BDK Deficiency in Cerebral Cortex Neurons Causes Neurological Abnormalities and Affects Endurance Capacity

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Western Blotting
2.3. Amino Acid Profiling
2.4. Assessment of Neurological Abnormalities
2.5. Endurance Exercise
2.6. Statistical Analysis
3. Results
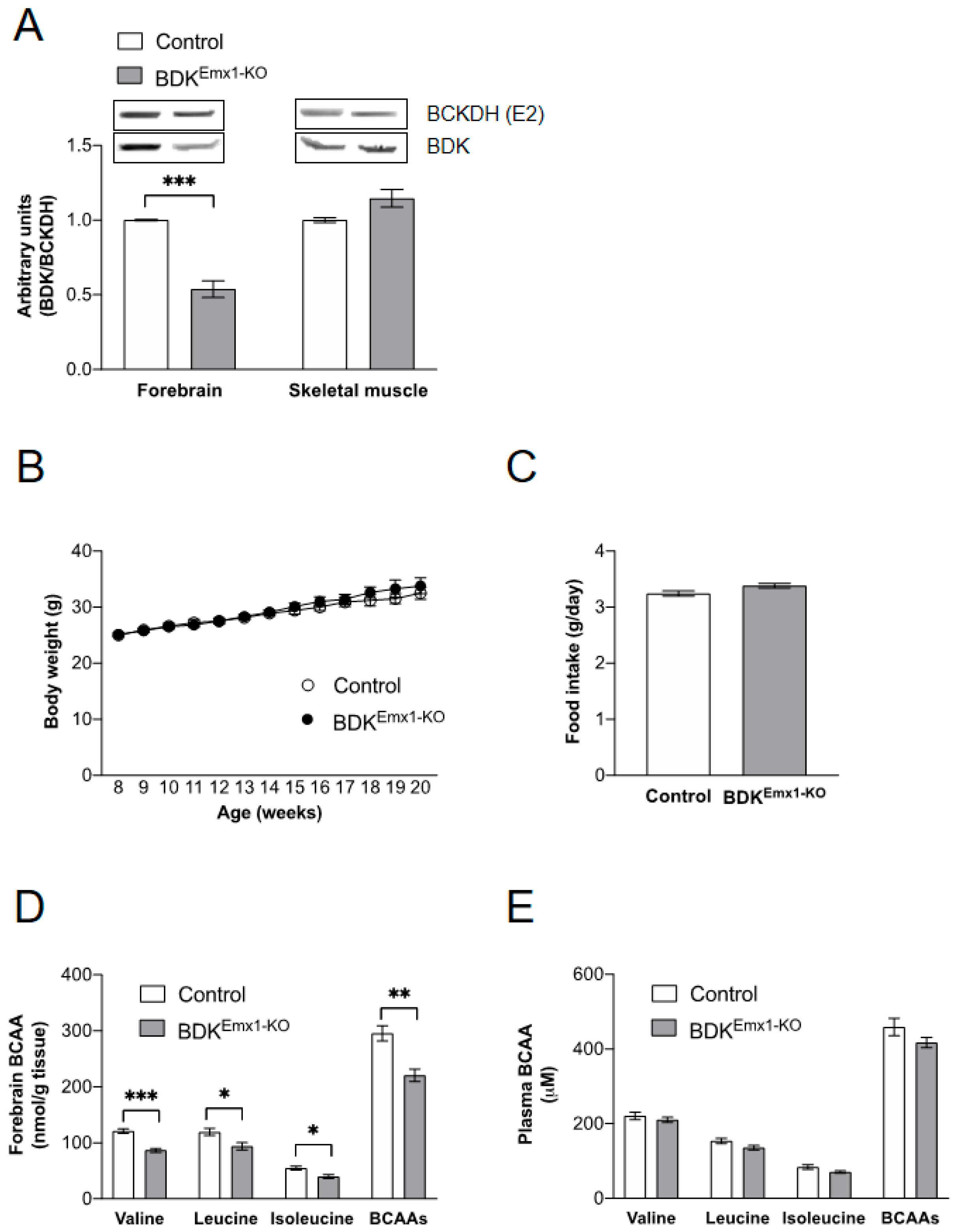
3.1. Characteristics of Mice with BDK Deficiency in Cerebral Cortex Neurons (BDKEmx1-KO Mice)
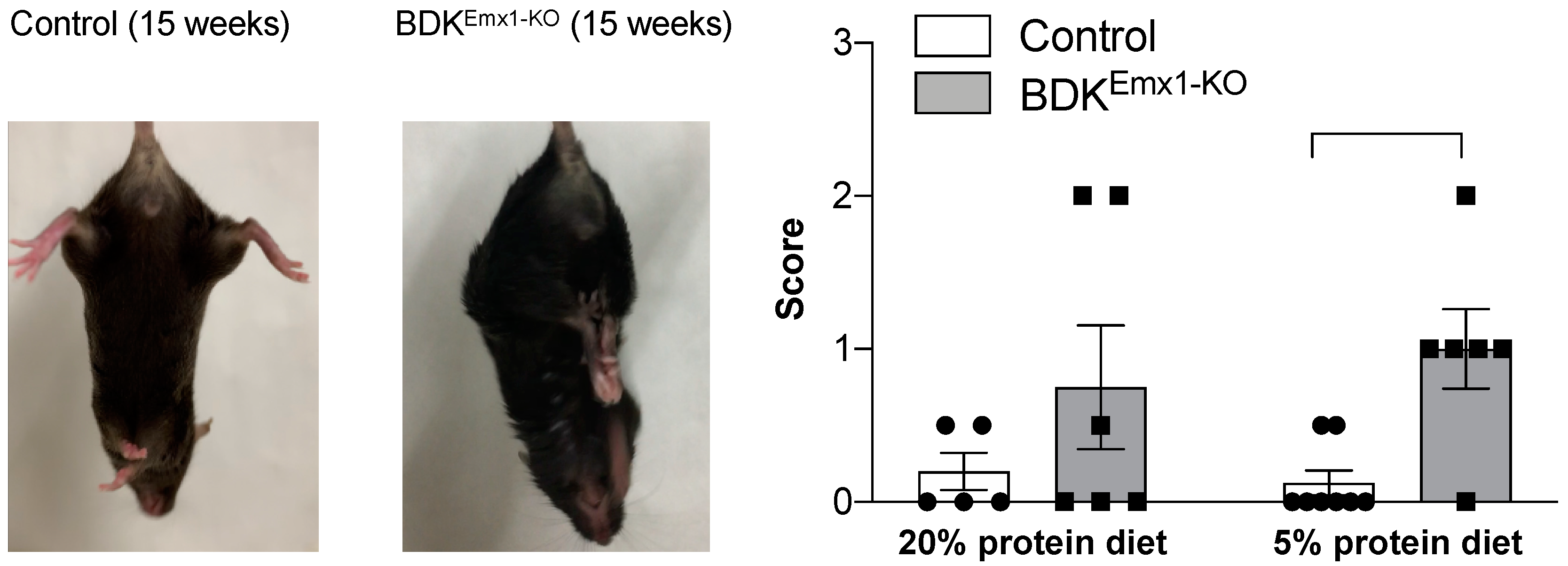
3.2. Neurological Abnormalities in BDKEmx1-KO Mice
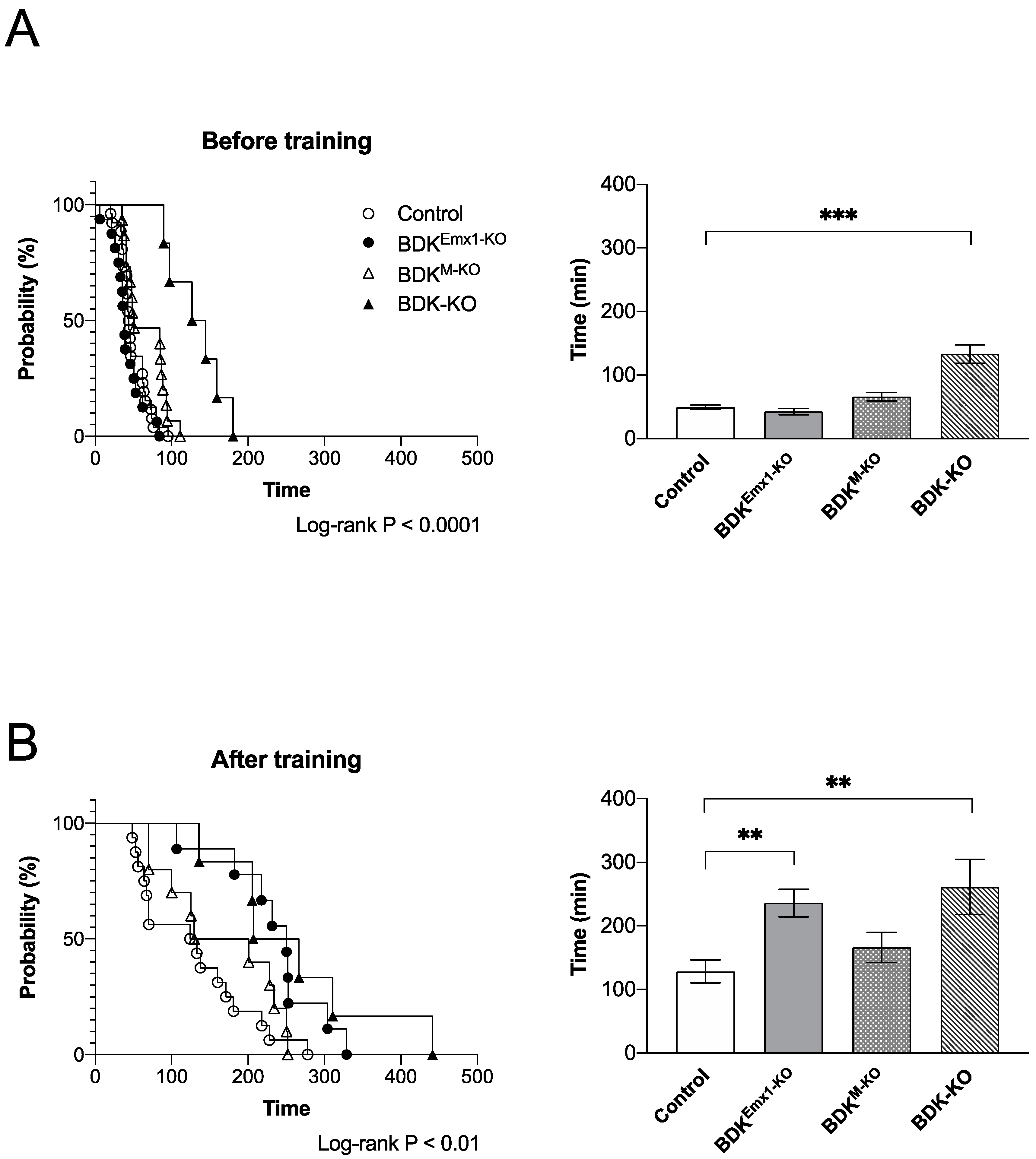
3.3. Endurance Exercise Capacity in BDKEmx1-KO Mice
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Neinast, M.; Murashige, D.; Arany, Z. Branched Chain Amino Acids. Ann. Rev. Physiol. 2019, 81, 139–164. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Kitaura, Y. Physiological and pathological roles of branched-chain amino acids in the regulation of protein and energy metabolism and neurological functions. Pharmacol Res. 2018, 133, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Kimball, S.R.; Jefferson, L.S. New functions for amino acids: Effects on gene transcription and translation. Am. J. Clin. Nutr. 2006, 83, 500s–507s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernstrom, J.D. Branched-Chain Amino Acids and Brain Function. J. Nutr. 2005, 135, 1539S–1546S. [Google Scholar] [CrossRef]
- Hawkins, R.A.; O’Kane, R.L.; Simpson, I.A.; Viña, J.R. Structure of the Blood–Brain Barrier and Its Role in the Transport of Amino Acids. J. Nutr. 2006, 136, 218S–226S. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.E.; Miller, R.H.; Block, K.P. Branched-chain amino acid metabolism. Annu. Rev. Nutr. 1984, 4, 409–454. [Google Scholar] [CrossRef]
