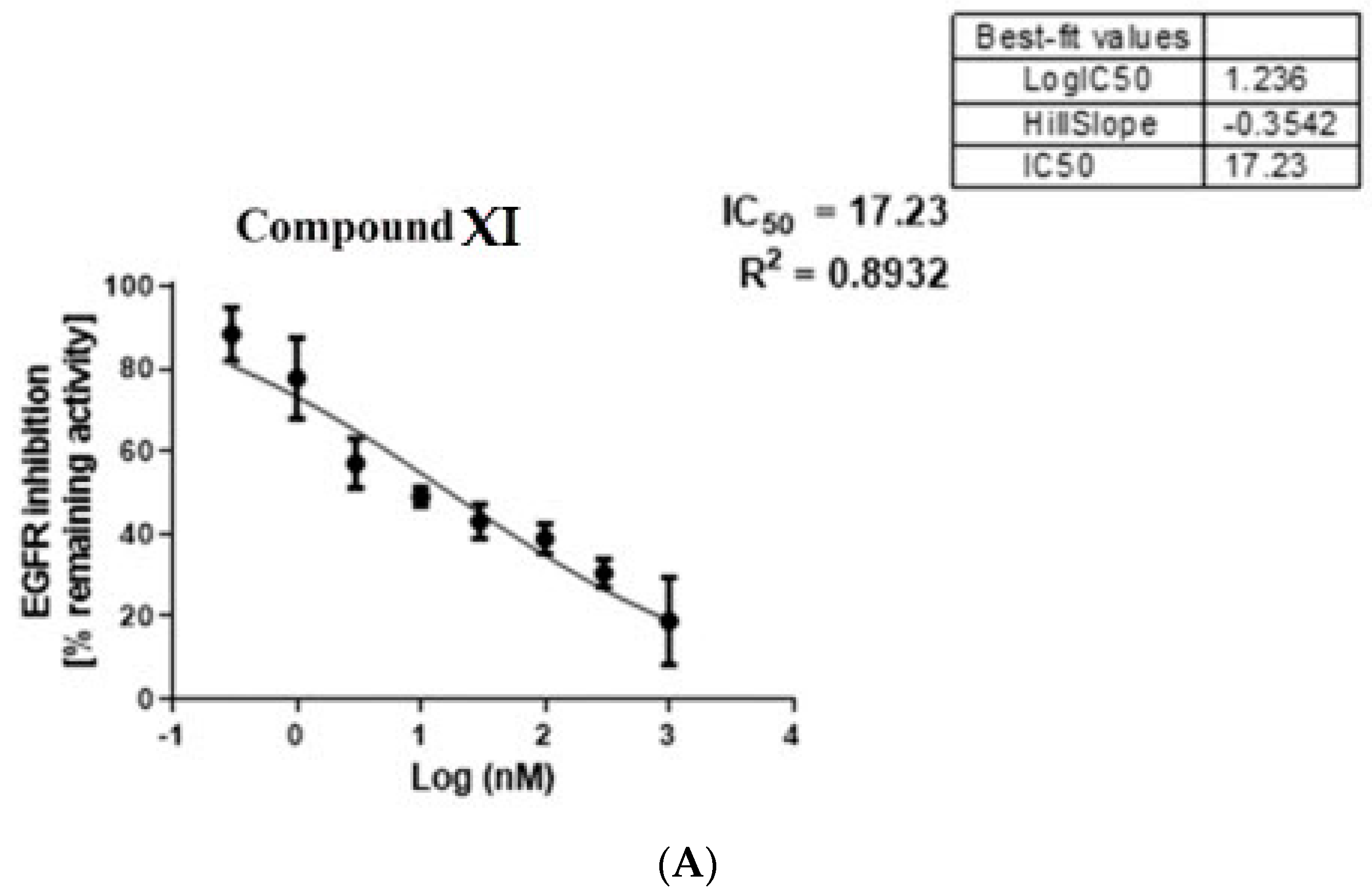
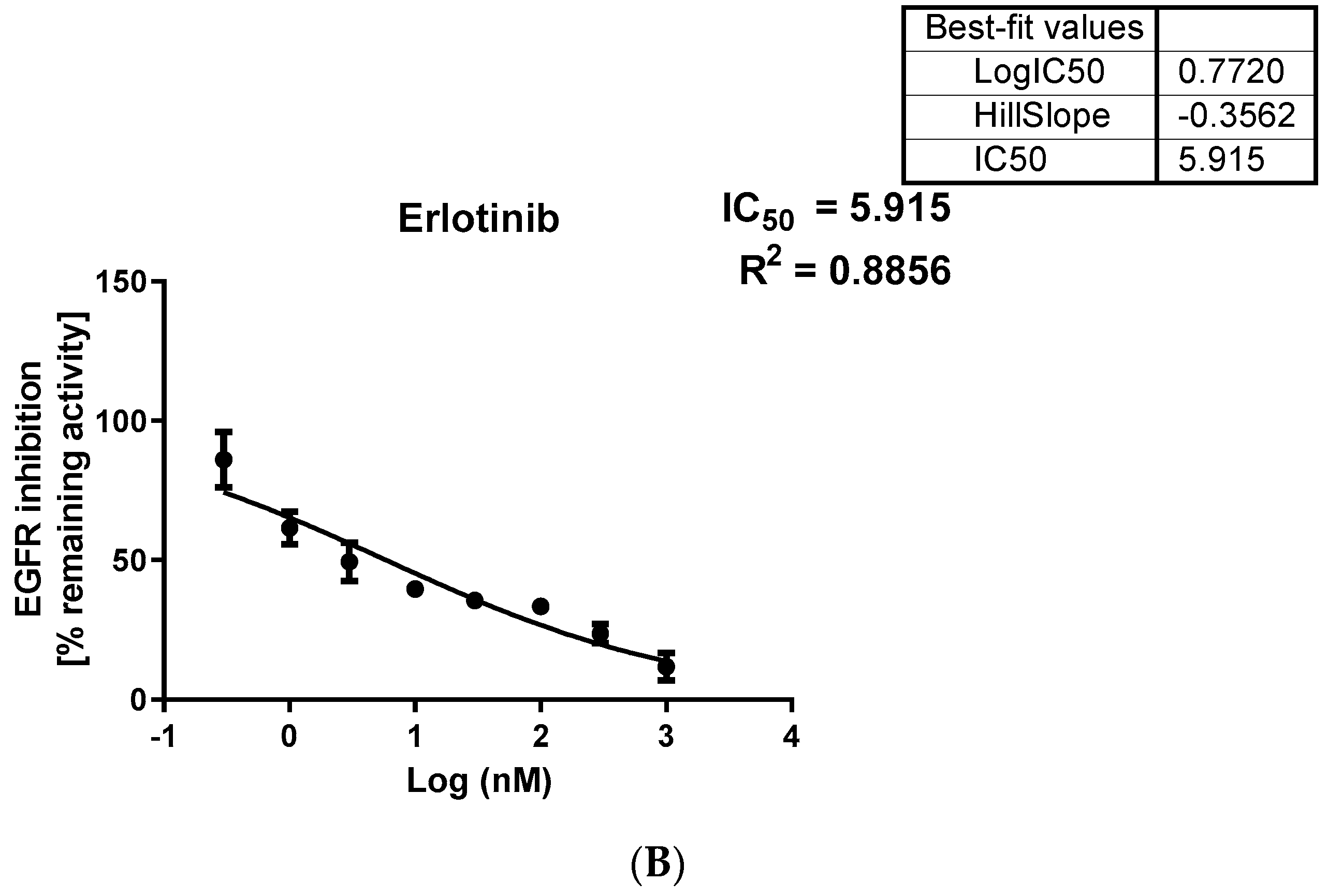
2.3.1. EGFR Inhibition
2.3.2. Cytotoxicity and Safety
2.3.3. Cell Cycle Analysis and Apoptosis Assay
3. Experimental
3.1. In Silico Studies
3.1.1. Docking Studies
3.1.2. MD Simulations
3.1.3. MM-GBSA
3.1.4. DFT
3.1.5. ADMET Studies
3.1.6. Toxicity Studies
3.2. Chemistry
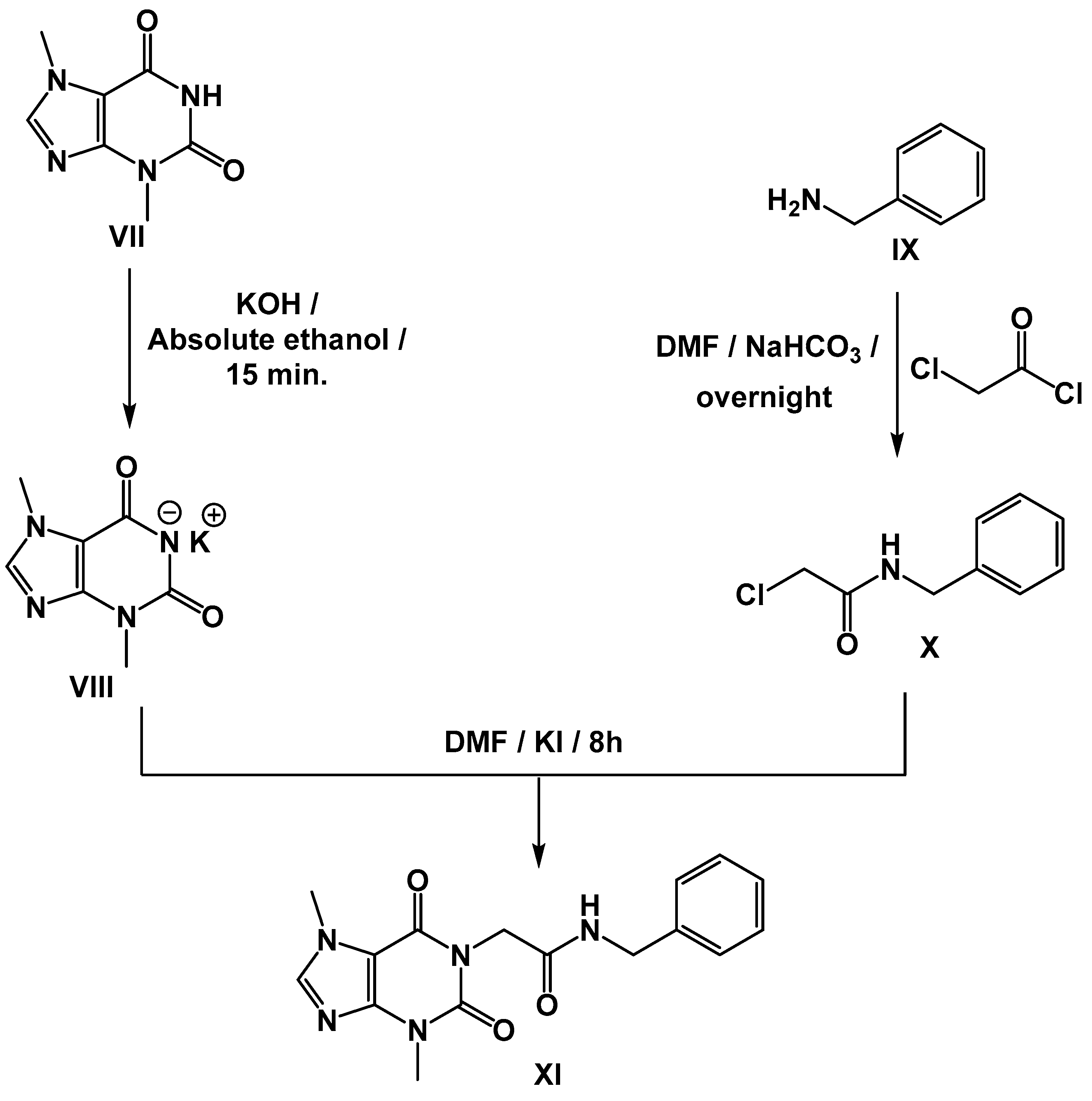
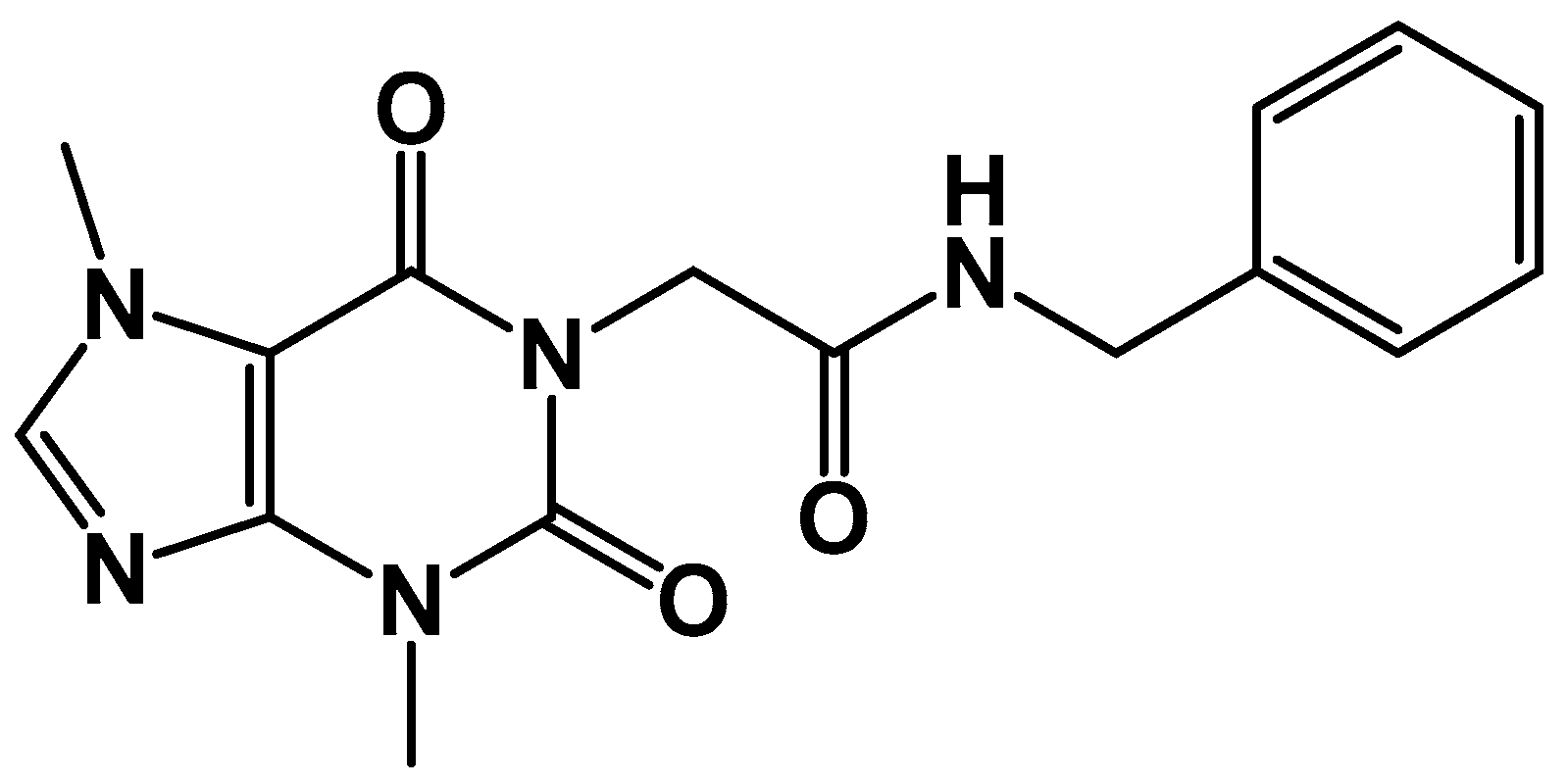
Synthesis of Compound XI
3.3. Biological Studies
3.3.1. In Vitro Egfr Inhibition
3.3.2. In Vitro Antiproliferative Activity
3.3.3. Safety Assay
3.3.4. Cell Cycle Analysis and Apoptosis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Abd El-Mageed, M.M.; Eissa, A.A.; Farag, A.E.-S.; Osman, E.E.A. Design and synthesis of novel furan, furo [2, 3-d] pyrimidine and furo [3, 2-e][1,2,4] triazolo [1, 5-c] pyrimidine derivatives as potential VEGFR-2 inhibitors. Bioorganic Chem. 2021, 116, 105336. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, P.; Bari, S.; Surana, S.; Shirkhedkar, A.; Wakode, S.; Shelar, S.; Racharla, S.; Ugale, V.; Ghodke, M. Logical synthetic strategies and structure-activity relationship of indolin-2-one hybrids as small molecule anticancer agents: An Overview. J. Mol. Struct. 2021, 1247, 131280. [Google Scholar] [CrossRef]
- El-Dash, Y.; Elzayat, E.; Abdou, A.M.; Hassan, R.A. Novel thienopyrimidine-aminothiazole hybrids: Design, synthesis, antimicrobial screening, anticancer activity, effects on cell cycle profile, caspase-3 mediated apoptosis and VEGFR-2 inhibition. Bioorganic Chem. 2021, 114, 105137. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.I.; Gee, J.M.W.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Spano, J.-P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.-F. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorganic Chem. 2020, 99, 103811. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Ghoneim, M.M.; Eissa, I.H.; Elsehemy, I.A.; Mostafa, A.E.; Hegazy, M.M.; Afifi, W.M.; Dou, D. Traditional ancient Egyptian medicine: A review. Saudi J. Biol. Sci. 2021, 28, 5823–5832. [Google Scholar] [CrossRef]
- Han, X.; Yang, Y.; Metwaly, A.M.; Xue, Y.; Shi, Y.; Dou, D. The Chinese herbal formulae (Yitangkang) exerts an antidiabetic effect through the regulation of substance metabolism and energy metabolism in type 2 diabetic rats. J. Ethnopharmacol. 2019, 239, 111942. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Barcz, E.; Sommer, E.; Janik, P.; Marianowski, L.; Skopinska-Rozewska, E. Adenosine receptor antagonism causes inhibition of angiogenic activity of human ovarian cancer cells. Oncol. Rep. 2000, 7, 1285–1376. [Google Scholar] [CrossRef] [PubMed]
- Kakuyamanee Iwazaki, A.; Sadzuka, Y. Effect of methylxanthine derivatives on doxorubicin transport and antitumor activity. Curr. Drug Metab. 2001, 2, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Sultani, H.N.; Ghazal, R.A.; Hayallah, A.M.; Abdulrahman, L.K.; Abu-Hammour, K.; AbuHammad, S.; Taha, M.O.; Zihlif, M.A. Inhibitory effects of new mercapto xanthine derivatives in human mcf7 and k562 cancer cell lines. J. Heterocycl. Chem. 2017, 54, 450–456. [Google Scholar] [CrossRef]
- Woskresensky, A. Ueber das Theobromin. Justus Liebigs Ann. Der Chem. 1842, 41, 125–127. [Google Scholar] [CrossRef]
- Smit, H.J. Theobromine and the pharmacology of cocoa. Methylxanthines 2011, 200, 201–234. [Google Scholar]
