A Low Glycaemic Index Diet in Pregnancy Induces DNA Methylation Variation in Blood of Newborns: Results from the ROLO Randomised Controlled Trial

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Dietary Intervention
2.3. DNA Extraction and Genome-Wide Methylation Detection
2.4. Statistical Analysis
2.5. Replication
3. Results
3.1. Cohort Characteristics and the Dietary Intervention
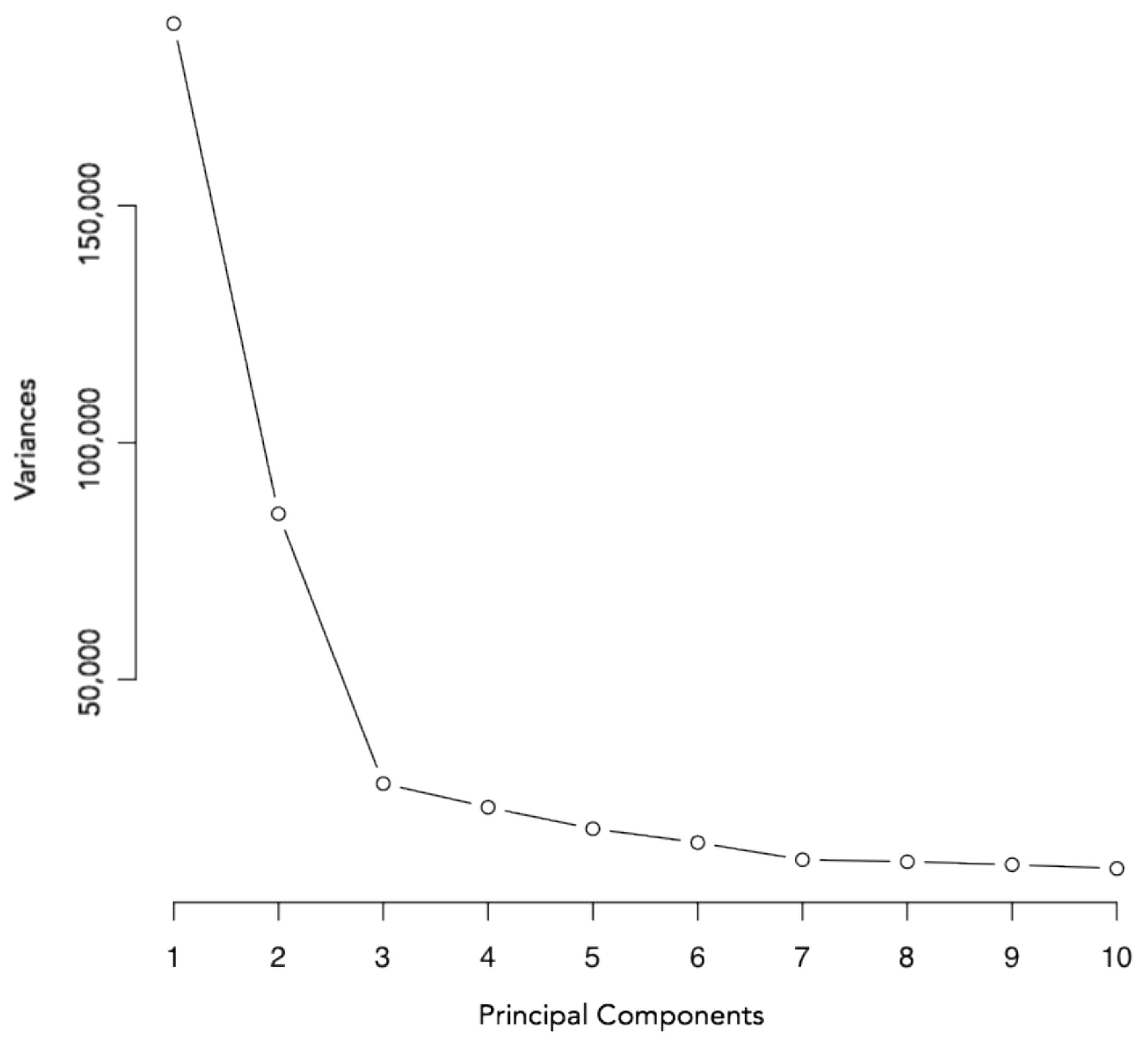
3.2. Principal Component Analysis
3.3. Linear Regression Analysis
3.4. Pathway Analysis
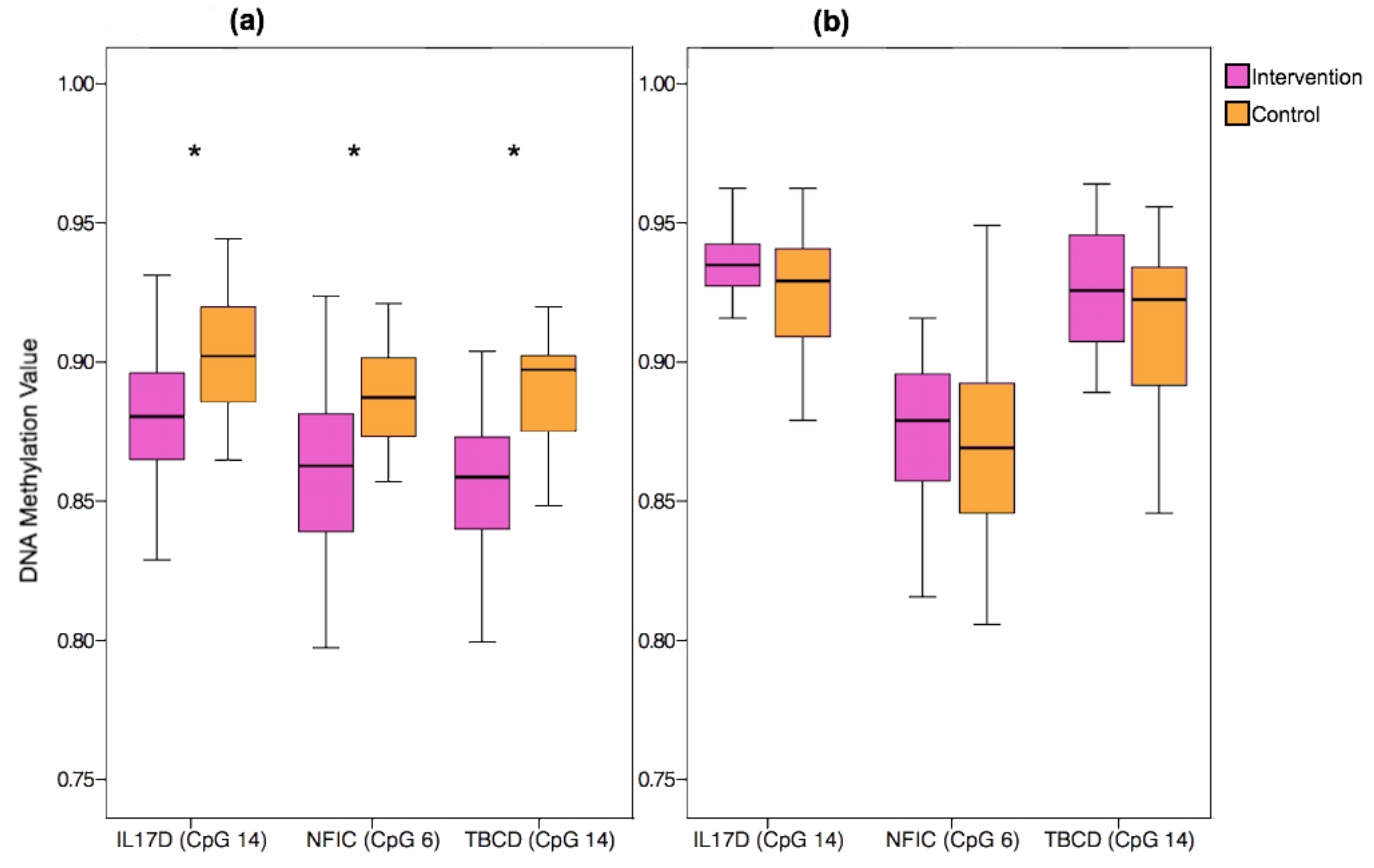
3.5. Replication
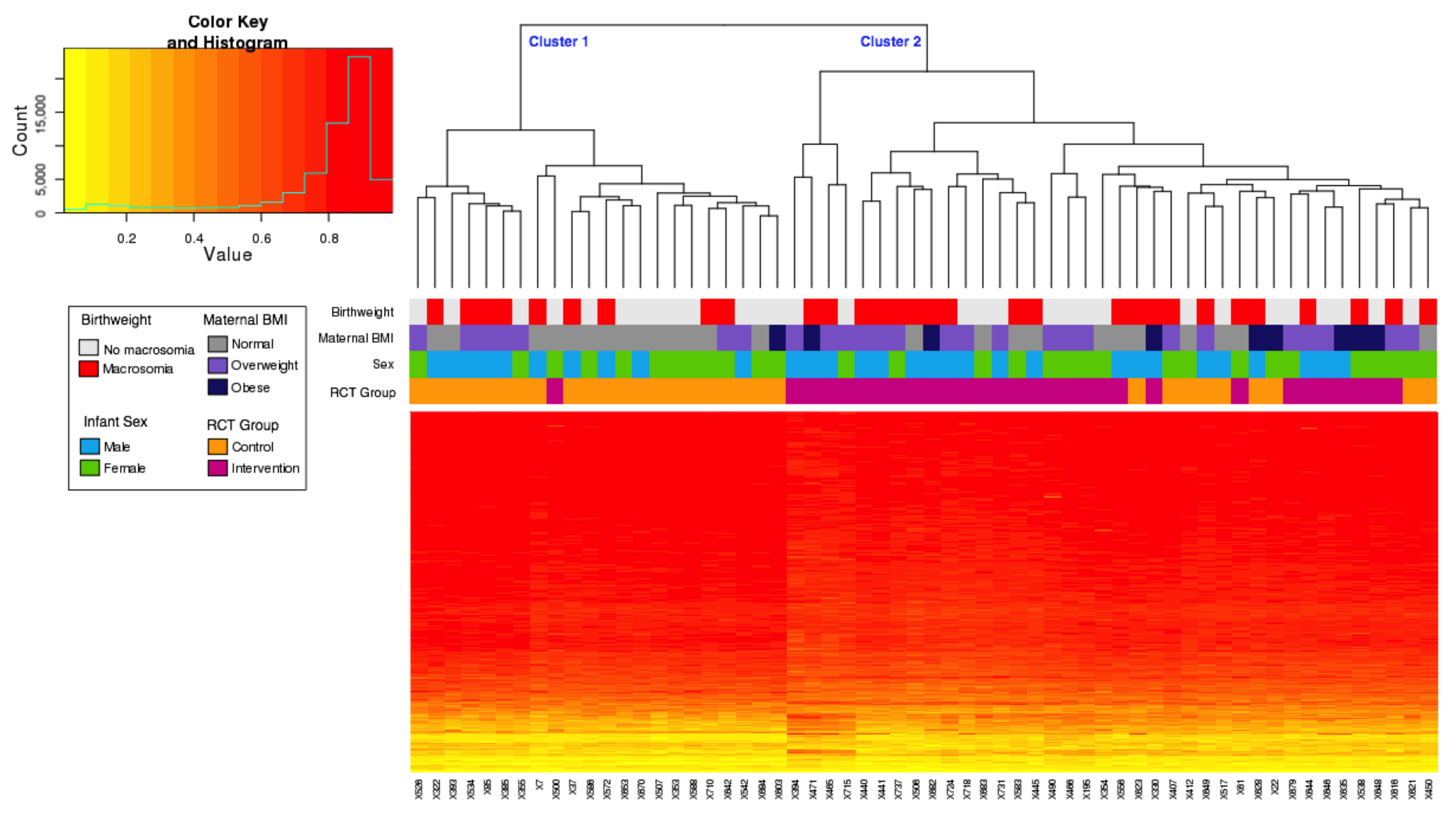
3.6. Cell-Type Analysis
4. Discussions
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Godfrey, K.M.; Costello, P.M.; Lillycrop, K.A. The developmental environment, epigenetic biomarkers and long-term health. J. Orig. Health Dis. 2015, 6, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Reichetzeder, C.; Dwi Putra, S.E.; Li, J.; Hocher, B. Developmental origins of disease-crisis precipitates change. Cell. Physiol. Biochem. 2016, 39, 919–938. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Ryckman, K.K. Epigenetic and developmental influences on the risk of obesity, diabetes, and metabolic syndrome. Diabetes Metab. Syndr. Obes. Targets Ther. 2015, 8, 295–302. [Google Scholar]
- Sharp, G.C.; Lawlor, D.A.; Richmond, R.C.; Fraser, A.; Simpkin, A.; Suderman, M.; Shihab, H.A.; Lyttleton, O.; McArdle, W.; Ring, S.M.; et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: Findings from the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 2015, 44, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lehnen, H.; Zechner, U.; Haaf, T. Epigenetics of gestational diabetes mellitus and offspring health: The time for action is in early stages of life. Mol. Hum. Reprod. 2013, 19, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Finer, S.; Mathews, C.; Lowe, R.; Smart, M.; Hillman, S.; Foo, L.; Sinha, A.; Williams, D.; Rakyan, V.K.; Hitman, G.A. Maternal gestational diabetes is associated with genome-wide DNA methylation variation in placenta and cord blood of exposed offspring. Hum. Mol. Genet. 