- Yudkoff, M.; Nissim, I.; Kim, S.; Pleasure, D.; Hummeler, K.; Segal, S. [15N]leucine as a source of [15N]glutamate in organotypic cerebellar explants. Biochem. Biophys. Res. Commun. 1983, 115, 174–179. [Google Scholar] [CrossRef]
- Bixel, M.; Shimomura, Y.; Hutson, S.; Hamprecht, B. Distribution of key enzymes of branched-chain amino acid metabolism in glial and neuronal cells in culture. J. Histochem. Cytochem. 2001, 49, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, M.E.; Hutson, S.M. BCAA Metabolism and NH3 Homeostasis. Adv. Neurobiol. 2016, 13, 99–132. [Google Scholar] [CrossRef] [PubMed]
- She, P.; Reid, T.M.; Bronson, S.K.; Vary, T.C.; Hajnal, A.; Lynch, C.J.; Hutson, S.M. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab. 2007, 6, 181–194. [Google Scholar] [CrossRef] [Green Version]
- She, P.; Zhou, Y.; Zhang, Z.; Griffin, K.; Gowda, K.; Lynch, C.J. Disruption of BCAA metabolism in mice impairs exercise metabolism and endurance. J. Appl. Physiol. 2010, 108, 941–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.L.; Li, C.J.; Xing, Y.; Yang, Y.H.; Jia, J.P. Hypervalinemia and hyperleucine-isoleucinemia caused by mutations in the branched-chain-amino-acid aminotransferase gene. J. Inherit. Metab. Dis. 2015, 38, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Elitsa, A.A.; Chirag, H.P.; Charles, H.D.; Jonathan, D.P.; Susan, M.H. Cytosolic branched chain aminotransferase (BCATc) regulates mTORC1 signaling and glycolytic metabolism in CD4+ T cells. J. Biol. Chem. 2014, 289, 18793–18804. [Google Scholar]
- Chuang, D.T. Maple Syrup Urine Disease: It Has Come a Long Way. J. Pediatr. 1998, 132, S17–S23. [Google Scholar] [CrossRef]