- Fischer, E. Synthese des Theobromins. In Untersuchungen in der Puringruppe; Springer: Berlin/Heidelberg, Germany, 1907; pp. 265–272. [Google Scholar]
- Carla Cadoná, F.; Kolinski Machado, A.; Farina Azzolin, V.; Barbisan, F.; Bortoluzzi Dornelles, E.; Glanzner, W.; Bayard Gonçalves, P.D.; Elias Assmann, C.; Esteves Ribeiro, E.; Beatrice Mânica da Cruz, I. Guaraná a caffeine-rich food increases oxaliplatin sensitivity of colorectal HT-29 cells by apoptosis pathway modulation. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem.-Anti-Cancer Agents) 2016, 16, 1055–1065. [Google Scholar] [CrossRef]
- Shojaei-Zarghani, S.; Khosroushahi, A.Y.; Rafraf, M. Oncopreventive effects of theanine and theobromine on dimethylhydrazine-induced colon cancer model. Biomed. Pharmacother. 2021, 134, 111140. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, K.W.; Kang, K.S.; Kim, D.Y.; Park, H.H.; Lee, M.J.; Kim, H.S.; Kwon, I.B. Theobromine with an Anti-Carcinogenic Activity. European Patent EP1304046A1, 18 October 2001. [Google Scholar]
- Sugimoto, N.; Miwa, S.; Hitomi, Y.; Nakamura, H.; Tsuchiya, H.; Yachie, A. Theobromine, the primary methylxanthine found in Theobroma cacao, prevents malignant glioblastoma proliferation by negatively regulating phosphodiesterase-4, extracellular signal-regulated kinase, Akt/mammalian target of rapamycin kinase, and nuclear factor-kappa B. Nutr. Cancer 2014, 66, 419–423. [Google Scholar]
- Gil, M.; Skopińska-Rózewska, E.; Radomska, D.; Demkow, U.; Skurzak, H.; Rochowska, M.; Beuth, J.; Roszkowski, K. Effect of purinergic receptor antagonists suramin and theobromine on tumor-induced angiogenesis in BALB/c mice. Folia Biol. 1993, 39, 63–68. [Google Scholar]
- Barcz, E.; Sommer, E.; Sokolnicka, I.; Gawrychowski, K.; Roszkowska-Purska, K.; Janik, P.; Skopinska-Rózewska, E. The influence of theobromine on angiogenic activity and proangiogenic cytokines production of human ovarian cancer cells. Oncol. Rep. 1998, 5, 517–537. [Google Scholar] [CrossRef]
- da Rosa, R.; Schenkel, E.P.; Bernardes, L.S.C. Semisynthetic and newly designed derivatives based on natural chemical scaffolds: Moving beyond natural products to fight Trypanosoma cruzi. Phytochem. Rev. 2020, 19, 105–122. [Google Scholar] [CrossRef]
- Goh, G.B.; Hodas, N.O.; Vishnu, A. Deep learning for computational chemistry. J. Comput. Chem. 2017, 38, 1291–1307. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; Alesawy, M.S.; Saleh, A.M.; Elkaeed, E.B.; Alsfouk, B.A.; El-Attar, A.-A.M.; Metwaly, A.M. Ligand and Structure-Based In Silico Determination of the Most Promising SARS-CoV-2 nsp16-nsp10 2′-o-Methyltransferase Complex Inhibitors among 3009 FDA Approved Drugs. Molecules 2022, 27, 2287. [Google Scholar] [CrossRef] [PubMed]
- Elkady, H.; Elwan, A.; El-Mahdy, H.A.; Doghish, A.S.; Ismail, A.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Dahab, M.A.; Mahdy, H.A. New benzoxazole derivatives as potential VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, anti-proliferative evaluation, flowcytometric analysis, and in silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Yousef, R.G.; Elkady, H.; Gobaara, I.M.M.; Alsfouk, A.A.; Husein, D.Z.; Ibrahim, I.M.; Metwaly, A.M.; Eissa, I.H. The Assessment of Anticancer and VEGFR-2 Inhibitory Activities of a New 1H-Indole Derivative: In Silico and In Vitro Approaches. Processes 2022, 10, 1391. [Google Scholar] [CrossRef]
