
As(III, V) Uptake from Nanostructured Iron Oxides and Oxyhydroxides: The Complex Interplay between Sorbent Surface Chemistry and Arsenic Equilibria

 , , , ,
, , , ,  and
and
Abstract
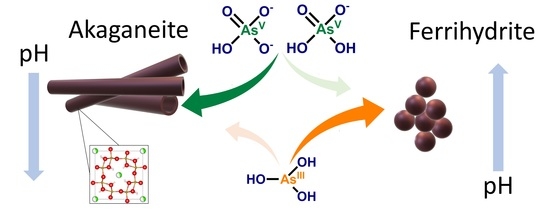
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis of the Sorbents
2.3. Adsorption Tests
2.4. Isotherm Models
2.5. Kinetic Models
2.6. Characterization Techniques
3. Results and Discussion
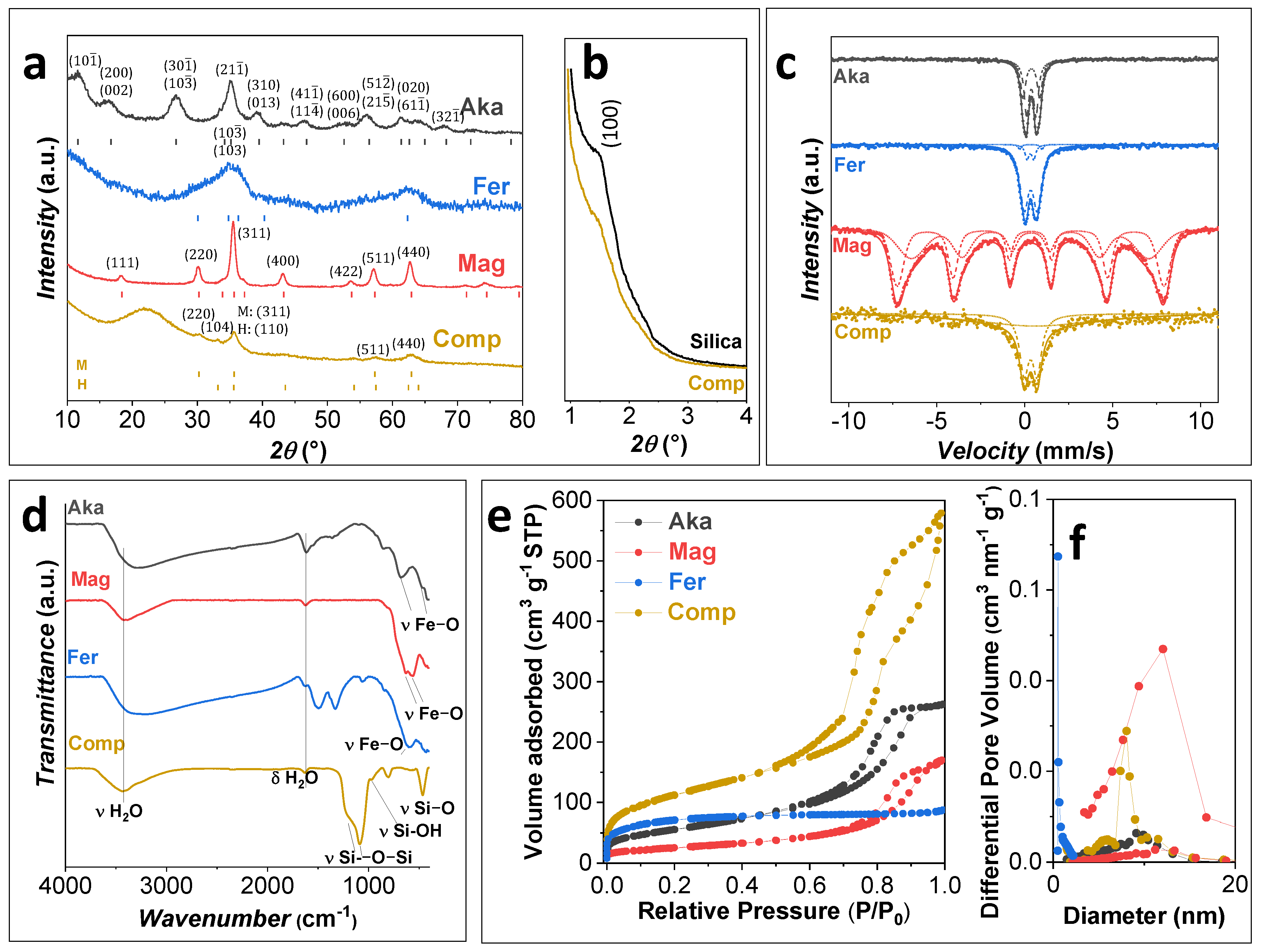
3.1. Characterization of the Sorbents
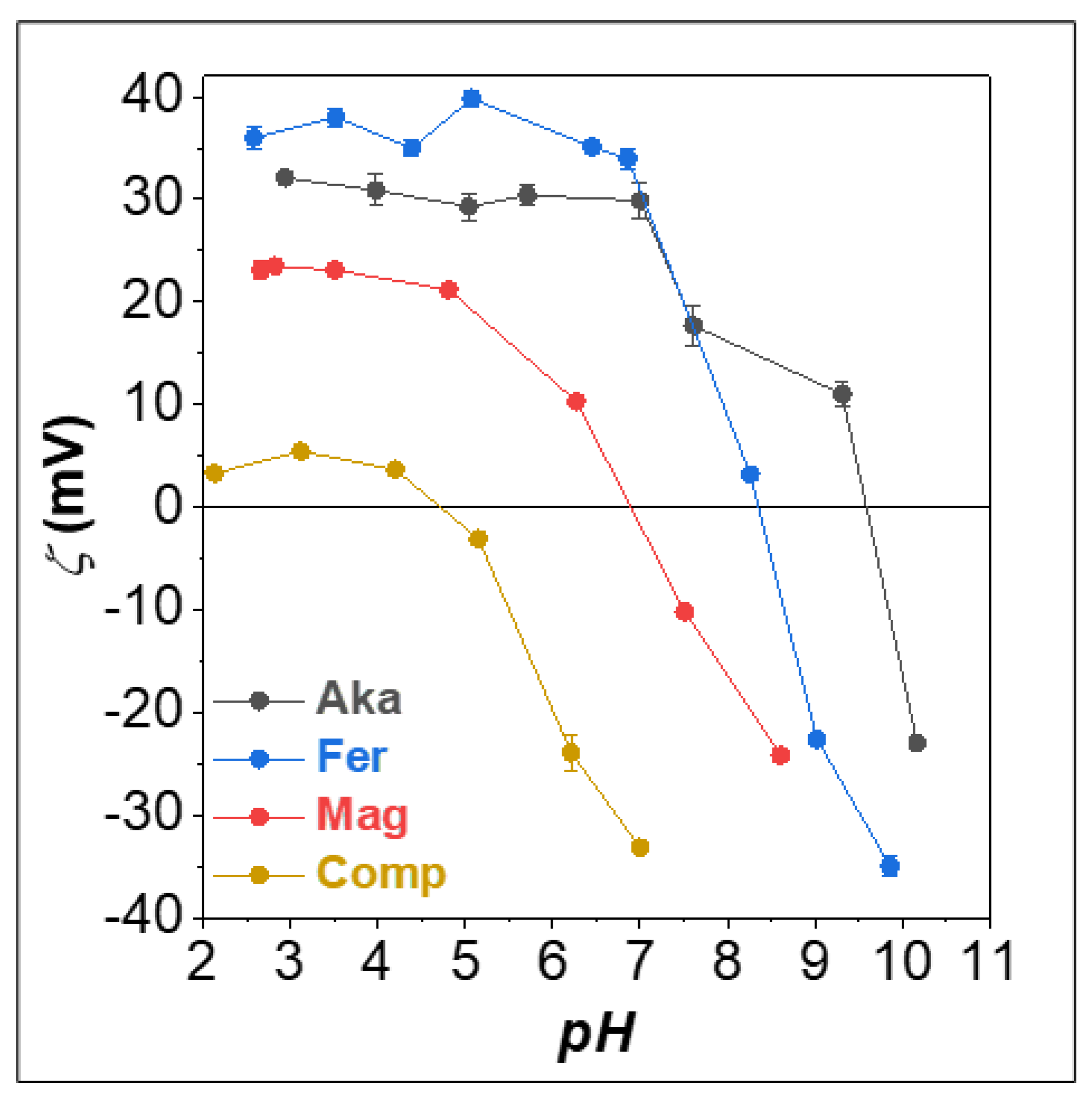
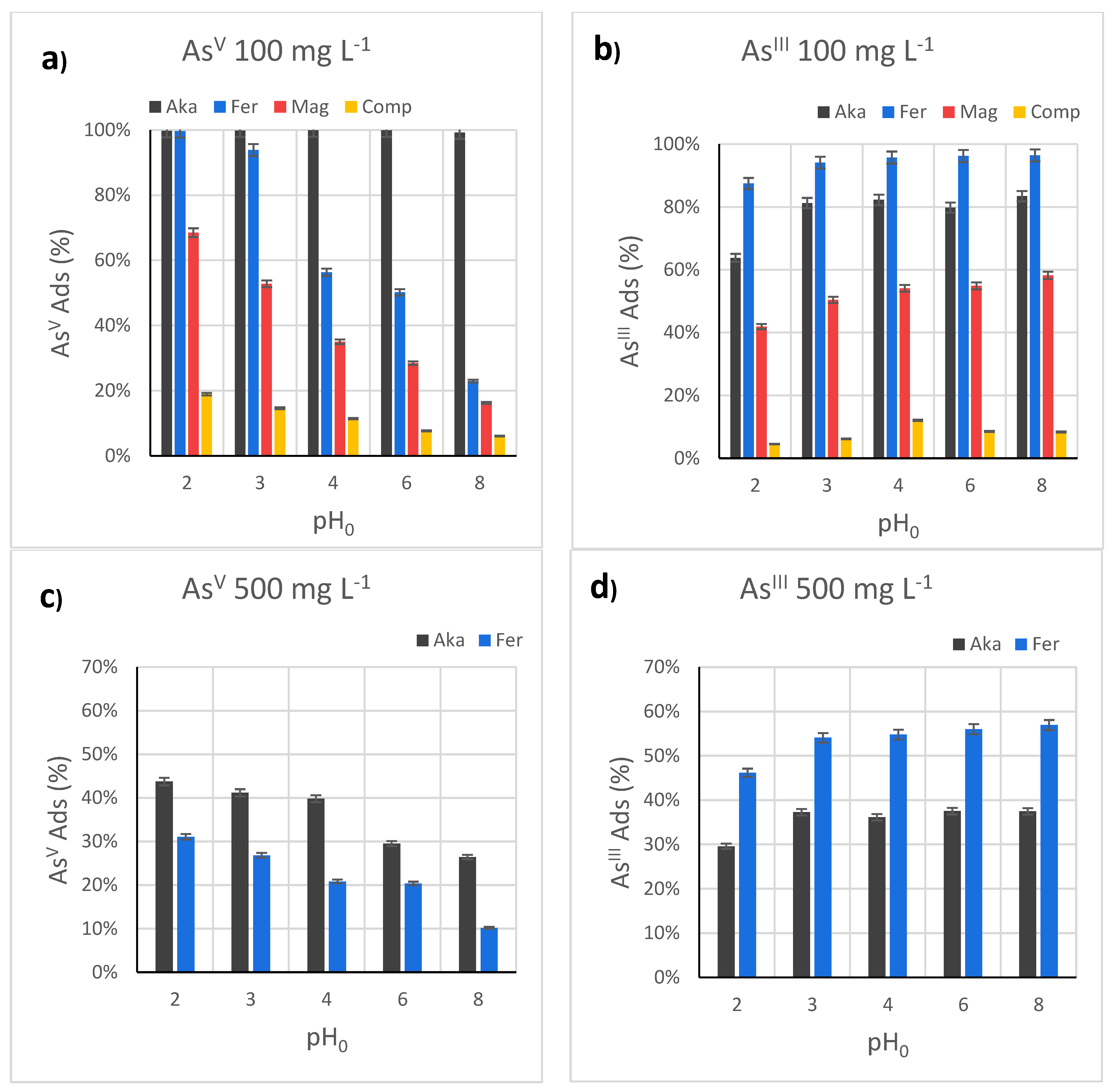
3.2. Effect of the pH in the AsV and AsIII Test Removal by FeIII-Based Sorbents
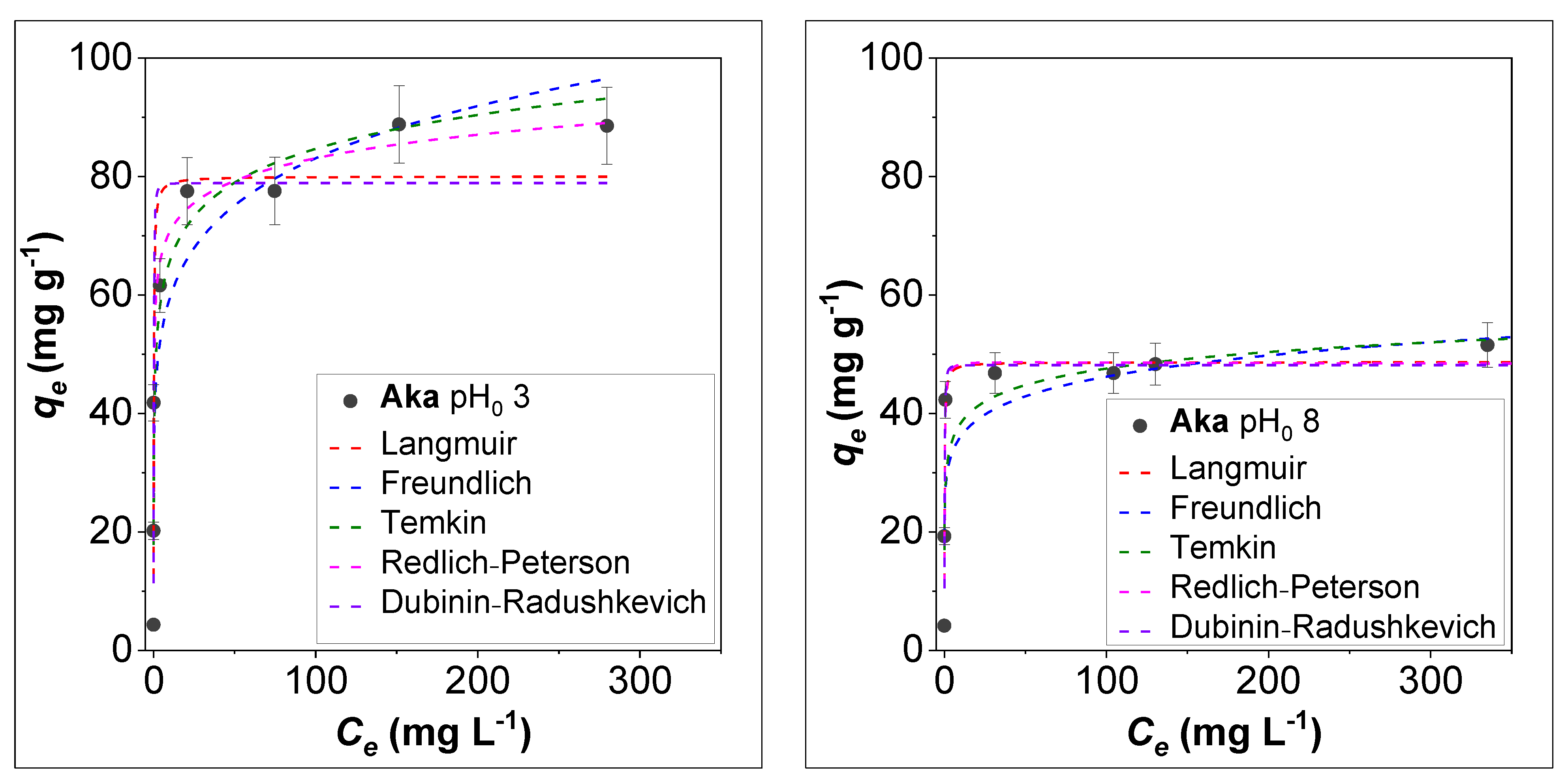
3.3. Effect of Initial Concentration and Isotherm Modelling on the Adsorption of AsV by Akaganeite
3.4. Effect of Contact Time and Kinetic Modelling on the Adsorption of AsV by Akaganeite
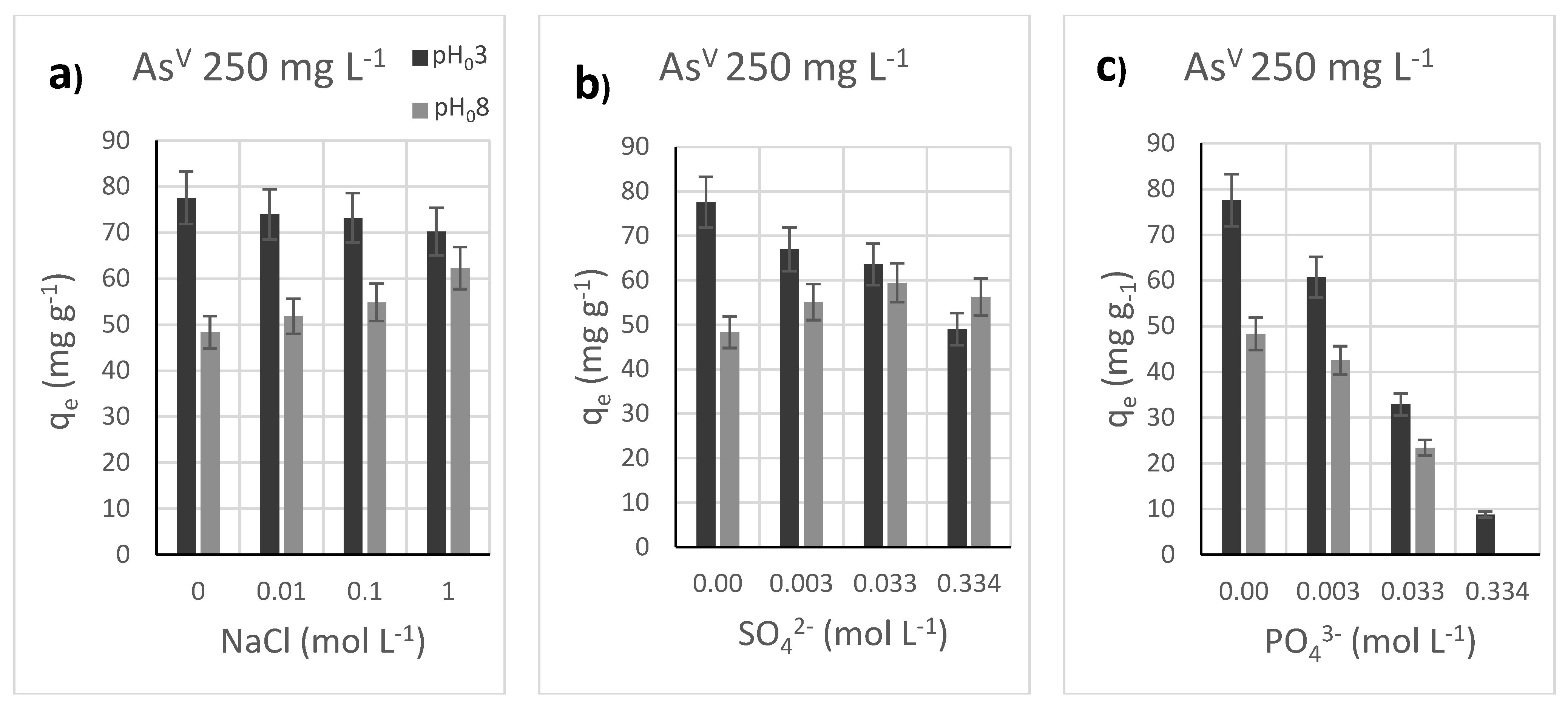
3.5. Effect of Added Salts as Competitors the Adsorption of AsV by Akaganeite
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. 2017 Guidelines for Drinking-Water Quality, 4th ed.; World Health Organization, Ed.; World Health Organization: Geneva, Switzerland, 2017; ISBN 9789241549950. [Google Scholar]
- Liu, B.; Kim, K.H.; Kumar, V.; Kim, S. A review of functional sorbents for adsorptive removal of arsenic ions in aqueous systems. J. Hazard. Mater. 2020, 388, 121815. [Google Scholar] [CrossRef] [PubMed]
- O’Day, P.A. Chemistry and Mineralogy of Arsenic. Elements 2006, 2, 77–83. [Google Scholar] [CrossRef]
- RoyChowdhury, A.; Sarkar, D.; Datta, R. Remediation of Acid Mine Drainage-Impacted Water. Curr. Pollut. Rep. 2015, 1, 131–141. [Google Scholar] [CrossRef]
- Bolisetty, S.; Peydayesh, M.; Mezzenga, R. Sustainable technologies for water purification from heavy metals: Review and analysis. Chem. Soc. Rev. 2019, 48, 463–487. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.S.; Selvarajan, E.; Sarswat, A.; Muthukumar, H.; Jacob, J.M.; Mukesh, M.; Pugazhendhi, A. Nanomaterials as adsorbents for As(III) and As(V) removal from water: A review. J. Hazard. Mater. 2022, 424, 127572. [Google Scholar] [CrossRef] [PubMed]
- Luong, V.T.; Cañas Kurz, E.E.; Hellriegel, U.; Luu, T.L.; Hoinkis, J.; Bundschuh, J. Iron-based subsurface arsenic removal technologies by aeration: A review of the current state and future prospects. Water Res. 2018, 133, 110–122. [Google Scholar] [CrossRef]