- Novarino, G.; El-Fishawy, P.; Kayserili, H.; Meguid, N.A.; Scott, E.M.; Schroth, J.; Jennifer, L.S.; Majdi, K.; Rehab, O.K.; Tawfeg, B.-O.; et al. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science (NY) 2012, 338, 394–397. [Google Scholar] [CrossRef] [Green Version]
- García-Cazorla, A.; Oyarzabal, A.; Fort, J.; Robles, C.; Castejón, E.; Ruiz-Sala, P.; Bodoy, S.; Merinero, B.; Lopez-Sala, A.; Dopazo, J.; et al. Two novel mutations in the BCKDK (branched-chain keto-acid dehydrogenase kinase) gene are responsible for a neurobehavioral deficit in two pediatric unrelated patients. Hum. Mutat. 2014, 35, 470–477. [Google Scholar] [CrossRef]
- Joshi, M.A.; Jeoung, N.H.; Obayashi, M.; Hattab, E.M.; Brocken, E.G.; Liechty, E.A.; Michael, J.K.; Krishna, M.V.; Ronald, C.W.; Robert, A.H. Impaired growth and neurological abnormalities in branched-chain alpha-keto acid dehydrogenase kinase-deficient mice. Biochem. J. 2006, 400, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Kitaura, Y.; Kadota, Y.; Morishita, Y.; Ota, M.; Yamanaka, F.; Minjun, X.; Masahito, I.; Naokazu, I.; Fuminori, K.; et al. Muscle-specific deletion of BDK amplifies loss of myofibrillar protein during protein undernutrition. Sci. Rep. 2017, 7, 39825. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Kitaura, Y.; Ishikawa, T.; Kadota, Y.; Terai, C.; Shindo, D.; Takashi, M.; Miki, O.; Yukako, M.; Kengo, I.; et al. Endurance performance and energy metabolism during exercise in mice with a muscle-specific defect in the control of branched-chain amino acid catabolism. PLoS ONE 2017, 12, e0180989. [Google Scholar] [CrossRef]
- Xu, M.; Kitaura, Y.; Shindo, D.; Shimomura, Y. Branched-chain amino acid (BCAA) supplementation enhances adaptability to exercise training of mice with a muscle-specific defect in the control of BCAA catabolism. Biosci. Biotechnol. Biochem. 2018, 82, 896–899. [Google Scholar] [CrossRef]