2015, 44, 1–31. [Google Scholar] [CrossRef] [PubMed]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.A.; Mathers, J.C. Diet induced epigenetic changes and their implications for health. Acta Physiol. 2011, 202, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Burdge, G.C. Maternal diet as a modifier of offspring epigenetics. J. Dev. Orig. Health Dis. 2015, 6, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S. Impact of maternal diet on the epigenome during in utero life and the developmental programming of diseases in childhood and adulthood. Nutrients 2015, 7, 9492–9507. [Google Scholar] [CrossRef] [PubMed]
- Navarro, E.; Funtikova, A.N.; Fíto, M.; Schröder, H. Prenatal nutrition and the risk of adult obesity: Long-term effects of nutrition on epigenetic mechanisms regulating gene expression. J. Nutr. Biochem. 2017, 39, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, A.A.; Lindsay, K.L.; Alberdi, G.; McAuliffe, F.M.; Gibney, E.R. Nutrition During Pregnancy Impacts Offspring Epigenetic Status; Evidence from Human and Animal Studies. Nutr. Metab. Insights 2015, 8, 41. [Google Scholar] [PubMed]
- Amarasekera, M.; Noakes, P.; Strickland, D.; Saffery, R.; Martino, D.J.; Prescott, S.L. Epigenome-wide analysis of neonatal CD4+ T-cell DNA methylation sites potentially affected by maternal fish oil supplementation. Epigenetics 2014, 9, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.M.; McGowan, C.A.; Mahony, R.; Foley, M.E.; McAuliffe, F.M. Low glycaemic index diet in pregnancy to prevent macrosomia (ROLO study): Randomised control trial. Br. Med. J. 2012, 345, e5605. [Google Scholar] [CrossRef] [PubMed]
- Food Standards Agency. McCance and Widdowson’s The Composition of Foods, 6th ed.; Royal Society of Chemistry: Cambridge, UK, 2002; ISBN 0854044280. [Google Scholar]