- Kobya, M.; Soltani, R.D.C.; Omwene, P.I.; Khataee, A. A review on decontamination of arsenic-contained water by electrocoagulation: Reactor configurations and operating cost along with removal mechanisms. Environ. Technol. Innov. 2020, 17, 100519. [Google Scholar] [CrossRef]
- Kamegawa, T.; Ishiguro, Y.; Seto, H.; Yamashita, H. Enhanced photocatalytic properties of TiO2-loaded porous silica with hierarchical macroporous and mesoporous architectures in water purification. J. Mater. Chem. A 2015, 3, 2323–2330. [Google Scholar] [CrossRef]
- Benhamou, A.; Basly, J.P.; Baudu, M.; Derriche, Z.; Hamacha, R. Amino-functionalized MCM-41 and MCM-48 for the removal of chromate and arsenate. J. Colloid Interface Sci. 2013, 404, 135–139. [Google Scholar] [CrossRef]
- Haldar, D.; Duarah, P.; Purkait, M.K. MOFs for the treatment of arsenic, fluoride and iron contaminated drinking water: A review. Chemosphere 2020, 251, 126388. [Google Scholar] [CrossRef]
- Sarkar, A.; Paul, B. The global menace of arsenic and its conventional remediation—A critical review. Chemosphere 2016, 158, 37–49. [Google Scholar] [CrossRef]
- Wu, X.; Hu, J.; Wu, F.; Zhang, X.; Wang, B.; Yang, Y.; Shen, G.; Liu, J.; Tao, S.; Wang, X. Application of TiO2 nanoparticles to reduce bioaccumulation of arsenic in rice seedlings (Oryza sativa L.): A mechanistic study. J. Hazard. Mater. 2021, 405, 124047. [Google Scholar] [CrossRef]
- Pereira, F.J.; Vázquez, M.D.; Debán, L.; Aller, A.J. Determination of arsenic by ICP-MS after retention on thoria nanoparticles. Anal. Methods 2015, 7, 598–606. [Google Scholar] [CrossRef]
- Jain, A.; Raven, K.P.; Loeppert, R.H. Arsenite and arsenate adsorption on ferrihydrite: Surface charge reduction and net OH- release stoichiometry. Environ. Sci. Technol. 1999, 33, 1179–1184. [Google Scholar] [CrossRef]
- Wang, S.; Mulligan, C.N. Speciation and surface structure of inorganic arsenic in solid phases: A review. Environ. Int. 2008, 34, 867–879. [Google Scholar] [CrossRef]
- Lin, S.; Lu, D.; Liu, Z. Removal of arsenic contaminants with magnetic γ-Fe2O3 nanoparticles. Chem. Eng. J. 2012, 211–212, 46–52. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Yanful, E.K.; Pratt, A.R. Arsenic removal from aqueous solutions by mixed magnetite-maghemite nanoparticles. Environ. Earth Sci. 2011, 64, 411–423. [Google Scholar] [CrossRef]
