
Two New Cytotoxic Sesquiterpene-Amino Acid Conjugates and a Coumarin-Glucoside from Crossostephium chinense
 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Structure Determination of Crossoseamines A and B (1 and 2) and a Novel Coumarin Glucoside (3)
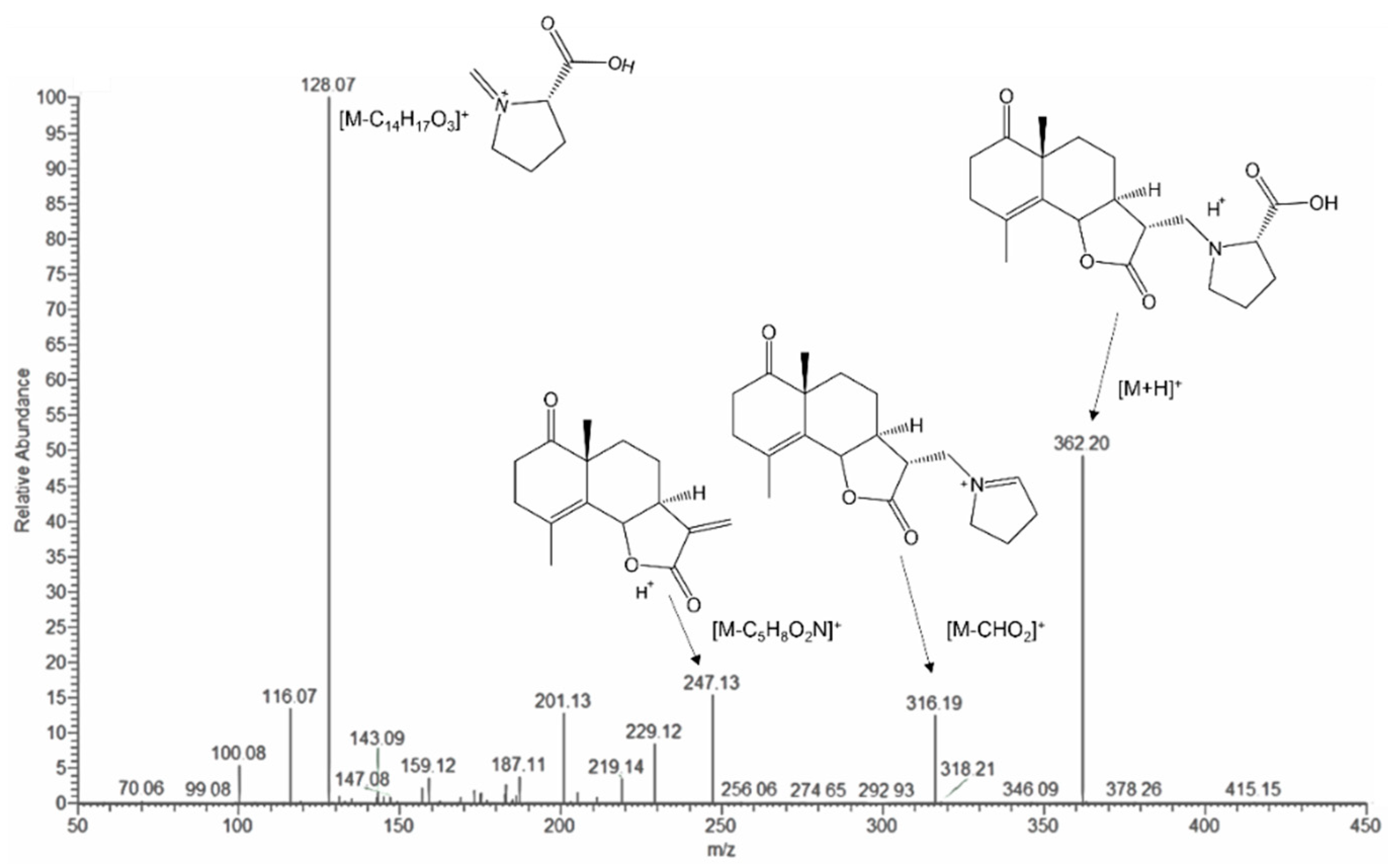
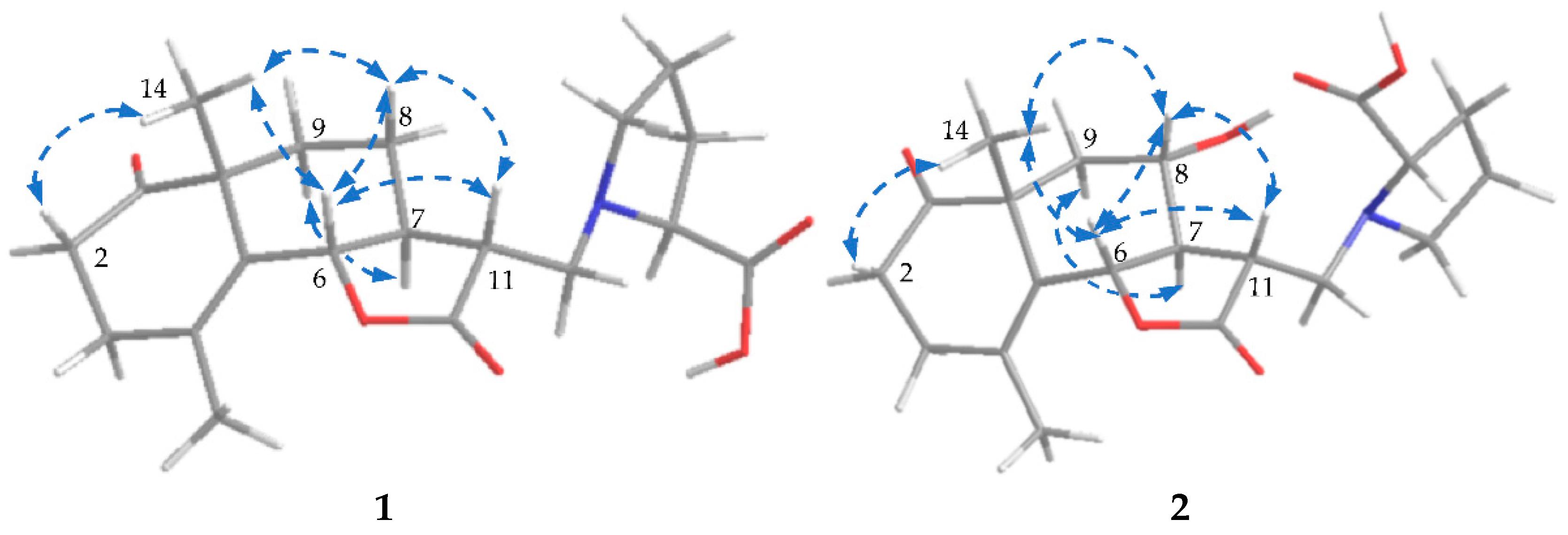
2.1.1. Structure of Crossoseamine A (1)
2.1.2. Structure of Crossoseamine B (2)
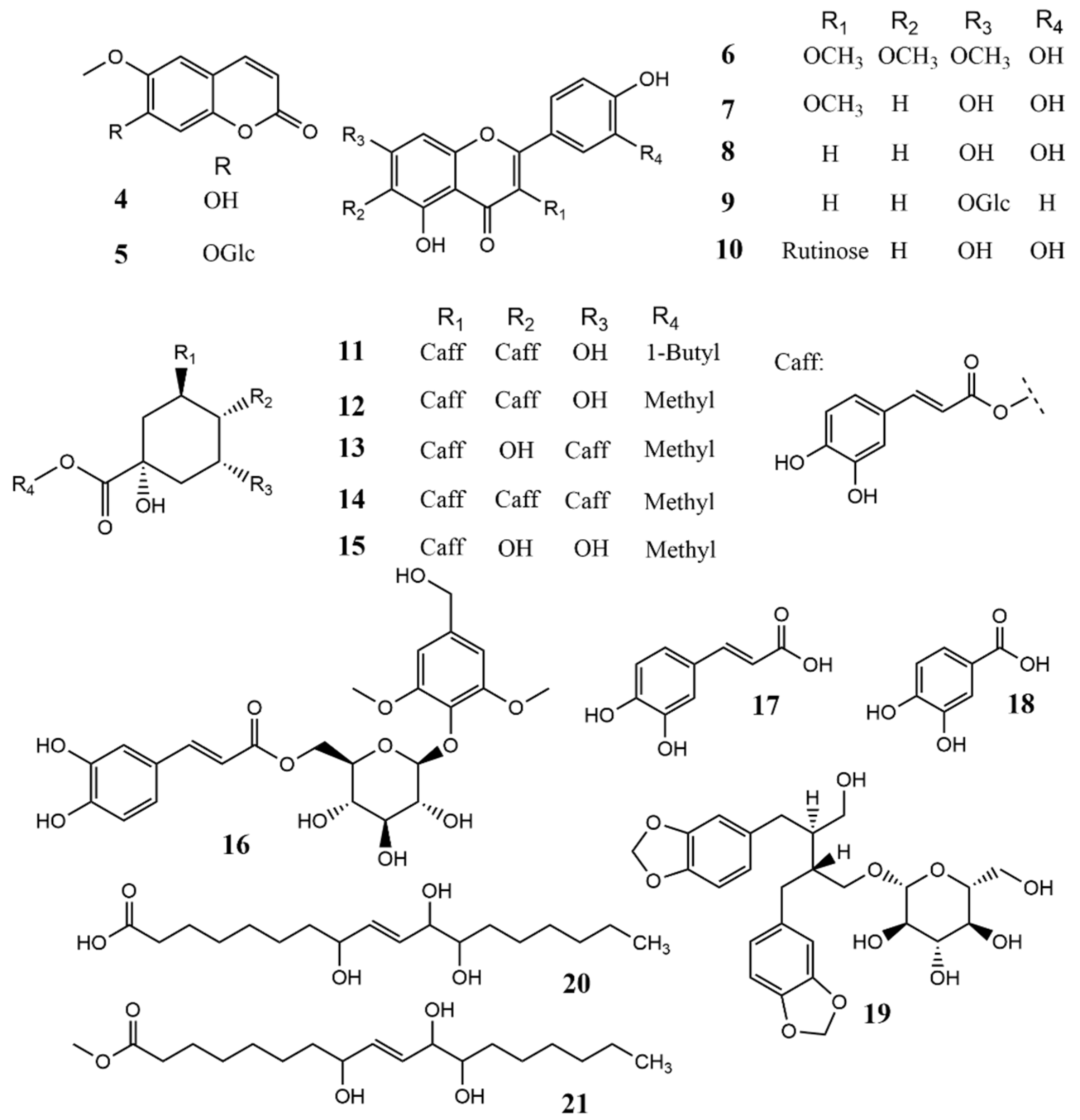
2.1.3. Structure of 6′-O-Caffeoyl Scopolin (3)
2.1.4. Plausible Biosynthetic Pathway of 1 and 2
2.2. Cytotoxic Activities of the Isolated Compounds
3. Experiments
3.1. General Experimental Procedure
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Acid Hydrolysis of Compounds 1 and 2
3.5. Sugar Analysis of Compound 3
3.6. Growth Inhibition Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tang, F.; Chen, F.; Chen, S.; Wang, X.-E.; Zhao, H. Molecular cytogenetic identification and relationship of the artificial intergeneric hybrid between Dendranthema indica and Crossostephium chinense by GISH. Plant Syst. Evol. 2010, 289, 91–99. [Google Scholar] [CrossRef]
- Wu, Q.; Zou, L.; Yang, X.-W.; Fu, D.-X. Novel sesquiterpene and coumarin constituents from the whole herbs of Crossostephium chinense. J. Asian Nat. Prod. Res. 2009, 11, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.-H.; YLin, P.; Wang, J.-Y.; Huang, H.-Y.; Huang, S.-C.; Lo, J.-M.; Wu, C.-H. Preventive Effect and Mechanism of Crossostephium chinense Extract on Balloon Angioplasty-Induced Neointimal Hyperplasia. Evid. Based Complement. Alternat. Med. 2021, 2021, 8466543. [Google Scholar] [CrossRef]
- Wang, S.-W.; Kuo, H.-C.; Hsu, H.-F.; Houng, J.-Y. Inhibitory effect of Crossostephium chinense extract on RANKL-activating osteoclastogenesis in patients with tophaceous gout. Bone Abstr. 2014, 3, PP167. [Google Scholar] [CrossRef]
- Huang, H.-T.; Lin, C.-C.; Kuo, T.-C.; Chen, S.-J.; Huang, R.-N. Phytochemical composition and larvicidal activity of essential oils from herbal plants. Planta 2019, 250, 59–68. [Google Scholar] [CrossRef]