- Levis, S.P.; McGowan, C.A.; McAuliffe, F.M. Methodology for adding and amending glycaemic index values to a nutrition analysis package. Br. J. Nutr. 2011, 105, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- McGowan, C.A.; Walsh, J.M.; Byrne, J.; Curran, S.; McAuliffe, F.M. The influence of a low glycemic index dietary intervention on maternal dietary intake, glycemic index and gestational weight gain during pregnancy: A randomized controlled trial. Nutr. J. 2013, 12, 140. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, J.; Gordon, L.; Oshlack, A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Gen. Biol. 2012, 13, R44. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2013, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl. Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Benajmini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucl. Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.; Hou, L.; Lin, S.M. Comparison of beta-value and m-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.; Mahony, R.M.; Canty, G.; Foley, M.E.; McAuliffe, F.; Walsh, J.M. Identification of those most likely to benefit from a low-glycaemic index dietary intervention in pregnancy. Br. J. Nutr. 2014, 112, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.M.; Matthews, P.; Poston, L. Maternal metabolism and obesity: Modifiable determinants of pregnancy outcome. Hum. Reprod. Update 2009, 16, 255–275. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, J.M.; Walsh, J.M.; Byrne, J.; Molloy, E.J.; McAuliffe, F.M. Impact of maternal diet on neonatal anthropometry: A randomized controlled trial. Pediatr. Obes. 2015, 10, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Dodd, J.M.; Turnbull, D.; McPhee, A.J.; Deussen, A.R.; Grivell, R.M.; Yelland, L.N.; McPhee, A.J.; Grivell, R.M.; Owens, J.A.; Gillman, M.W.; et al. Antenatal lifestyle advice for women who are overweight or obese: LIMIT randomised trial. Br. Med. J. 2014, 348, g1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, J.M.; Deussen, A.R.; Mohamad, I.; Rifas-Shiman, S.L.; Yelland, L.N.; Louise, J.; McPhee, A.J.; Grivell, R.M.; Owens, J.A.; Gillman, M.W.; et al. The effect of antenatal lifestyle advice for women who are overweight or obese on secondary measures of neonatal body composition: The LIMIT randomised trial. BJOG Int. J. Obstet. Gynaecol. 