- Tuutijärvi, T.; Lu, J.; Sillanpää, M.; Chen, G. As(V) adsorption on maghemite nanoparticles. J. Hazard. Mater. 2009, 166, 1415–1420. [Google Scholar] [CrossRef]
- Ramos-Guivar, J.A.; Flores-Cano, D.A.; Passamani, E.C. Differentiating nanomaghemite and nanomagnetite and discussing their importance in arsenic and lead removal from contaminated effluents: A critical review. Nanomaterials 2021, 11, 2310. [Google Scholar] [CrossRef] [PubMed]
- Memon, A.Q.; Ahmed, S.; Bhatti, Z.A.; Maitlo, G.; Shah, A.K.; Mazari, S.A.; Muhammad, A.; Jatoi, A.S.; Kandhro, G.A. Experimental investigations of arsenic adsorption from contaminated water using chemically activated hematite (Fe2O3) iron ore. Environ. Sci. Pollut. Res. 2021, 28, 12898–12908. [Google Scholar] [CrossRef]
- Freitas, E.T.F.; Montoro, L.A.; Gasparon, M.; Ciminelli, V.S.T. Natural attenuation of arsenic in the environment by immobilization in nanostructured hematite. Chemosphere 2015, 138, 340–347. [Google Scholar] [CrossRef]
- Hajji, S.; Montes-Hernandez, G.; Sarret, G.; Tordo, A.; Morin, G.; Ona-Nguema, G.; Bureau, S.; Turki, T.; Mzoughi, N. Arsenite and chromate sequestration onto ferrihydrite, siderite and goethite nanostructured minerals: Isotherms from flow-through reactor experiments and XAS measurements. J. Hazard. Mater. 2019, 362, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Deliyanni, E.I.; Bakoyannakis, D.N.; Zouboulis, A.I.; Matis, K.A. Sorption of As(V) ions by akaganéite-type nanocrystals. Chemosphere 2003, 50, 155–163. [Google Scholar] [CrossRef]
- Samanta, A.; Das, S.; Jana, S. Exploring β-FeOOH Nanorods as an Efficient Adsorbent for Arsenic and Organic Dyes. ChemistrySelect 2018, 3, 2467–2473. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Jia, Y. A facile solution approach for the synthesis of akaganéite (β-FeOOH) nanorods and their ion-exchange mechanism toward As(V) ions. Appl. Surf. Sci. 2014, 290, 102–106. [Google Scholar] [CrossRef]
- Kolbe, F.; Weiss, H.; Morgenstern, P.; Wennrich, R.; Lorenz, W.; Schurk, K.; Stanjek, H.; Daus, B. Sorption of aqueous antimony and arsenic species onto akaganeite. J. Colloid Interface Sci. 2011, 357, 460–465. [Google Scholar] [CrossRef]
- Giles, D.E.; Mohapatra, M.; Issa, T.B.; Anand, S.; Singh, P. Iron and aluminium based adsorption strategies for removing arsenic from water. J. Environ. Manag. 2011, 92, 3011–3022. [Google Scholar] [CrossRef]
- Deliyanni, E.A.; Peleka, E.N.; Matis, K.A. Effect of cationic surfactant on the adsorption of arsenites onto akaganeite nanocrystals. Sep. Sci. Technol. 2007, 42, 993–1012. [Google Scholar] [CrossRef]