- Mohammed, S.O.; El Ashry, E.S.H.; Khalid, A.; Amer, M.R.; Metwaly, A.M.; Eissa, I.H.; Elkaeed, E.B.; Elshobaky, A.; Hafez, E.E. Expression, Purification, and Comparative Inhibition of Helicobacter pylori Urease by Regio-Selectively Alkylated Benzimidazole 2-Thione Derivatives. Molecules 2022, 27, 865. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Elkady, H.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Alanazi, M.M.; Alharbi, M.A.; Eissa, I.H.; Dahab, M.A. New quinoxaline-based VEGFR-2 inhibitors: Design, synthesis, and antiproliferative evaluation with in silico docking, ADMET, toxicity, and DFT studies. RSC Adv. 2021, 11, 30315–30328. [Google Scholar] [CrossRef]
- Suleimen, Y.M.; Jose, R.A.; Suleimen, R.N.; Arenz, C.; Ishmuratova, M.Y.; Toppet, S.; Dehaen, W.; Alsfouk, B.A.; Elkaeed, E.B.; Eissa, I.H. Jusanin, a New Flavonoid from Artemisia commutata with an In Silico Inhibitory Potential against the SARS-CoV-2 Main Protease. Molecules 2022, 27, 1636. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Elkady, H.; Alsaif, N.A.; Obaidullah, A.J.; Alanazi, W.A.; Al-Hossaini, A.M.; Alharbi, M.A.; Eissa, I.H.; Dahab, M.A. Discovery of new quinoxaline-based derivatives as anticancer agents and potent VEGFR-2 inhibitors: Design, synthesis, and in silico study. J. Mol. Struct. 2021, 1253, 132220. [Google Scholar] [CrossRef]
- Suleimen, Y.M.; Jose, R.A.; Suleimen, R.N.; Ishmuratova, M.Y.; Toppet, S.; Dehaen, W.; Alsfouk, A.A.; Elkaeed, E.B.; Eissa, I.H.; Metwaly, A.M. Isolation and In Silico SARS-CoV-2 Main Protease Inhibition Potential of Jusan Coumarin, a New Dicoumarin from Artemisia glauca. Molecules 2022, 27, 2281. [Google Scholar] [CrossRef]
- Eissa, I.H.; Khalifa, M.M.; Elkaeed, E.B.; Hafez, E.E.; Alsfouk, A.A.; Metwaly, A.M. In Silico Exploration of Potential Natural Inhibitors against SARS-CoV-2 nsp10. Molecules 2021, 26, 6151. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Elkady, H.; Belal, A.; Alsfouk, B.A.; Ibrahim, T.H.; Abdelmoaty, M.; Arafa, R.K.; Metwaly, A.M.; Eissa, I.H. Multi-Phase In Silico Discovery of Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors among 3009 Clinical and FDA-Approved Related Drugs. Processes 2022, 10, 530. [Google Scholar] [CrossRef]
- Suleimen, Y.M.; Jose, R.A.; Suleimen, R.N.; Arenz, C.; Ishmuratova, M.; Toppet, S.; Dehaen, W.; Alsfouk, A.A.; Elkaeed, E.B.; Eissa, I.H. Isolation and In Silico Anti-SARS-CoV-2 Papain-Like Protease Potentialities of Two Rare 2-Phenoxychromone Derivatives from Artemisia spp. Molecules 2022, 27, 1216. [Google Scholar] [CrossRef] [PubMed]
- Alesawy, M.S.; Elkaeed, E.B.; Alsfouk, A.A.; Metwaly, A.M.; Eissa, I.H. In silico screening of semi-synthesized compounds as potential inhibitors for SARS-CoV-2 papain-like protease: Pharmacophoric features, molecular docking, ADMET, toxicity and DFT studies. Molecules 2021, 26, 6593. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, P. Erlotinib: A new therapeutic approach for non-small cell lung cancer. Expert Opin. Investig. Drugs 2003, 12, 1395–1401. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Muhsin, M.; Graham, J.; Kirkpatrick, P. Fresh from the pipeline: Gefitinib. Nat. Rev. Cancer 2003, 3, 556–557. [Google Scholar] [CrossRef]
- Singh, H.; Walker, A.J.; Amiri-Kordestani, L.; Cheng, J.; Tang, S.; Balcazar, P.; Barnett-Ringgold, K.; Palmby, T.R.; Cao, X.; Zheng, N. US Food and Drug Administration Approval: Neratinib for the Extended Adjuvant Treatment of Early-Stage HER2-Positive Breast CancerFDA Approval Summary: Neratinib. Clin. Cancer Res. 2018, 24, 3486–3491. [Google Scholar] [CrossRef]