2016, 123, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Poston, L.; Bell, R.; Croker, H.; Flynn, A.C.; Godfrey, K.M.; Goff, L.; Hayes, L.; Khazaezadeh, N.; Nelson, S.M.; Oteng-Ntim, E.; et al. Effect of a behavioural intervention in obese pregnant women (the UPBEAT study): A multicentre, randomised controlled trial. Lancet Diabetes Endocrinol. 2015, 3, 767–777. [Google Scholar] [CrossRef]
- Patel, N.; Godfrey, K.M.; Pasupathy, D.; Levin, J.; Flynn, A.C.; Hayes, L.; Briley, A.L.; Bell, R.; Lawlor, D.A.; Oteng-Ntim, E.; et al. Infant adiposity following a randomised controlled trial of a behavioural intervention in obese pregnancy. Int. J. Obes. 2017, 41, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Barclay, A.W.; Petocz, P.; Mcmillan-Price, J.; Flood, V.M.; Prvan, T.; Mitchell, P.; Brand-Miller, J.C. Glycemic index, glycemic load and chronic disease risk—A meta-analysis of observational studies. Am. J. Clin. Nutr. 2008, 87, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, I.; Granström, C.; Haldorsson, T.; Rytter, D.; Hammer Bech, B.; Henriksen, T.B.; Vaag, A.A.; Olsen, S.F. Dietary glycemic index during pregnancy is associated with biomarkers of the metabolic syndrome in offspring at age 20 years. PLoS ONE 2013, 8, 10–13. [Google Scholar] [CrossRef]
- Kizirian, N.V.; Kong, Y.; Muirhead, R.; Brodie, S.; Garnett, S.P.; Petocz, P.; Sim, K.A.; Celermajer, D.S.; Louie, J.C.; Markovic, T.P.; et al. Effects of a low-glycemic index diet during pregnancy on offspring growth, body composition, and vascular health: A pilot randomized controlled trial. Am. J. Clin. Nutr. 2016, 103, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, L.; Thibault, S.; Guay, S.P.; Santure, M.; Monpetit, A.; St-Pierre, J.; Perron, P.; Brisson, D. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care 2010, 33, 2436–2441. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. Principles and challenges of genome wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Gen. Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- Harper, K.; Peters, B.; Gamble, M. Batch Effects and Pathway Analysis: Two Potential Perils in Cancer Studies Involving DNA Methylation Array Analysis. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1052–1060. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Intervention | Control | p | |||