- Pepper, R.A.; Couperthwaite, S.J.; Millar, G.J. A novel akaganeite sorbent synthesised from waste red mud: Application for treatment of arsenate in aqueous solutions. J. Environ. Chem. Eng. 2018, 6, 6308–6316. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Tsang, Y.F.; Wang, Y.-N.; Sun, Y.; Zhang, D.; Pan, X. Adsorption capacities of poorly crystalline Fe minerals for antimonate and arsenate removal from water: Adsorption properties and effects of environmental and chemical conditions. Clean Technol. Environ. Policy 2018, 20, 2169–2179. [Google Scholar] [CrossRef]
- Raven, K.P.; Jain, A.; Loeppert, R.H. Arsenite and arsenate adsorption on ferrihydrite: Kinetics, equilibrium, and adsorption envelopes. Environ. Sci. Technol. 1998, 32, 344–349. [Google Scholar] [CrossRef]
- Jain, A.; Loeppert, R.H. Effect of Competing Anions on the Adsorption of Arsenate and Arsenite by Ferrihydrite. J. Environ. Qual. 2000, 29, 1422–1430. [Google Scholar] [CrossRef]
- Qi, P.; Pichler, T. Competitive Adsorption of As(III) and As(V) by Ferrihydrite: Equilibrium, Kinetics, and Surface Complexation. Water Air Soil Pollut. 2016, 227, 387. [Google Scholar] [CrossRef]
- Hao, L.; Liu, M.; Wang, N.; Li, G. A critical review on arsenic removal from water using iron-based adsorbents. RSC Adv. 2018, 8, 39545–39560. [Google Scholar] [CrossRef]
- Richmond, W.R.; Cowley, J.M.; Parkinson, G.M.; Saunders, M. An electron microscopy study of β-FeOOH (akaganéite) nanorods and nanotubes. CrystEngComm 2006, 8, 36–40. [Google Scholar] [CrossRef] [Green Version]
- Rémazeilles, C.; Refait, P. On the formation of β-FeOOH (akaganéite) in chloride-containing environments. Corros. Sci. 2007, 49, 844–857. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides; Wiley: New York, NY, USA, 2003; Volume 39, ISBN 9783527302741. [Google Scholar]
- Sanna Angotzi, M.; Mameli, V.; Cara, C.; Borchert, K.B.L.; Steinbach, C.; Boldt, R.; Schwarz, D.; Cannas, C. Meso- and macroporous silica-based arsenic adsorbents: Effect of pore size, nature of the active phase, and silicon release. Nanoscale Adv. 2021, 3, 6100–6113. [Google Scholar] [CrossRef]
- Langmuir, I. The constitution and fundamental properties of solids and liquids. Part I. Solids. J. Am. Chem. Soc. 1916, 38, 2221–2295. [Google Scholar] [CrossRef] [Green Version]