- Brosnan, M.E.; Lowry, A.; Wasi, Y.; Lowry, M.; Brosnan, J.T. Regional and subcellular distribution of enzymes of branched-chain amino acid metabolism in brains of normal and diabetic rats. Can. J. Physiol. Pharmacol. 1985, 63, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.T.; Sweatt, A.J.; Hutson, S.M. Expression of mitochondrial branched-chain aminotransferase and alpha-keto-acid dehydrogenase in rat brain: Implications for neurotransmitter metabolism. Front. Neuroanat. 2012, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasato, T.; Nomura, R.; Ando, R.; Ikeda, T.; Tanaka, M.; Itohara, S. Dorsal telencephalon-specific expression of Cre recombinase in PAC transgenic mice. Genesis 2004, 38, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Nanaumi, N.; Suzuki, M.; Popov, K.M.; Harris, R.A. Purification and partial characterization of branched-chain alpha-ketoacid dehydrogenase kinase from rat liver and rat heart. Arch. Biochem. Biophys. 1990, 283, 293–299. [Google Scholar] [CrossRef]
- Xu, M.; Nagasaki, M.; Obayashi, M.; Sato, Y.; Tamura, T.; Shimomura, Y. Mechanism of activation of branched-chain alpha-keto acid dehydrogenase complex by exercise. Biochem. Biophys. Res. Commun. 2001, 287, 752–756. [Google Scholar] [CrossRef]
- Uji, M.; Yokoyama, Y.; Ohbuchi, K.; Tsuchiya, K.; Sadakane, C.; Shimobori, C.; Yamamoto, M.; Nagino, M. Exploration of Serum Biomarkers for Predicting the Response to Inchinkoto (ICKT), a Japanese Traditional Herbal Medicine. Metabolomics 2017, 13, 155. [Google Scholar] [CrossRef] [Green Version]
- Guezennec, C.; Abdelmalki, A.; Serrurier, B.; Merino, D.; Bigard, X.; Berthelot, M.; Pierard, C.; Peres, M. Effects of prolonged exercise on brain ammonia and amino acids. Int. J. Sports Med. 1998, 19, 323–327. [Google Scholar] [CrossRef]