- Uehara, A.; Kitajima, J.; Kokubugata, G.; Iwashina, T. Further characterization of foliar flavonoids in Crossostephium chinense and their geographic variation. Nat. Prod. Commun. 2014, 9, 1934578X1400900207. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, Q.; Yang, X.W.; Yang, X.; Wang, K. The membrane transport of flavonoids from Crossostephium chinense across the Caco2 monolayer. Biopharm. Drug Dispos. 2011, 32, 16–24. [Google Scholar] [CrossRef]
- Yang, X.-W.; Zou, L.; Wu, Q.; Fu, D.-X. Studies on chemical constituents from whole plants of Crossostephium chinense. China J. Chin. Mater. Med. 2008, 33, 905–908. [Google Scholar]
- Sasaki, S.-I.; Aoyagi, S.; Hsii, H.-Y. The isolation of taraxerol, taraxeryl acetate, and taraxerone from Crossostephium chinense Makino (Compositae). Chem. Pharm. Bull. 1965, 13, 87–88. [Google Scholar] [CrossRef] [Green Version]
- Alesha, A.T.; Benjamin, J.S.; Lecia, V.S.; Justin, F.G.; Rebecca, S.H. Lung cancer. Lancet 2021, 398, 535–554. [Google Scholar]
- Asaumi, S.; Kawakami, S.; Sugimoto, S.; Matsunami, K.; Otsuka, H.; Shinzato, T. Alkylated Benzoquinones: Ardisiaquinones A–H from the Leaves of Ardisia quinquegona and Their Anti-Leishmania Activity. Chem. Pharm. Bull. 2018, 66, 757–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nok, A.J. Arsenicals (melarsoprol), pentamidine and suramin in the treatment of human African trypanosomiasis. Parasitol. Res. 2003, 90, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kuzoe, F.A. Current situation of African trypanosomiasis. Acta Trop. 1993, 54, 153–162. [Google Scholar] [CrossRef]
- Balikagala, B.; Fukuda, N.; Ikeda, M.; Katuro, O.T.; Tachibana, S.-I.; Yamauchi, M.; Opio, W.; Emoto, S.; Anywar, D.A.; Kimura, E.; et al. Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med. 2021, 385, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Woodrow, C.J.; White, N.J. The clinical impact of artemisinin resistance in Southeast Asia and the potential for future spread. FEMS Microbiol. Rev. 2017, 41, 34–48. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.; Omar, H.; Michael, B.; Srikkanth, B.; Tobias, A.O.; Ulrike, H. Improving anti-trypanosomal activity of alkamides isolated from Achillea fragrantissima. Fitoterapia 2018, 125, 191–198. [Google Scholar]