- Freundlich, H. Über die Adsorption in Lösungen. Z. Phys. Chem. 1907, 57U, 385–470. [Google Scholar] [CrossRef]
- Temkin, M.I.; Pyzhev, V. Kinetics of ammonia synthesis over a promoted iron catalyst. Acta Phys. Chim. URSS 1940, 12, 327. [Google Scholar]
- Redlich, O.; Peterson, D.L. A Useful Adsorption Isotherm. J. Phys. Chem. 1959, 63, 1024. [Google Scholar] [CrossRef]
- Dubinin, M.M.; Radushkevich, L.V. On the characteristic curve equation for active charcoals. Doklay Akad. Nauk 1947, 15, 327–329. [Google Scholar]
- Lutterotti, L.; Scardi, P. Simultaneous structure and size–strain refinement by the Rietveld method. J. Appl. Crystallogr. 1990, 23, 246–252. [Google Scholar] [CrossRef]
- Post, J.E.; Heaney, P.J.; Von Dreele, R.B.; Hanson, J.C. Neutron and temperature-resolved synchrotron X-ray powder diffraction study of akaganéite. Am. Mineral. 2003, 88, 782–788. [Google Scholar] [CrossRef]
- Michel, F.M.; Ehm, L.; Antao, S.M.; Lee, P.L.; Chupas, P.J.; Liu, G.; Strongin, D.R.; Schoonen, M.A.A.; Phillips, B.L.; Parise, J.B. The Structure of Ferrihydrite, a Nanocrystalline Material. Science 2007, 316, 1726–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecharromán, C.; González-Carreño, T.; Iglesias, J. The infrared dielectric properties of maghemite, γ-Fe2O3, from reflectance measurement on pressed powders. Phys. Chem. Miner. 1995, 22, 21–29. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Horváth, G.; Kawazoe, K. Method for the calculation of effective pore size distribution in molecular sieve carbon. J. Chem. Eng. Japan 1983, 16, 470–475. [Google Scholar] [CrossRef] [Green Version]
- Hoy, G.R.; Long, G.J. Mössbauer Spectroscopy Applied to Inorganic Chemistry; Long, G.J., Ed.; Springer: Boston, MA, USA, 1984; Volume 2, ISBN 0306416476. [Google Scholar]
- Sanna Angotzi, M.; Mameli, V.; Cara, C.; Peddis, D.; Xin, H.L.; Sangregorio, C.; Mercuri, M.L.; Cannas, C. On the synthesis of bi-magnetic manganese ferrite-based core–shell nanoparticles. Nanoscale Adv. 2021, 3, 1612–1623. [Google Scholar] [CrossRef]
- Sanna Angotzi, M.; Mameli, V.; Musinu, A.; Nizňnanský, D. 57 Fe Mössbauer Spectroscopy for the Study of Nanostructured Mixed Mn–Co Spinel Ferrites. J. Nanosci. Nanotechnol. 2019, 19, 5008–5013. [Google Scholar] [CrossRef]
- Sanna Angotzi, M.; Mameli, V.; Cara, C.; Ardu, A.; Nizňnanský, D.; Musinu, A. Oleate-Based Solvothermal Approach for Size Control of MIIFe2IIIO4 (MII = MnII, FeII) Colloidal Nanoparticles. J. Nanosci. Nanotechnol. 2019, 19, 4954–4963. [Google Scholar] [CrossRef] [PubMed]