- Sequist, L.V.; Besse, B.; Lynch, T.J.; Miller, V.A.; Wong, K.K.; Gitlitz, B.; Eaton, K.; Zacharchuk, C.; Freyman, A.; Powell, C. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: Results of a phase II trial in patients with advanced non–small-cell lung cancer. J. Clin. Oncol. 2010, 28, 3076–3083. [Google Scholar] [CrossRef]
- Kim, Y.; Ko, J.; Cui, Z.; Abolhoda, A.; Ahn, J.S.; Ou, S.-H.; Ahn, M.-J.; Park, K. The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor. Mol. Cancer Ther. 2012, 11, 784–791. [Google Scholar] [CrossRef]
- Carroll, J. Following Lethal Tox Report, Boehringer Scraps Plans for High-Speed Development, Kills $730M Hanmi Deal. Available online: https://endpts.com/following-lethal-tox-report-boehringer-scraps-plans-for-high-speed-development-kills-730m-hanmi-deal/ (accessed on 20 May 2019).
- Traxler, P.; Bold, G.; Frei, J.; Lang, M.; Lydon, N.; Mett, H.; Buchdunger, E.; Meyer, T.; Mueller, M.; Furet, P. Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4-(phenylamino) pyrazolo [3, 4-d] pyrimidines. J. Med. Chem. 1997, 40, 3601–3616. [Google Scholar] [CrossRef] [PubMed]
- Ducray, R.; Ballard, P.; Barlaam, B.C.; Hickinson, M.D.; Kettle, J.G.; Ogilvie, D.J.; Trigwell, C.B. Novel 3-alkoxy-1H-pyrazolo [3, 4-d] pyrimidines as EGFR and erbB2 receptor tyrosine kinase inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Gaber, A.A.; Bayoumi, A.H.; El-Morsy, A.M.; Sherbiny, F.F.; Mehany, A.B.; Eissa, I.H. Design, synthesis and anticancer evaluation of 1H-pyrazolo [3, 4-d] pyrimidine derivatives as potent EGFRWT and EGFRT790M inhibitors and apoptosis inducers. Bioorganic Chem. 2018, 80, 375–395. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Ferrarese, A.; Dalla Via, M.; Marzano, C.; Chilin, A.; Marzaro, G. Targeting kinases with anilinopyrimidines: Discovery of N-phenyl-N’-[4-(pyrimidin-4-ylamino) phenyl] urea derivatives as selective inhibitors of class III receptor tyrosine kinase subfamily. Sci. Rep. 2015, 5, 16750. [Google Scholar] [CrossRef]
- Traxler, P.; Furet, P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 1999, 82, 195–206. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; El Hassab, M.A.; Elsayed, Z.M.; Al-Warhi, T.; Elkady, H.; Abo-Ashour, M.F.; Abourehab, M.A.S.; Eissa, I.H.; Abdel-Aziz, H.A. Design, synthesis, in vitro biological assessment and molecular modeling insights for novel 3-(naphthalen-1-yl)-4,5-dihydropyrazoles as anticancer agents with potential EGFR inhibitory activity. Sci. Rep. 2022, 12, 12821. [Google Scholar] [CrossRef]
- Mowafy, S.; Galanis, A.; Doctor, Z.M.; Paranal, R.M.; Lasheen, D.S.; Farag, N.A.; Jänne, P.A.; Abouzid, K.A. Toward discovery of mutant EGFR inhibitors; Design, synthesis and in vitro biological evaluation of potent 4-arylamino-6-ureido and thioureido-quinazoline derivatives. Biorg. Med. Chem. 2016, 24, 3501–3512. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, H.; Wang, L.; Liu, Y.; Knapp, S.; Liu, Q.; Gray, N.S. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS Chem. Biol. 2014, 9, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Caravatti, G.; Lydon, N.; Priestle, J.P.; Sowadski, J.M.; Trinks, U.; Traxler, P. Modelling study of protein kinase inhibitors: Binding mode of staurosporine and origin of the selectivity of CGP 52411. J. Comput.-Aided Mol. Des. 1995, 9, 465–472. [Google Scholar] [CrossRef]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef]