- Takeara, S.R.; Lopes, A.N.P.; Lopes, J.L.C. Trypanocidal activity of Lychnophora staavioides Mart. (Vernonieae, Asteraceae). Phytomedicine 2003, 10, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Taleb-Contini, S.H.; Salvador, M.J.; Balanco, J.M.F.; Albuquerque, S.; de Oliveira, D.C.R. Antiprotozoal effect of crude extracts and flavonoids isolated from Chromolaena hirsuta (asteraceae). Phytother. Res. 2004, 18, 250–254. [Google Scholar] [CrossRef]
- Stipan, N.; Buyankhishig, B.; Suganuma, K.; Ishikawa, Y.; Kutsuma, M.; Abe, M.; Sasaki, K.; Davaapurev, B.O.; Batkhuu, J.; Murata, T. Phytochemical investigation of Scutellaria scordiifolia and its trypanocidal activity. Phytochemistry 2023, 209, 11361. [Google Scholar]
- Mamadalieva, N.Z.; Herrmann, F.; El-Readi, M.Z.; Tahrani, A.; Hamoud, R.; Egamberdieva, D.R.; Azimova, S.S.; Wink, M. Flavonoids in Scutellaria immaculata and S. ramosissima (Lamiaceae) and their biological activity. J. Pharm. Pharmacol. 2011, 63, 1346–1357. [Google Scholar] [CrossRef]
- Hashim, Y.; Toume, K.; Mizukami, S.; Kitami, T.; Taniguchi, M.; Teklemichael, A.A.; Tayama, Y.; Huy, N.T.; Lami, J.N.; Bodi, J.M.; et al. Phenylpropanoid-conjugated iridoid glucosides from leaves of Morinda morindoides. J. Nat. Med. 2022, 76, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.C.; Yang, S.L.; Roberts, M.F.; Elford, B.C.; Phillipson, J.D. Antimalarial activity of Artemisia annua flavonoids from whole plants and cell cultures. Plant Cell Rep. 1992, 11, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Ogunlana, O.O.; Kim, H.S.; Wataya, Y.; Olagunju, J.O.; Akindahunsi, A.; Tan, N.H. Antiplasmodial flavonoid from young twigs and leaves of Caesalpinia bonduc (Linn) Roxb. J. Chem. Pharm. Res. 2015, 7, 931–937. [Google Scholar]
- Vitalini, S.; Beretta, G.; Iriti, M.; Orsenigo, S.; Basilico, N.; Dall’Acqua, S.; Iorizzi, M.; Fico, G. Phenolic compounds from Achillea millefolium L. and their bioactivity. Acta Biochim. Pol. 2011, 58, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Sangsopha, W.; Lekphrom, R.; Kanokmedhakul, S.; Kanokmedhakul, K. Cytotoxic and antimalarial constituents from aerial parts of Sphaeranthus indicus. Phytochem. Lett. 2016, 17, 278–281. [Google Scholar] [CrossRef]