- Sanna Angotzi, M.; Mameli, V.; Khanal, S.; Veverka, M.; Vejpravova, J.; Cannas, C. Effect of Different Molecular Coating on the Heating Properties of Maghemite Nanoparticles. Nanoscale Adv. 2022, 4, 408–420. [Google Scholar] [CrossRef]
- Sanna Angotzi, M.; Mameli, V.; Cara, C.; Grillo, V.; Enzo, S.; Musinu, A.; Cannas, C. Defect-assisted synthesis of magneto-plasmonic silver-spinel ferrite heterostructures in a flower-like architecture. Sci. Rep. 2020, 10, 17015. [Google Scholar] [CrossRef] [PubMed]
- Sanna Angotzi, M.; Mameli, V.; Zákutná, D.; Kubániová, D.; Cara, C.; Cannas, C. Evolution of the Magnetic and Structural Properties with the Chemical Composition in Oleate-Capped MnxCo1−xFe2O4 Nanoparticles. J. Phys. Chem. C 2021, 125, 20626–20638. [Google Scholar] [CrossRef]
- Knyazev, Y.V.; Balaev, D.A.; Stolyar, S.V.; Krasikov, A.A.; Bayukov, O.A.; Volochaev, M.N.; Yaroslavtsev, R.N.; Ladygina, V.P.; Velikanov, D.A.; Iskhakov, R.S. Interparticle magnetic interactions in synthetic ferrihydrite: Mössbauer spectroscopy and magnetometry study of the dynamic and static manifestations. J. Alloys Compd. 2022, 889, 161623. [Google Scholar] [CrossRef]
- Vacca, M.A.; Cara, C.; Mameli, V.; Sanna Angotzi, M.; Scorciapino, M.A.; Cutrufello, M.G.; Musinu, A.; Tyrpekl, V.; Pala, L.; Cannas, C. Hexafluorosilicic Acid (FSA): From Hazardous Waste to Precious Resource in Obtaining High Value-Added Mesostructured Silica. ACS Sustain. Chem. Eng. 2020, 8, 14286–14300. [Google Scholar] [CrossRef]
- Cara, C.; Mameli, V.; Rombi, E.; Pinna, N.; Sanna Angotzi, M.; Nižňanský, D.; Musinu, A.; Cannas, C. Anchoring ultrasmall FeIII-based nanoparticles on silica and titania mesostructures for syngas H2S purification. Microporous Mesoporous Mater. 2020, 298, 110062. [Google Scholar] [CrossRef]
- Villacorta, V.; Barrero, C.A.; Turrión, M.-B.; Lafuente, F.; Greneche, J.-M.; García, K.E. Removal of As³⁺, As⁵⁺, Sb³⁺, and Hg²⁺ ions from aqueous solutions by pure and co-precipitated akaganeite nanoparticles: Adsorption kinetics studies. RSC Adv. 2020, 10, 42688–42698. [Google Scholar] [CrossRef]
- Sanna Angotzi, M.; Musinu, A.; Mameli, V.; Ardu, A.; Cara, C.; Niznansky, D.; Xin, H.L.; Cannas, C. Spinel Ferrite Core–Shell Nanostructures by a Versatile Solvothermal Seed-Mediated Growth Approach and Study of Their Nanointerfaces. ACS Nano 2017, 11, 7889–7900. [Google Scholar] [CrossRef]