|---|---|---|---|---|---|
| n | 30 | 30 | |||
| Maternal Characteristics | Mean | SD | Mean | SD | |
| Mother Age (years) | 32.78 | 4.48 | 33.91 | 4.16 | 0.31 |
| Mother Weight (14 weeks, kg) | 75.89 | 12.03 | 70.93 | 11.06 | 0.10 |
| Maternal BMI (14 weeks, kg/m2) | 27.72 | 4.26 | 25.66 | 7.74 | 0.05 |
| 3rd Level Education (n (%)) | 15 (50) | 20 (66.7) | 0.37 | ||
| Smoking During Pregnancy (n (%)) | 0 (0) | 0 (0) | - | ||
| Gestational Weight Gain (kg) | 11.82 | 3.50 | 13.93 | 5.01 | 0.09 |
| Daily GI Trimester 1 | 57.74 | 3.38 | 57.51 | 3.47 | 0.79 |
| Energy intake Trimester 1 (kcals) | 1803.19 | 308.64 | 1970.90 | 468.81 | 0.11 |
| Daily GI Trimester 2 | 56.42 | 3.60 | 57.35 | 3.28 | 0.30 |
| Energy intake Trimester 2 (kcals) | 1795.54 | 440.82 | 1960.72 | 383.73 | 0.13 |
| Daily GI Trimester 3 | 55.34 | 3.80 | 57.37 | 2.99 | 0.03 * |
| Energy intake Trimester 2 (kcals) | 1826.38 | 400.64 | 2023.79 | 409.63 | 0.06 |
| Maternal BMI Category at Booking Visit (n (%)) | |||||
| Normal (18.5–24.9 kg/m2) | 8 (26.7) | 17 (56.7) | 0.04 * | ||
| Overweight (25–29.9 kg/m2) | 16 (53.3) | 10 (33.3) | 0.19 | ||
| Obese (≥30 kg/m2) | 6 (20.0) | 3 (10) | 0.47 | ||
| Neonatal Characteristics | |||||
| Birth Weight (kg) | 4.20 | 0.62 | 3.98 | 0.43 | 0.12 |
| Macrosomic Neonate (n (%)) | 16 (53.3) | 14 (46.7) | 0.80 | ||
| Gestational Age (weeks) | 40.33 | 1.07 | 40.10 | 1.17 | 0.55 |
| Individual | Chip | Chip Position | Infant Sex | RCT Group | Maternal BMI | Maternal Weight (kg) | Maternal Age (years) | Birth Weight (kg) | Gestational Age (weeks) | |
|---|---|---|---|---|---|---|---|---|---|---|
| PC 1 correlation | 0.062 | 0.005 | 0.405 | −0.015 | −0.049 | 0.130 | 0.097 | 0.031 | 0.146 | −0.097 |
| PC 1 p value | 0.638 | 0.970 | 0.001 * | 0.912 | 0.713 | 0.322 | 0.461 | 0.813 | 0.264 | 0.463 |
| PC 2 correlation | 0.198 | −0.029 | −0.080 | −0.228 | −0.407 | 0.098 | 0.090 | 0.076 | 0.113 | 0.101 |
