

Exploring the Diversity of Plant-Associated Viruses and Related Viruses in Riverine Freshwater Samples Collected in Berlin, Germany


Abstract
:1. Introduction
2. Materials and Methods
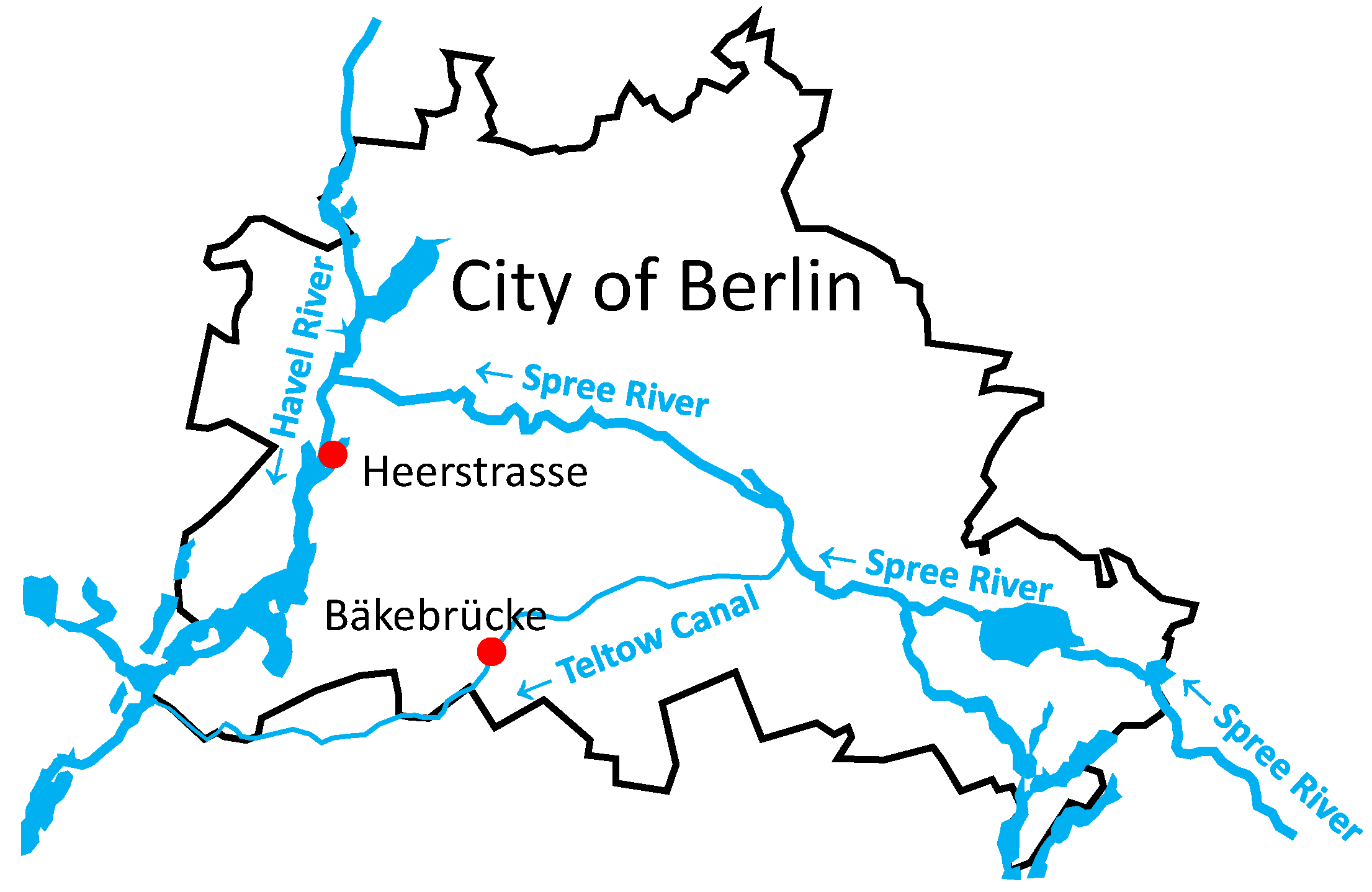
2.1. Sample Collection and Virus Enrichment
2.2. RNA Preparation, Illumina Next-Generation