- Lagnika, L.; Weniger, B.; Attioua, B.; Jensen, O.; Anthaume, C.; Sanni, A.; Kaiser, M.; Lobstein, A.; Vonthron-Senecheau, C. Trypanocidal activity of diarylheptanoids from Schrankia leptocarpa DC. S. Afr. J. Bot. 2012, 83, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Aldulaimi, O.; Uche, F.I.; Hameed, H.; Mbye, H.; Ullah, I.; Drijfhout, F.; Claridge, T.D.W.; Horrocks, P.; Li, W.W. A characterization of the antimalarial activity of the bark of Cylicodiscus gabunensis Harms. J. Ethnopharmacol. 2017, 198, 221–225. [Google Scholar] [CrossRef]
- Vasconcelos, J.M.J.; Silva, A.M.S.; Cavaleiro, J.A.S. Chromones and flavanones from artemisia campestris subsp. maritima. Phytochemistry 1998, 49, 1421–1424. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Hisada, S.; Nishibe, S. Coumarin and Secoiridoid Glucosides from Bark of Olea africana and Olea capensis. Chem. Pharm. Bull. 1985, 33, 396–399. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Zhang, L.-X.; Zhao, Y.; Huang, G.-D. Unusual sesquiterpene lactones with a new carbon skeleton and new acetylenes from Ajania przewalskii. Food Chem. 2010, 118, 228–238. [Google Scholar] [CrossRef]
- Marco, J.A.; Barberá, O.; Rodríguez, S.; Domingo, C.; Adell, J. Flavonoids and other phenolics from Artemisia hispanica. Phytochemistry 1988, 27, 3155–3159. [Google Scholar] [CrossRef]
- Lee, E.H.; Kim, H.J.; Song, Y.S.; Jin, C.; Lee, K.-T.; Cho, J.; Lee, Y.S. Constituents of the stems and fruits of Opuntia ficus-indica var. saboten. Arch. Pharm. Res. 2003, 26, 1018–1023. [Google Scholar] [CrossRef]
- Jung, M.J.; Kang, S.S.; Jung, H.A.; Kim, G.J.; Choi, J.S. Isolation of flavonoids and a cerebroside from the stem bark of Albizzia julibrissin. Arch. Pharm. Res. 2004, 27, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, C.; Formentini, L.; Santaniello, E. Apigenin 7-glucoside diacetates in ligulate flowers of Matricaria chamomilla. Phytochemistry 1982, 21, 1828–1830. [Google Scholar] [CrossRef]
- Kazuma, K.; Noda, N.; Suzuki, M. Malonylated flavonol glycosides from the petals of Clitoria ternatea. Phytochemistry 2003, 62, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Xiao-Li, M.A.; Lin, W.B. Chemical Constituents of Osmanthus yunnanensis. Nat. Prod. Res. Dev. 2009, 21, 593–599. [Google Scholar]