- Lutterotti, L.; Scardi, P. Profile Fitting by the Interference Function. Adv. X-ray Anal. 1991, 35, 577–584. [Google Scholar] [CrossRef]
- Kokunešoski, M.; Gulicovski, J.; Matović, B.; Logar, M.; Milonjić, S.K.; Babić, B. Synthesis and surface characterization of ordered mesoporous silica SBA-15. Mater. Chem. Phys. 2010, 124, 1248–1252. [Google Scholar] [CrossRef]
- Kersten, M.; Karabacheva, S.; Vlasova, N.; Branscheid, R.; Schurk, K.; Stanjek, H. Surface complexation modeling of arsenate adsorption by akagenéite (β-FeOOH)-dominant granular ferric hydroxide. Colloids Surf. Physicochem. Eng. Asp. 2014, 448, 73–80. [Google Scholar] [CrossRef]
- Brinker, C.J. Sol-Gel Science: The Physics and Chemistry of Sol–Gel Processing; Academic Press: Cambridge, MA, USA, 1990; ISBN 9780080571034. [Google Scholar]
- Mohan, D.; Pittman, C.U. Arsenic removal from water/wastewater using adsorbents—A critical review. J. Hazard. Mater. 2007, 142, 1–53. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Equation # | Equation | Parameters | References |
|---|---|---|---|---|
| Langmuir | Equation (2) | qm = maximum adsorption capacity (mg g−1) KL = Langmuir constant (L mg−1) Ce = equilibrium concentration (mg L−1) | [40] | |
| Frendlich | Equation (3) | KF = Freundlich constant (mg1−1/n L1/n g−1) | [41] | |
| Temkin | Equation (4) | R = universal gas constant (J mol−1 K−1) T = temperature (K) bT = Temkin parameter (J g mol−1 mg−1) KT = Temkin constant (L mg−1) | [42] | |
| Redlich–Peterson | Equation (5) | KRP = Redlich–Peterson constant (L g−1) αRP = Redlich–Peterson parameter 1 (L mg−1) βRP = Redlich–Peterson parameter 2 | [43] | |
| Dubinin–Radushkevich | Equation (6) | KDR = Dubinin–Radushkevich constant (mol2 kJ−2) εDR = Dubinin–Radushkevich variable (kJ mol−1) | [44] |
| Model | Equation # | Equation | Parameters |
|---|---|---|---|
| Pseudo 1st-Order | Equation (9) | K′ = pseudo-1st order constant (min−1) | |
| Pseudo 2nd-Order | Equation (10) | K″ = pseudo-2nd order constant (g mg−1 min−1) | |
| Intraparticle diffusion model | Equation (12) | ki = intraparticle diffusion constant (mg g−1 min−1/2) |
| Sample | Phase | a (Å) | b (Å) | c (Å) | DXRD 1 (nm) | DXRD 2 (nm) | DTEM 1 (nm) | DTEM 2 (nm) | SBET (m2 g−1) | Vp (cm3 g−1) | Dp (nm) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Aka | Isotropic akaganeite | 10.57 (1) | 3.030 (1) | 10.48 (1) | 5.3 (1) | - | 57 (16) | 4.3 (8) | 202 (4) | 0.327 (3) | 9.4 (2) |