- Nasser, A.A.; Eissa, I.H.; Oun, M.R.; El-Zahabi, M.A.; Taghour, M.S.; Belal, A.; Saleh, A.M.; Mehany, A.B.; Luesch, H.; Mostafa, A.E. Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFR WT and EGFR T790M. Org. Biomol. Chem. 2020, 18, 7608–7634. [Google Scholar] [CrossRef] [PubMed]
- Elzahabi, H.S.; Nossier, E.S.; Alasfoury, R.A.; El-Manawaty, M.; Sayed, S.M.; Elkaeed, E.B.; Metwaly, A.M.; Hagras, M.; Eissa, I.H. Design, synthesis, and anti-cancer evaluation of new pyrido [2, 3-d] pyrimidin-4 (3H)-one derivatives as potential EGFRWT and EGFRT790M inhibitors and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2022, 37, 1053–1076. [Google Scholar] [CrossRef] [PubMed]
- Elmetwally, S.A.; Saied, K.F.; Eissa, I.H.; Elkaeed, E.B. Design, synthesis and anticancer evaluation of thieno [2, 3-d] pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers. Bioorganic Chem. 2019, 88, 102944. [Google Scholar] [CrossRef] [PubMed]
- Belal, A.; Abdel Gawad, N.M.; Mehany, A.B.; Abourehab, M.A.; Elkady, H.; Al-Karmalawy, A.A.; Ismael, A.S. Design, synthesis and molecular docking of new fused 1 H-pyrroles, pyrrolo [3, 2-d] pyrimidines and pyrrolo [3, 2-e][1,4] diazepine derivatives as potent EGFR/CDK2 inhibitors. J. Enzym. Inhib. Med. Chem. 2022, 37, 1884–1902. [Google Scholar] [CrossRef]
- Belal, A.; Elanany, M.A.; Santali, E.Y.; Al-Karmalawy, A.A.; Aboelez, M.O.; Amin, A.H.; Abdellattif, M.H.; Mehany, A.B.; Elkady, H. Screening a Panel of Topical Ophthalmic Medications against MMP-2 and MMP-9 to Investigate Their Potential in Keratoconus Management. Molecules 2022, 27, 3584. [Google Scholar] [CrossRef]
- Abdallah, A.E.; Alesawy, M.S.; Eissa, S.I.; El-Fakharany, E.M.; Kalaba, M.H.; Sharaf, M.H.; Shama, N.M.A.; Mahmoud, S.H.; Mostafa, A.; Al-Karmalawy, A.A. Design and synthesis of new 4-(2-nitrophenoxy) benzamide derivatives as potential antiviral agents: Molecular modeling and in vitro antiviral screening. New J. Chem. 2021, 45, 16557–16571. [Google Scholar] [CrossRef]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J 2012, 448, 417–423. [Google Scholar] [CrossRef]
- Sogabe, S.; Kawakita, Y.; Igaki, S.; Iwata, H.; Miki, H.; Cary, D.R.; Takagi, T.; Takagi, S.; Ohta, Y.; Ishikawa, T. Structure-based approach for the discovery of pyrrolo [3, 2-d] pyrimidine-based EGFR T790M/L858R mutant inhibitors. ACS Med. Chem. Lett. 2012, 4, 201–205. [Google Scholar] [CrossRef]
- Husein, D.Z.; Hassanien, R.; Khamis, M. Cadmium oxide nanoparticles/graphene composite: Synthesis, theoretical insights into reactivity and adsorption study. RSC Adv. 2021, 11, 27027–27041. [Google Scholar] [CrossRef]
- Wang, T.; Husein, D.Z. Novel synthesis of multicomponent porous nano-hybrid composite, theoretical investigation using DFT and dye adsorption applications: Disposing of waste with waste. Environ. Sci. Pollut. Res. 2022, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Illustrated Glossary of Organic Chemistry. Electrostatic Potential Map. Available online: http://www.chem.ucla.edu/~harding/IGOC/E/electrostatic_potential_map.html (accessed on 20 August 2022).
- Norinder, U.; Bergström, C.A. Prediction of ADMET properties. ChemMedChem Chem. Enabling Drug Discov. 2006, 1, 920–937. [Google Scholar] [CrossRef] [PubMed]