- Nugroho, A.; Lee, K.R.; Alam, M.B.; Choi, J.S.; Park, H.J. Isolation and quantitative analysis of peroxynitrite scavengers from Artemisia princeps var. orientalis. Arch. Pharm. Res. 2010, 33, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Cho, J.-Y.; Ma, Y.-K.; Park, K.Y.; Lee, S.-H.; Ham, K.-S.; Lee, H.J.; Park, K.-H.; Moon, J.-H. Dicaffeoylquinic acid derivatives and flavonoid glucosides from glasswort (Salicornia herbacea L.) and their antioxidative activity. Food Chem. 2011, 125, 55–62. [Google Scholar]
- Merfort, I. Caffeoylquinic acids from flowers of Arnica montana and Arnica chamissonis. Phytochemistry 1992, 31, 2111–2113. [Google Scholar] [CrossRef]
- Zhu, X.; Dong, X.; Wang, Y.; Ju, P.; Luo, S. Phenolic Compounds from Viburnum cylindricum. Helv. Chim. Acta 2005, 88, 339–342. [Google Scholar] [CrossRef]
- Yang, P.-F.; Yang, Y.-N.; Feng, Z.-M.; Jiang, J.-S.; Zhang, P.-C. Six new compounds from the flowers of Chrysanthemum morifolium and their biological activities. Bioorg. Chem. 2019, 82, 139–144. [Google Scholar] [CrossRef]
- Nakazawa, T.; Ohsawa, K. Metabolism of Rosmarinic Acid in Rats. J. Nat. Prod. 1998, 61, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Nagatsu, A.; Okuyama, H.; Mizukami, H.; Sakakibara, J. Sesquiterpene glycosides from cotton oil cake. Phytochemistry 1998, 48, 665–668. [Google Scholar] [CrossRef]
- Yi, J.-H.; Zhang, G.-L.; Li, B.-G.; Chen, Y.-Z. Two glycosides from the stem bark of Tetracentron sinense. Phytochemistry 2000, 53, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wong, M.; Wang, N.; Chan, A.S.C.; Yao, X. A New Eudesmane Derivative and a New Fatty Acid Ester from Sambucus williamsii. Chem. Pharm. Bull. 2006, 54, 676–678. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, H.; Kageura, T.; Inoue, Y.; Morikawa, T.; Yoshikawa, M. Absolute Stereostructures and Syntheses of Saussureamines A, B, C, D and E, Amino Acid–Sesquiterpene Conjugates with Gastroprotective Effect, from the Roots of Saussurea lappa. Tetrahedron 2000, 56, 7763–7777. [Google Scholar] [CrossRef]
- Moiseeva, G.P.; Kasymov, S.Z.; Yagudaev, M.R.; Sidyakin, G.P. Determination of the absolute configuration of the asymmetric center at C11 in the eudesmanolides. Chem. Nat. Compd. 1980, 16, 254–256. [Google Scholar] [CrossRef]