| PC 2 p value | 0.130 | 0.824 | 0.545 | 0.080 | 0.001 * | 0.456 | 0.494 | 0.565 | 0.391 | 0.444 |
| PC 3 correlation | 0.153 | 0.435 | −0.487 | −0.150 | 0.048 | −0.294 | −0.282 | 0.198 | 0.137 | −0.057 |
| PC 3 p value | 0.243 | 0.001 * | 0.000 * | 0.251 | 0.718 | 0.022 | 0.029 | 0.129 | 0.297 | 0.663 |
| PC 4 correlation | 0.457 | −0.078 | 0.131 | 0.185 | 0.088 | 0.024 | 0.009 | 0.035 | −0.102 | 0.029 |
| PC 4 p value | 0.000 * | 0.555 | 0.320 | 0.156 | 0.502 | 0.858 | 0.946 | 0.792 | 0.437 | 0.824 |
| PC 5 correlation | −0.051 | −0.294 | −0.316 | 0.059 | 0.150 | −0.204 | −0.110 | 0.053 | −0.259 | 0.095 |
| PC 5 p value | 0.696 | 0.023 | 0.014 | 0.656 | 0.253 | 0.118 | 0.404 | 0.687 | 0.046 | 0.472 |
| PC 6 correlation | −0.085 | 0.179 | −0.047 | 0.054 | −0.152 | −0.027 | 0.051 | 0.023 | −0.212 | −0.142 |
| PC 6 p value | 0.518 | 0.172 | 0.724 | 0.684 | 0.247 | 0.840 | 0.700 | 0.864 | 0.105 | 0.281 |
| PC 7 correlation | −0.358 | 0.312 | 0.115 | −0.289 | 0.237 | 0.023 | 0.026 | −0.044 | −0.007 | 0.016 |
| PC 7 p value | 0.005 * | 0.015 | 0.380 | 0.025 | 0.068 | 0.862 | 0.841 | 0.741 | 0.958 | 0.901 |
| PC 8 correlation | 0.528 | −0.274 | 0.187 | 0.112 | −0.096 | 0.103 | 0.037 | 0.238 | −0.290 | −0.006 |
| PC 8 p value | 0.000 * | 0.034 | 0.153 | 0.394 | 0.464 | 0.435 | 0.778 | 0.067 | 0.025 | 0.966 |
| PC 9 correlation | −0.070 | −0.036 | −0.038 | 0.071 | 0.046 | −0.187 | −0.257 | 0.258 | −0.115 | 0.251 |
| PC 9 p value | 0.596 | 0.784 | 0.775 | 0.592 | 0.729 | 0.153 | 0.048 | 0.046 | 0.381 | 0.053 |
| PC 10 correlation | 0.103 | −0.208 | −0.132 | −0.564 | −0.028 | −0.012 | 0.062 | −0.079 | 0.289 | −0.071 |
| PC 10 p value | 0.434 | 0.111 | 0.314 | 0.000 * | 0.829 | 0.928 | 0.638 | 0.548 | 0.025 | 0.589 |
| Pathway | Function | Count | p | Benjamini | |
|---|---|---|---|---|---|
| Cluster 1: Cardiac Functioning | |||||
| ES: 0.78 | KEGG | Dilated cardiomyopathy | 7 | 0.11 | 0.72 |
| KEGG | Cardiac muscle contraction | 6 | 0.14 | 0.7 | |
| KEGG | Hypertrophic cardiomyopathy (HCM) | 6 | 0.18 | 0.74 | |
| KEGG | Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 5 | 0.28 | 0.79 | |