- Dearden, J.C. In silico prediction of drug toxicity. J. Comput.-Aided Mol. Des. 2003, 17, 119–127. [Google Scholar] [CrossRef]
- Idakwo, G.; Luttrell, J.; Chen, M.; Hong, H.; Zhou, Z.; Gong, P.; Zhang, C. A review on machine learning methods for in silico toxicity prediction. J. Environ. Sci. Health Part C 2018, 36, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Kruhlak, N.; Benz, R.; Zhou, H.; Colatsky, T. (Q) SAR modeling and safety assessment in regulatory review. Clin. Pharmacol. Ther. 2012, 91, 529–534. [Google Scholar] [CrossRef]
- Zygmunt, M.; Zmudzki, P.; Chlon-Rzepa, G.; Sapa, J.; Pawlowski, M. Synthesis and Analgesic Activity of 3, 7-dimethylpurine-2, 6-dion-1-yl Derivatives of Acetic and Butanoic Acid. Lett. Drug Des. Discov. 2014, 11, 1204–1213. [Google Scholar] [CrossRef]
- Elwan, A.; Abdallah, A.E.; Mahdy, H.A.; Dahab, M.A.; Taghour, M.S.; Elkaeed, E.B.; Mehany, A.B.; Nabeeh, A.; Adel, M.; Alsfouk, A.A. Modified Benzoxazole-Based VEGFR-2 Inhibitors and Apoptosis Inducers: Design, Synthesis, and Anti-Proliferative Evaluation. Molecules 2022, 27, 5047. [Google Scholar] [CrossRef]
- Nossier, E.S.; Alasfoury, R.A.; Hagras, M.; El-Manawaty, M.; Sayed, S.M.; Ibrahim, I.M.; Elkady, H.; Eissa, I.H.; Elzahabi, H.S.A. Modified pyrido[2,3-d]pyrimidin-4(3H)-one derivatives as EGFRWT and EGFRT790M inhibitors: Design, synthesis, and anti-cancer evaluation. J. Mol. Struct. 2022, 1270, 133971. [Google Scholar] [CrossRef]
- Alsaif, N.A.; Taghour, M.S.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Elwan, A.; Elkady, H. Discovery of new VEGFR-2 inhibitors based on bis ([1, 2, 4] triazolo)[4, 3-a: 3’, 4’-c] quinoxaline derivatives as anticancer agents and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1093–1114. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Eissa, I.H.; Alsaif, N.A.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.F.; Albassam, H.; Elkady, H.; Elwan, A. Design, synthesis, docking, ADMET studies, and anticancer evaluation of new 3-methylquinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1760–1782. [Google Scholar] [CrossRef]
- Taghour, M.S.; Mahdy, H.A.; Gomaa, M.H.; Aglan, A.; Eldeib, M.G.; Elwan, A.; Dahab, M.A.; Elkaeed, E.B.; Alsfouk, A.A.; Khalifa, M.M. Benzoxazole derivatives as new VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, in silico studies, and antiproliferative evaluation. J. Enzym. Inhib. Med. Chem. 2022, 37, 2063–2077. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Eissa, I.H.; Elkady, H.; Abdelalim, A.; Alqaisi, A.M.; Alsfouk, A.A.; Elwan, A.; Metwaly, A.M. A Multistage In Silico Study of Natural Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Int. J. Mol. Sci. 2022, 23, 8407. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Youssef, F.S.; Eissa, I.H.; Elkady, H.; Alsfouk, A.A.; Ashour, M.L.; El Hassab, M.A.; Abou-Seri, S.M.; Metwaly, A.M. Multi-step in silico discovery of natural drugs against COVID-19 targeting main protease. Int. J. Mol. Sci. 2022, 23, 6912. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Yousef, R.G.; Elkady, H.; Gobaara, I.M.M.; Alsfouk, B.A.; Husein, D.Z.; Ibrahim, I.M.; Metwaly, A.M.; Eissa, I.H. Design, Synthesis, Docking, DFT, MD Simulation Studies of a New Nicotinamide-Based Derivative: In Vitro Anticancer and VEGFR-2 Inhibitory Effects. Molecules 2022, 27, 4606. [Google Scholar] [CrossRef] [PubMed]
- Gaussian. Available online: https://gaussian.com/citation/ (accessed on 1 July 2022).