| Anisotropic akaganeite | 2.0 (6) | 25.1 (2) | |||||||||
| Mag | Maghemite | 8.379 (1) | =a | =a | 14.0 (1) | n.a. | 12 (3) | n.a. | 92 (2) | 0.156 (2) | 11.8 (2) |
| Fer | Ferrihydrite | 5.69 (5) | =a | 9.03 (9) | 1.7 (3) | n.a. | 4 (1) | n.a. | 260 (5) | 0.110 (2) | 0.73 (1) |
| Comp | Hematite 18(2)% | 5.052 (6) | =a | 13.74 (2) | 9.1 (6) | n.a. | 9 (2) | n.a. | 410 (9) | 0.594 (6) | 8.1 (2) |
| Maghemite 82(8)% | 8.357 (6) | =a | =a | 6.6 (2) | n.a. | n.a. | |||||
| Silica | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 457 (9) | 0.666 (7) | 7.7 (2) |
| Sample | pH0 | Isotherm | R2 | K | qm (mg g−1) | n | bT (kJ g mol−1 mg−1) | αRP (L mg−1) | βRP | Eads (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| Aka | 3 | L | 0.93 | 6 (2) | 80 (4) | - | - | - | - | - |
| F | 0.88 | 42 (6) | - | 0.15 (3) | - | - | - | - | ||
| T | 0.95 | 3 (2)∙102 | - | - | 0.30 (3) | - | - | - | ||
| RP | 0.97 | 7 (3)∙102 | - | - | - | 12 (6) | 0.93 (3) | - | ||
| DR | 0.92 | 2.5 (6) 10−2 | 79 (4) | - | - | - | - | 4(1) | ||
| 8 | L | 0.91 | 6 (2) | 49 (2) | - | - | - | - | - | |
| F | 0.68 | 28 (5) | - | 0.11 (4) | - | - | - | - | ||
| T | 0.75 | 1 (3)∙103 | - | - | 0.6 (1) | - | - | - | ||
| RP | 0.92 | 3 (1)∙102 | - | - | - | 6 (3) | 1.00 (4) | - | ||
| DR | 0.91 | 2.7 (6) 10−2 | 48 (2) | - | - | - | - | 4(1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanna Angotzi, M.; Mameli, V.; Fantasia, A.; Cara, C.; Secci, F.; Enzo, S.; Gerina, M.; Cannas, C. As(III, V) Uptake from Nanostructured Iron Oxides and Oxyhydroxides: The Complex Interplay between Sorbent Surface Chemistry and Arsenic Equilibria. Nanomaterials 2022, 12, 326. https://doi.org/10.3390/nano12030326
Sanna Angotzi M, Mameli V, Fantasia A, Cara C, Secci F, Enzo S, Gerina M, Cannas C. As(III, V) Uptake from Nanostructured Iron Oxides and Oxyhydroxides: The Complex Interplay between Sorbent Surface Chemistry and Arsenic Equilibria. Nanomaterials. 2022; 12(3):326. https://doi.org/10.3390/nano12030326
Chicago/Turabian StyleSanna Angotzi, Marco, Valentina Mameli, Alessandra Fantasia, Claudio Cara, Fausto Secci, Stefano Enzo, Marianna Gerina, and Carla Cannas. 2022. "As(III, V) Uptake from Nanostructured Iron Oxides and Oxyhydroxides: The Complex Interplay between Sorbent Surface Chemistry and Arsenic Equilibria" Nanomaterials 12, no. 3: 326. https://doi.org/10.3390/nano12030326