- Zhang, D.M.; Wang, L.L.; Li, J.; Hu, L.H. Two new coumarins from Fraxinus chinensis Rexb. J. Integr. Plant Biol. 2007, 49, 218–221. [Google Scholar] [CrossRef]
- He, R.; Huang, X.; Zhang, Y.; Wu, L.; Nie, H.; Zhou, D.; Liu, B.; Deng, S.; Yang, R.; Huang, S.; et al. Structural Characterization and Assessment of the Cytotoxicity of 2,3-Dihydro-1H-indene Derivatives and Coumarin Glucosides from the Bark of Streblus indicus. J. Nat. Prod. 2016, 79, 2472–2478. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Hatakeyama, S.; Inoue, Y.; Yamahara, J. Saussureamines a, b, c, d, and e, new anti-ulcer principles from chinese saussureae radix. Chem. Pharm. Bull. 1993, 41, 214–216. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, S.; Yamano, Y.; Desoukey, S.Y.; Katakawa, K.; Wanas, A.S.; Otsuka, H.; Matsunami, K. Isolation of Sesquiterpene–Amino Acid Conjugates, Onopornoids A–D, and a Flavonoid Glucoside from Onopordum alexandrinum. J. Nat. Prod. 2019, 82, 1471–1477. [Google Scholar] [CrossRef]
- Mahmoud, B.K.; Samy, M.N.; Hamed, A.N.E.; Abdelmohsen, U.R.; Hajjar, D.; Yamano, Y.; Sugimoto, S.; Matsunami, K.; Kamel, M.S. Bignanoside A “A new neolignan glucoside” and bignanoside B “A new iridoid glucoside” from Bignonia binata leaves. Phytochem. Lett. 2020, 35, 200–205. [Google Scholar] [CrossRef]
- Suganuma, K.; Allamanda, P.; Hakimi, H.; Zhou, M.; Angeles, J.M.; Kawazu, S.; Inoue, N. Establishment of ATP-based luciferase viability assay in 96-well plate for Trypanosoma congolense. J. Vet. Med. Sci. 2014, 76, 1437–1441. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Mizuta, S.; Kaneko, O.; Yahata, K. Fragment Molecular Orbital Study of the Interaction between Sarco/Endoplasmic Reticulum Ca2+-ATPase and its Inhibitor Thapsigargin toward Anti-Malarial Development. J. Phys. Chem. B 2018, 122, 7970–7977. [Google Scholar] [CrossRef]
- Walliker, D.; Quakyi, I.A.; Wellems, T.E.; McCutchan, T.F.; Szarfman, A.; London, W.T.; Corcoran, L.M.; Burkot, T.R.; Carter, R. Genetic Analysis of the Human Malaria Parasite Plasmodium falciparum. Science 1987, 236, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Khalid, S.A.; Farouk, A.; Geary, T.G.; Jensen, J.B. Potential anti-malarial candidates from African plants: An in vitro approach using Plasmodium falciparum. J. Ethnopharmacol. 1986, 15, 201–209. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| Position | 13C | 1H | 13C | 1H |