- Wilkinson, D.J.; Smeeton, N.J.; Watt, P.W. Ammonia metabolism, the brain and fatigue; revisiting the link. Prog. Neurobiol. 2010, 91, 200–219. [Google Scholar] [CrossRef]
- Newsholme, E.A.; Blomstrand, E. Branched-Chain Amino Acids and Central Fatigue. J. Nutr. 2006, 136, 274S–276S. [Google Scholar] [CrossRef]
- Schuler, V.; Lüscher, C.; Blanchet, C.; Klix, N.; Sansig, G.; Klebs, K.; Schmutz, M.; Heid, J.; Gentry, C.; Urban, L.; et al. Epilepsy, Hyperalgesia, Impaired Memory, and Loss of Pre- And Postsynaptic GABA(B) Responses in Mice Lacking GABA(B(1)). Neuron 2001, 31, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Marimoutou, M.; Springer, D.A.; Liu, C.; Kim, G.; Levine, R.L. Oxidation of Methionine 77 in Calmodulin Alters Mouse Growth and Behavior. Antioxidants (Basel) 2018, 7, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolisnyk, B.; Guzman, M.S.; Raulic, S.; Fan, J.; Magalhães, A.C.; Feng, G.; Gros, R.; Prado, V.F.; Prado, M.A. ChAT-ChR2-EYFP mice have enhanced motor endurance but show deficits in attention and several additional cognitive domains. J. Neurosci. 2013, 33, 10427–10438. [Google Scholar] [CrossRef] [PubMed]
- Holmstrand, E.C.; Lund, D.; Cherian, A.K.; Wright, J.; Martin, R.F.; Ennis, E.A.; Stanwood, G.D.; Sarter, M.; Blakely, R.D. Transgenic overexpression of the presynaptic choline transporter elevates acetylcholine levels and augments motor endurance. Neurochem. Int. 2014, 73, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Soya, M.; Soya, H. Endurance and Brain Glycogen: A Clue toward Understanding Central Fatigue. Adv. Neurobiol. 2019, 23, 331–346. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Tissue | Control | BDKEmx1-KO | ||||
|---|---|---|---|---|---|---|
| g/100 g Body Weight | ||||||