| Cluster 2: Cancer Formation | |||||
| ES: 0.72 | KEGG | ErbB signalling pathway | 7 | 0.087 | 0.75 |
| KEGG | Non-small cell lung cancer | 5 | 0.12 | 0.7 | |
| KEGG | Glioma | 3 | 0.66 | 0.94 | |
| Cluster 3: Immune Functioning | |||||
| ES: 0.69 | KEGG | T cell receptor signalling pathway | 8 | 0.087 | 0.71 |
| KEGG | Natural killer cell mediated cytotoxicity | 8 | 0.19 | 0.73 | |
| KEGG | Fc epsilon R1 signalling pathway | 4 | 0.53 | 0.92 | |
| Cell Type | Total Group | Intervention | Control | p | |||
|---|---|---|---|---|---|---|---|
| Median | IQR | Median | IQR | Median | IQR | ||
| B cells | 0.025 | 0.040 | 0.040 | 0.063 | 0.020 | 0.030 | 0.014 * |
| CD4T | 0.050 | 0.038 | 0.050 | 0.053 | 0.030 | 0.033 | 0.02 * |
| CD8T | 0.000 | 0.008 | 0.000 | 0.010 | 0.000 | 0.000 | 0.048 * |
| Granulocytes | 0.825 | 0.185 | 0.800 | 0.258 | 0.855 | 0.140 | 0.042 * |
| Monocytes | 0.015 | 0.048 | 0.030 | 0.063 | 0.010 | 0.020 | 0.123 |
| NK cells | 0.030 | 0.030 | 0.040 | 0.043 | 0.030 | 0.030 | 0.006 * |
| nRBC | 0.040 | 0.048 | 0.055 | 0.060 | 0.030 | 0.043 | 0.026 * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geraghty, A.A.; Sexton-Oates, A.; O’Brien, E.C.; Alberdi, G.; Fransquet, P.; Saffery, R.; McAuliffe, F.M. A Low Glycaemic Index Diet in Pregnancy Induces DNA Methylation Variation in Blood of Newborns: Results from the ROLO Randomised Controlled Trial. Nutrients 2018, 10, 455. https://doi.org/10.3390/nu10040455
Geraghty AA, Sexton-Oates A, O’Brien EC, Alberdi G, Fransquet P, Saffery R, McAuliffe FM. A Low Glycaemic Index Diet in Pregnancy Induces DNA Methylation Variation in Blood of Newborns: Results from the ROLO Randomised Controlled Trial. Nutrients. 2018; 10(4):455. https://doi.org/10.3390/nu10040455
Chicago/Turabian StyleGeraghty, Aisling A., Alexandra Sexton-Oates, Eileen C. O’Brien, Goiuri Alberdi, Peter Fransquet, Richard Saffery, and Fionnuala M. McAuliffe. 2018. "A Low Glycaemic Index Diet in Pregnancy Induces DNA Methylation Variation in Blood of Newborns: Results from the ROLO Randomised Controlled Trial" Nutrients 10, no. 4: 455. https://doi.org/10.3390/nu10040455