| 1 | 212.9 | 212.2 | ||
| 2 | 35.5 | 2.43 (ddd, 13.6, 7.8, 6.6) α | 35.2 | 2.40 (m) α |
| 2.55 (ddd, 13.6, 7.0, 6.2) β | 2.58 (m) β | |||
| 3 | 32.3 | 2.35 (br dt, 17.0, 6.2) α | 32.4 | 2.37 (m) α |
| 2.26 (br dt, 17.0, 6.9) β | 2.27 (m) β | |||
| 4 | 125.2 | 126.1 | ||
| 5 | 130.0 | 128.6 | ||
| 6 | 80.8 | 4.80 (dquint-like, 11.2, 1.3) | 77.9 | 4.87 (dquint-like, 11.4, 1.1) |
| 7 | 48.5 | 2.02 (m) | 57.6 | 1.88 (m) |
| 8 | 23.1 | 1.95 (m) α | 66.0 | 3.89 (td, 10.8, 4.5) |
| 1.63 (m) β | ||||
| 9 | 34.6 | 1.44 (td, 14.0, 5.1) α | 43.1 | 1.25 (br t, 11.8) α |
| 1.63 (m) β | 1.87 (m) β | |||
| 10 | 48.4 | 47.4 | ||
| 11 | 44.2 | 2.73 (dt-like, 12.1, 5.4) | 44.3 | 3.01 (m) |
| 12 | 177.0 | 175.8 | ||
| 13 | 51.2 | 2.86 (dd, 13.2, 5.1) | 52.8 | 2.92 (m) |
| 3.08 (dd, 13.2, 5.7) | 3.15 (m) | |||
| 14 | 23.0 | 1.25 (3H, s) | 24.1 | 1.24 (3H, s) |
| 15 | 19.4 | 1.86 (3H, br s) | 19.3 | 1.87 (3H, br s) |
| 2′ | 66.6 | 3.30 (dd, 8.7, 4.6) | 66.3 | 3.45 (dd, 9.0, 3.4) |
| 3′ | 28.6 | 1.81 (m) | 28.9 | 1.90 (m) |
| 2.03 (m) | 2.07 (dq-like, 12.8, 8.8) | |||
| 4′ | 23.4 | 1.68–1.79 (2H, m) | 23.1 | 1.74 (m) |
| 1.80 (m) | ||||
| 5′ | 53.2 | 2.58 (dd-like, 9.0, 7.8) | 52.8 | 2.84 (dt, 9.5, 7.5) |
| 2.99 (ddd, 9.0, 7.5, 3.9) | 3.00 (m) | |||
| 6′ | 173.9 | 173.6 | ||
| 3 | |||||
|---|---|---|---|---|---|
| Position | 13C | 1H | Position | 13C | 1H |
| 2 | 161.6 | 4′ | 71.6 | 4.24 (br t, 8.8) | |
| 3 | 114.5 | 6.32 (d, 9.5) | 5′ | 76.1 | 4.37 (ddd, 9.5, 6.5, 1.9) |
| 4 | 144.0 | 7.65 (d, 9.5) | 6′ | 64.6 | 4.91 (dd, 11.9, 6.5) |
| 4a | 113.5 | 5.11 (dd, 11.9, 1.9) | |||
| 5 | 110.2 | 7.01 (s) | 1″ | 127.1 | |
| 6 | 147.4 | 2″ | 116.5 | 7.570 (d, 1.8) | |
| 7 | 151.5 | 3″ | 147.8 | ||
| 8 | 104.6 | 7.568 (s) | 4″ | 149.8 | |
| 8a | 150.8 | 5″ | 117.3 | 7.31 (d, 8.3) | |
| 6-OCH3 | 56.5 | 3.69 (3H, s) | 6″ | 122.5 | 7.27 (dd, 8.3, 1.8) |
| 1′ | 102.3 | 5.80 (d, 7.3) | 7″ | 146.6 | 7.92 (d, 15.9) |
| 2′ | 74.9 | 4.42 (dd-like, 8.8, 7.3) | 8″ | 114.9 | 6.69 (d, 15.9) |
| 3′ | 78.7 | 4.44 (br t, 8.8) | 9″ | 167.9 | |
| Compounds | A549 | L. major | T. b. gambiense | T. b. rhodesiense | P. falciparum |