| Brain | 1.43 | ± | 0.06 | 1.36 | ± | 0.06 |
| Liver | 3.66 | ± | 0.11 | 3.89 | ± | 0.07 |
| Kidney | 1.04 | ± | 0.07 | 1.05 | ± | 0.04 |
| Heart | 0.38 | ± | 0.01 | 0.38 | ± | 0.02 |
| Adipose | 6.85 | ± | 0.37 | 7.52 | ± | 0.71 |
| Skeletal muscle | 1.02 | ± | 0.02 | 1.04 | ± | 0.03 |
| Control | BDKEmx1-KO | ||||||
|---|---|---|---|---|---|---|---|
| Tissue | Amino Acid | nmol/g Tissue | |||||
| Brain | Alanine | 671.7 | ± | 119.3 | 745.5 | ± | 138.9 |
| Asparate | 4840.2 | ± | 841.9 | 6718.5 | ± | 999.5 | |
| Glysine | 956.9 | ± | 131.8 | 1017.8 | ± | 143.4 | |
| Glutamate | 15,628.5 | ± | 3287.1 | 16,945.9 | ± | 2941.6 | |
| Lysine | 204.6 | ± | 35.7 | 486.7 | ± | 182.2 | |
| Methionine | 54.3 | ± | 8.6 | 51.5 | ± | 15.5 | |
| Phenylalanine | 88.4 | ± | 17.4 | 89.2 | ± | 16.5 | |
| Proline | 99.5 | ± | 20.6 | 107.3 | ± | 20.7 | |
| Serine | 1061.3 | ± | 162.2 | 1203.2 | ± | 209.7 | |
| Threonine | 336.5 | ± | 39.4 | 454.6 | ± | 82.9 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizusawa, A.; Watanabe, A.; Yamada, M.; Kamei, R.; Shimomura, Y.; Kitaura, Y. BDK Deficiency in Cerebral Cortex Neurons Causes Neurological Abnormalities and Affects Endurance Capacity. Nutrients 2020, 12, 2267. https://doi.org/10.3390/nu12082267
Mizusawa A, Watanabe A, Yamada M, Kamei R, Shimomura Y, Kitaura Y. BDK Deficiency in Cerebral Cortex Neurons Causes Neurological Abnormalities and Affects Endurance Capacity. Nutrients. 2020; 12(8):2267. https://doi.org/10.3390/nu12082267
Chicago/Turabian StyleMizusawa, Anna, Ayako Watanabe, Minori Yamada, Rina Kamei, Yoshiharu Shimomura, and Yasuyuki Kitaura. 2020. "BDK Deficiency in Cerebral Cortex Neurons Causes Neurological Abnormalities and Affects Endurance Capacity" Nutrients 12, no. 8: 2267. https://doi.org/10.3390/nu12082267