- Frisch, M.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Taghour, M.S.; Elkady, H.; Eldehna, W.M.; El-Deeb, N.M.; Kenawy, A.M.; Elkaeed, E.B.; Alsfouk, A.A.; Alesawy, M.S.; Metwaly, A.M.; Eissa, I.H. Design and synthesis of thiazolidine-2, 4-diones hybrids with 1, 2-dihydroquinolones and 2-oxindoles as potential VEGFR-2 inhibitors: In-vitro anticancer evaluation and in-silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1903–1917. [Google Scholar] [CrossRef]





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster Number | H-Is | Amino Acids in EGFR | H-Bs | Amino Acids in EGFR |
|---|---|---|---|---|
| C1 | 1 | F832 | 3 | Lys721-Thr830-Asp831 |
| C2 | 4 | Leu764-Thr766-Phe832-Leu834 | 2 | Lys721-Asp831 |
| C3 | 3 | Leu764-Phe832-Leu834 | 2 | Lys721-Thr830 |
| IP | EA | μ (eV) | χ (eV) | η (eV) | σ (eV) | ω (eV) | µ (D) | TE (eV) | ∆Nmax | ∆E (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| −1.512 | −6.607 | −4.059 | 4.059 | −2.548 | −0.393 | −20.987 | 5.816 | −30469.8 | −1.593 | 20.987 |
| Comp. | FDA Rodent Carcinogenicity (Mouse-Male) | Carcinogenic Potency TD50 (Mouse) a | Ames Mutagenicity | Rat Maximum Tolerated Dose (Feed) b | Rat Oral LD50 b | Rat Chronic LOAEL b | Skin Irritancy | Ocular Irritancy |
|---|---|---|---|---|---|---|---|---|
| The designed compound | Non-Carcinogen | 70.942 | Non-Mutagen | 0.024 | 1.575 | 0.039 | Non-Irritant | Mild |
| Erlotinib | 39.771 | 0.083 | 0.662 | 0.036 |
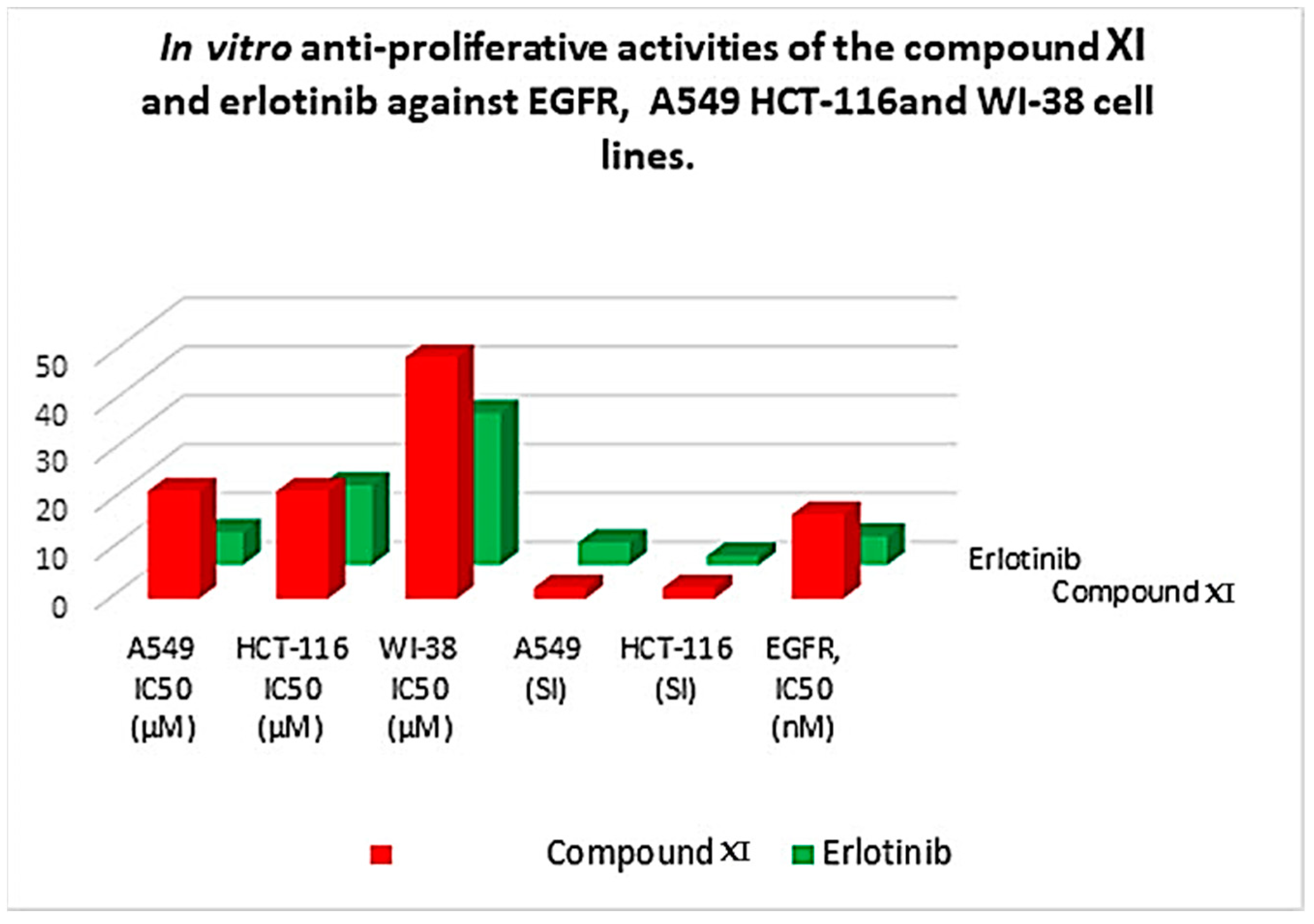
| Comp. | In Vitro Cytotoxicity IC50 (µM) a | A549 (SI) | HCT-116 (SI) | EGFR, IC50 (nM) | ||
|---|---|---|---|---|---|---|
| A549 | HCT-116 | WI-38 | ||||
| Compound XI | 21.99 | 22.02 | 49.44 | 2.2 | 2.2 | 17.23 |