|---|---|---|---|---|---|
| 1 | 3.3 ± 0.3 | 6.9 ± 0.6 | 26.3 ± 8.5 | 27.8 ± 10.4 | 12.1 ± 1.1 |
| 2 | 12.3 ± 1.0 | 24.9 ± 2.2 | 39.5 ± 6.9 | 41.0 ± 8.9 | 15.6 ± 1.2 |
| 6 | 4.4 ± 1.5 | 5.4 ± 0.8 | 2.6 ± 0.1 | 1.7 ± 0.3 | 6.0 ± 1.1 |
| 7 | 17.7 ± 4.4 | 17.5 ± 1.2 | 4.0 ± 0.2 | 3.2 ± 0.3 | 16.5 ± 1.1 |
| 8 | 19.9 ± 5.5 | 26.9 ± 2.8 | 4.9 ± 0.3 | 3.2 ± 0.4 | 11.0 ±1.3 |
| 9 | 78.3 ± 15.6 | 58.9 ± 7.5 | 16.9 ± 0.7 | 8.6 ± 1.2 | 8.9 ± 1.0 |
| 10 | >100 | 90.9 ± 5.5 | >100 | >100 | 37.0 ± 1.1 |
| 11 | 73.5 ± 13.0 | >100 | 47.4 ± 2.7 | 34.8 ± 4.6 | 5.4 ± 1.1 |
| 12 | 115.0 ± 19.3 | >100 | 97.1 ± 0.2 | 68.9 ± 14.2 | 4.6 ± 1.1 |
| 13 | 118.2 ± 19.7 | >100 | >100 | 80.5 ± 18.4 | 10.4 ± 1.2 |
| 14 | 42.6 ± 6.9 | 58.3 ± 6.6 | 11.0 ± 0.1 | 12.4 ± 0.8 | 3.6 ± 1.1 |
| 15 | 79.4 ± 8.7 | >100 | 83.8 ± 13.3 | 53.6 ± 2.2 | 10.2 ± 1.1 |
| 16 | >100 | >100 | >100 | >100 | 35.5 ± 1.0 |
| 17 | 42.5 ± 0.1 | >100 | 53.2 ± 7.5 | 42.3 ±2.9 | 25.0 ± 1.1 |
| 18 | >100 | >100 | 19.6 ± 2.7 | 24.8 ± 1.2 | 15.9 ± 1.2 |
| 19 | >100 | >100 | >100 | 55.2 ± 0.8 | 30.6 ± 1.1 |
| 20 | >100 | >100 | 44.1 ± 3.0 | 32.8 ± 1.8 | 26.1 ± 1.1 |
| 21 | >100 | >100 | 58.7 ± 3.0 | 36.0 ± 0.9 | 27.1 ± 1.1 |
| PC | 17.5 ± 0.3 | 7.4 ± 0.7 | 1.3 ± 0.7 | 1.3 ± 0.5 | 0.0023 ± 0.0003 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Chitama, B.-Y.A.; Suganuma, K.; Yamano, Y.; Sugimoto, S.; Kawakami, S.; Kaneko, O.; Otsuka, H.; Matsunami, K. Two New Cytotoxic Sesquiterpene-Amino Acid Conjugates and a Coumarin-Glucoside from Crossostephium chinense. Molecules 2023, 28, 4696. https://doi.org/10.3390/molecules28124696
Wang Z, Chitama B-YA, Suganuma K, Yamano Y, Sugimoto S, Kawakami S, Kaneko O, Otsuka H, Matsunami K. Two New Cytotoxic Sesquiterpene-Amino Acid Conjugates and a Coumarin-Glucoside from Crossostephium chinense. Molecules. 2023; 28(12):4696. https://doi.org/10.3390/molecules28124696
Chicago/Turabian StyleWang, Zhichao, Ben-Yeddy Abel Chitama, Keisuke Suganuma, Yoshi Yamano, Sachiko Sugimoto, Susumu Kawakami, Osamu Kaneko, Hideaki Otsuka, and Katsuyoshi Matsunami. 2023. "Two New Cytotoxic Sesquiterpene-Amino Acid Conjugates and a Coumarin-Glucoside from Crossostephium chinense" Molecules 28, no. 12: 4696. https://doi.org/10.3390/molecules28124696