| Erlotinib | 6.73 | 16.35 | 31.17 | 4.6 | 1.9 | 5.91 |
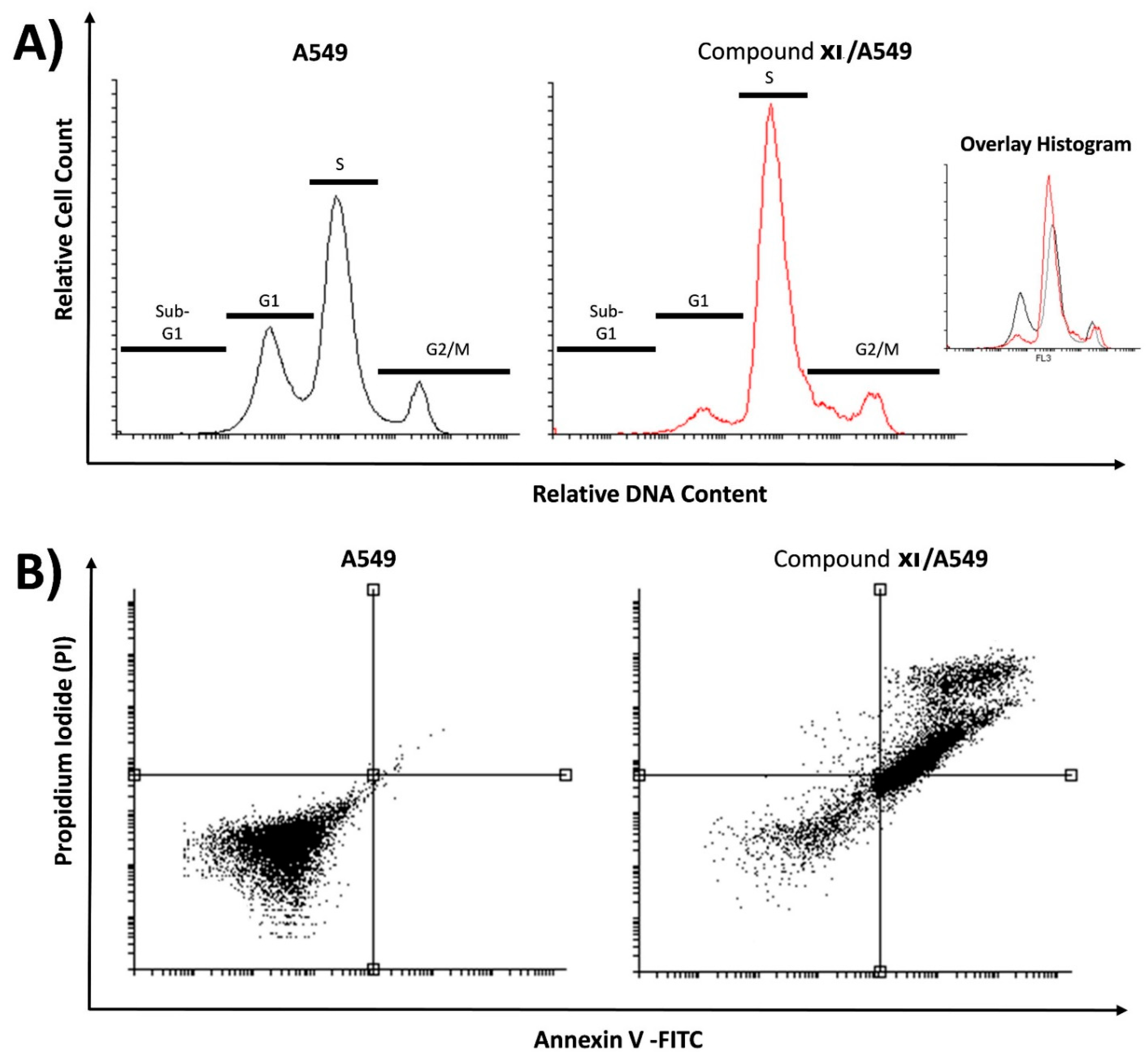
| Sample | Cell Cycle Distribution (%) a | |||
|---|---|---|---|---|
| % Sub-G1 | % G1 | % S | % G2/M | |
| A549 | 0.87 ± 0.35 | 43.47 ± 3.99 | 43.56 ± 2.31 | 12.10 ± 2.04 |
| Compound XI/A549 | 0.80 ± 0.17 | 16.01 ± 5.00 | 63.35 ± 2.50 * | 19.84 ± 2.69 |
| Sample | Viable a (Left Bottom) | Apoptosis a | Necrosis a (Left Top) | |
|---|---|---|---|---|
| Early (Right Bottom) | Late (Right Top) | |||
| A549 | 99.44 ± 0.46 | 0.05 ± 0.02 | 0.49 ± 0.46 | 0.02 ± 0.01 |
| Compound XI/A549 | 32.55 ± 2.98 | 24.02 ± 1.52 ** | 41.70 ± 1.76 ** | 1.73 ± 0.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkaeed, E.B.; Yousef, R.G.; Elkady, H.; Alsfouk, A.A.; Husein, D.Z.; Ibrahim, I.M.; Metwaly, A.M.; Eissa, I.H. New Anticancer Theobromine Derivative Targeting EGFRWT and EGFRT790M: Design, Semi-Synthesis, In Silico, and In Vitro Anticancer Studies. Molecules 2022, 27, 5859. https://doi.org/10.3390/molecules27185859
Elkaeed EB, Yousef RG, Elkady H, Alsfouk AA, Husein DZ, Ibrahim IM, Metwaly AM, Eissa IH. New Anticancer Theobromine Derivative Targeting EGFRWT and EGFRT790M: Design, Semi-Synthesis, In Silico, and In Vitro Anticancer Studies. Molecules. 2022; 27(18):5859. https://doi.org/10.3390/molecules27185859
Chicago/Turabian StyleElkaeed, Eslam B., Reda G. Yousef, Hazem Elkady, Aisha A. Alsfouk, Dalal Z. Husein, Ibrahim M. Ibrahim, Ahmed M. Metwaly, and Ibrahim H. Eissa. 2022. "New Anticancer Theobromine Derivative Targeting EGFRWT and EGFRT790M: Design, Semi-Synthesis, In Silico, and In Vitro Anticancer Studies" Molecules 27, no. 18: 5859. https://doi.org/10.3390/molecules27185859