Sequencing, Sequence Data Processing
2.3. Sequence Data Analysis
3. Results
3.1. Study Approach
3.2. Genetic Analysis of Viruses from Teltow Canal and Havel River
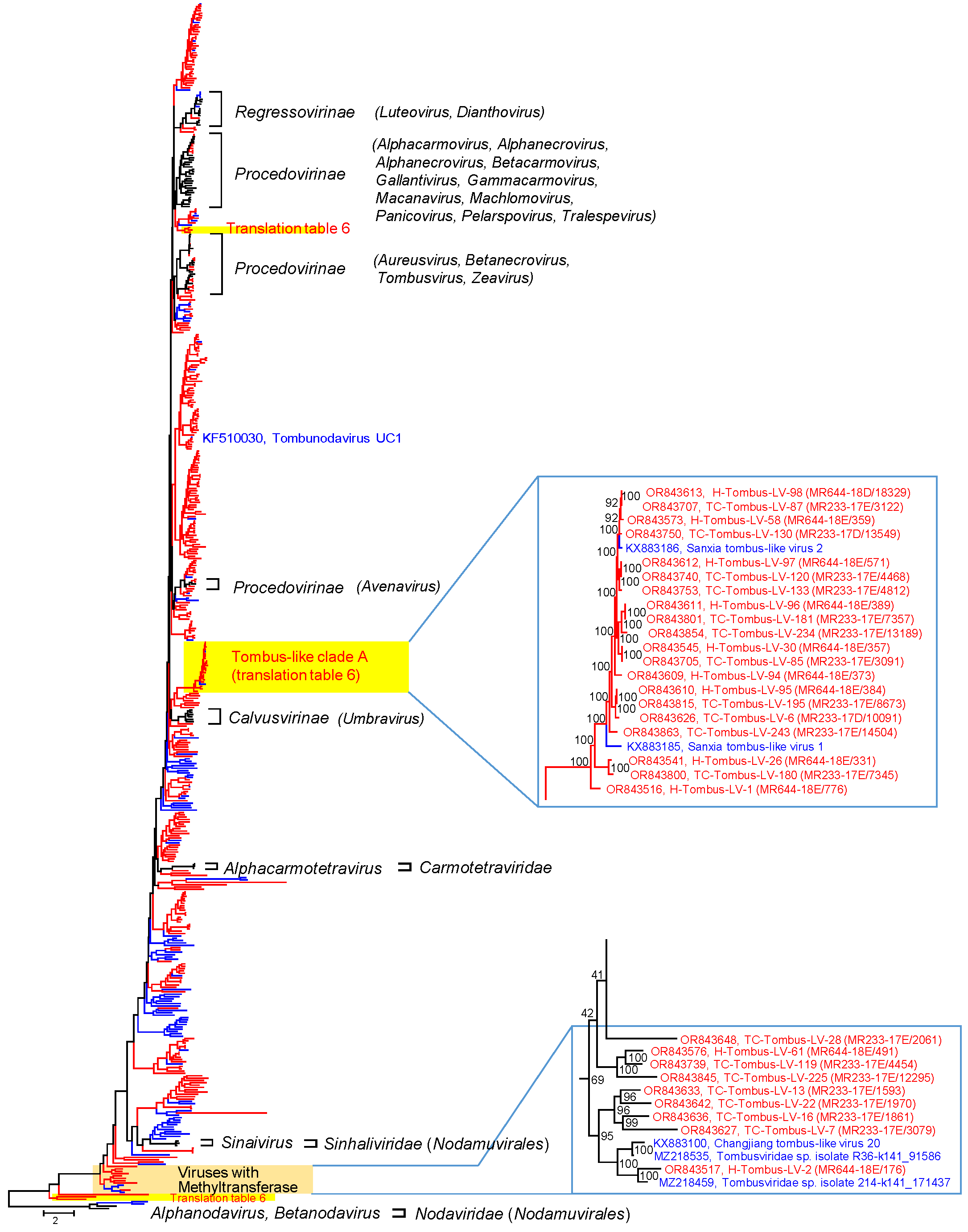
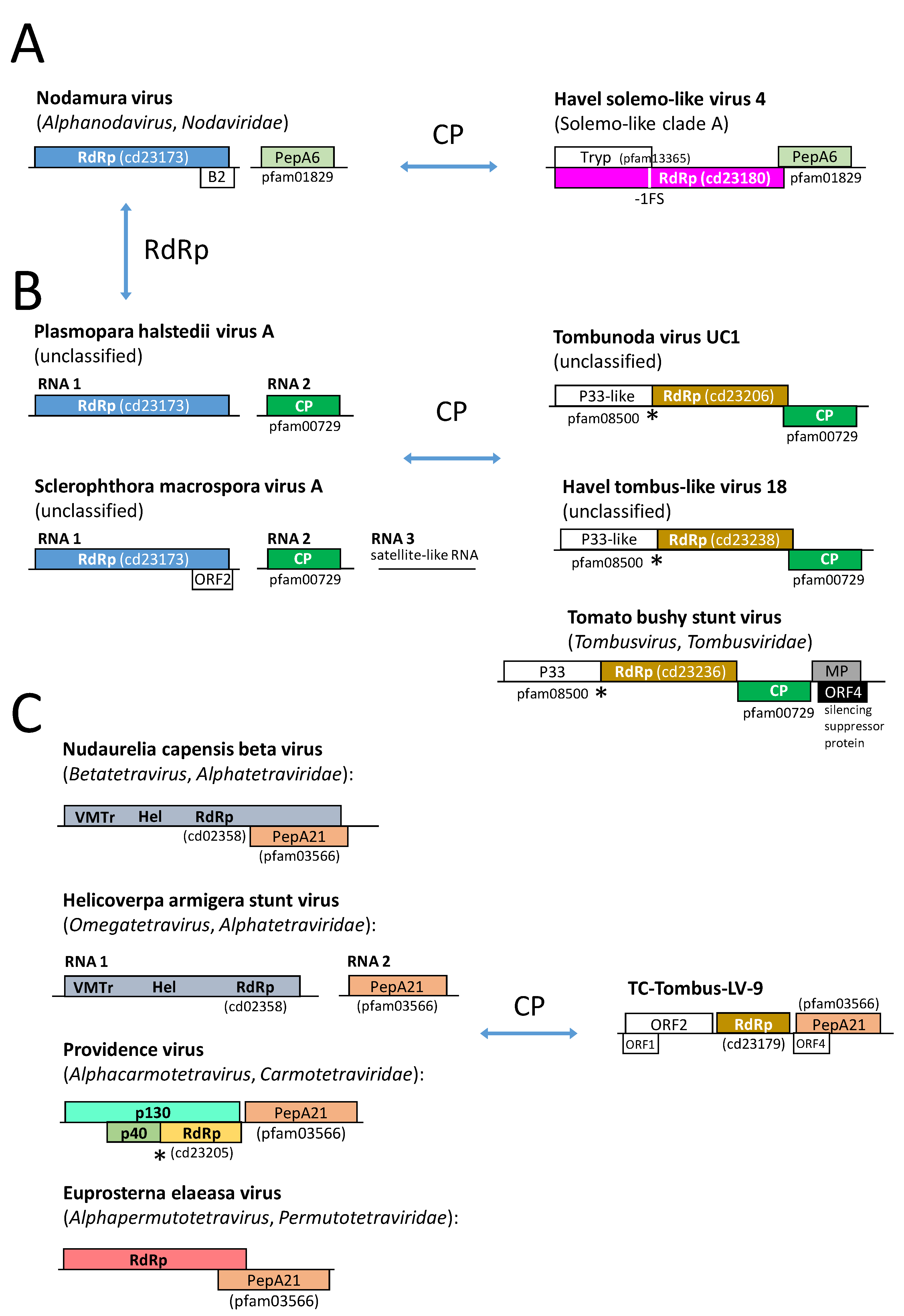
3.2.1. Tombusviridae
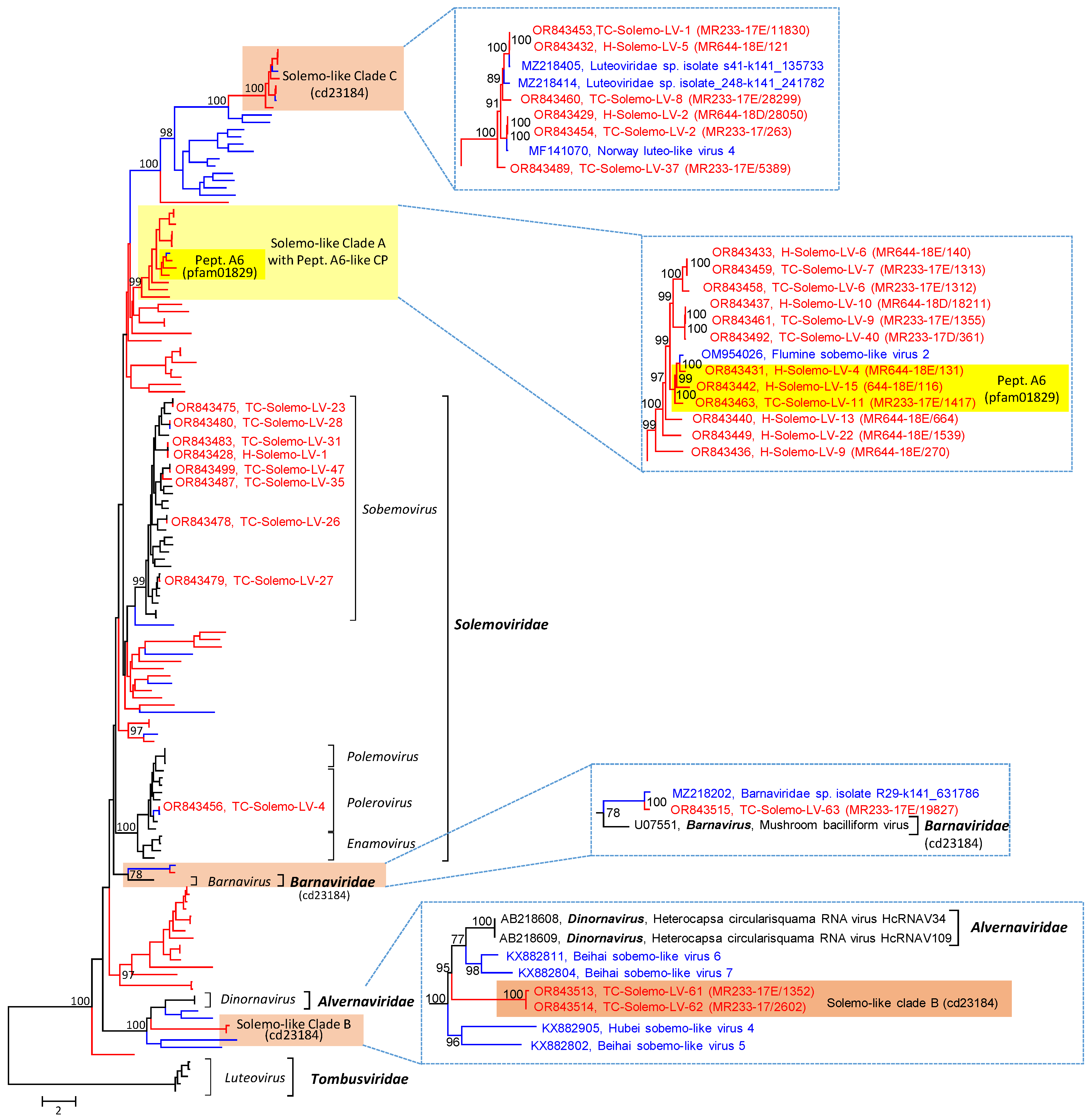
3.2.2. Solemoviridae
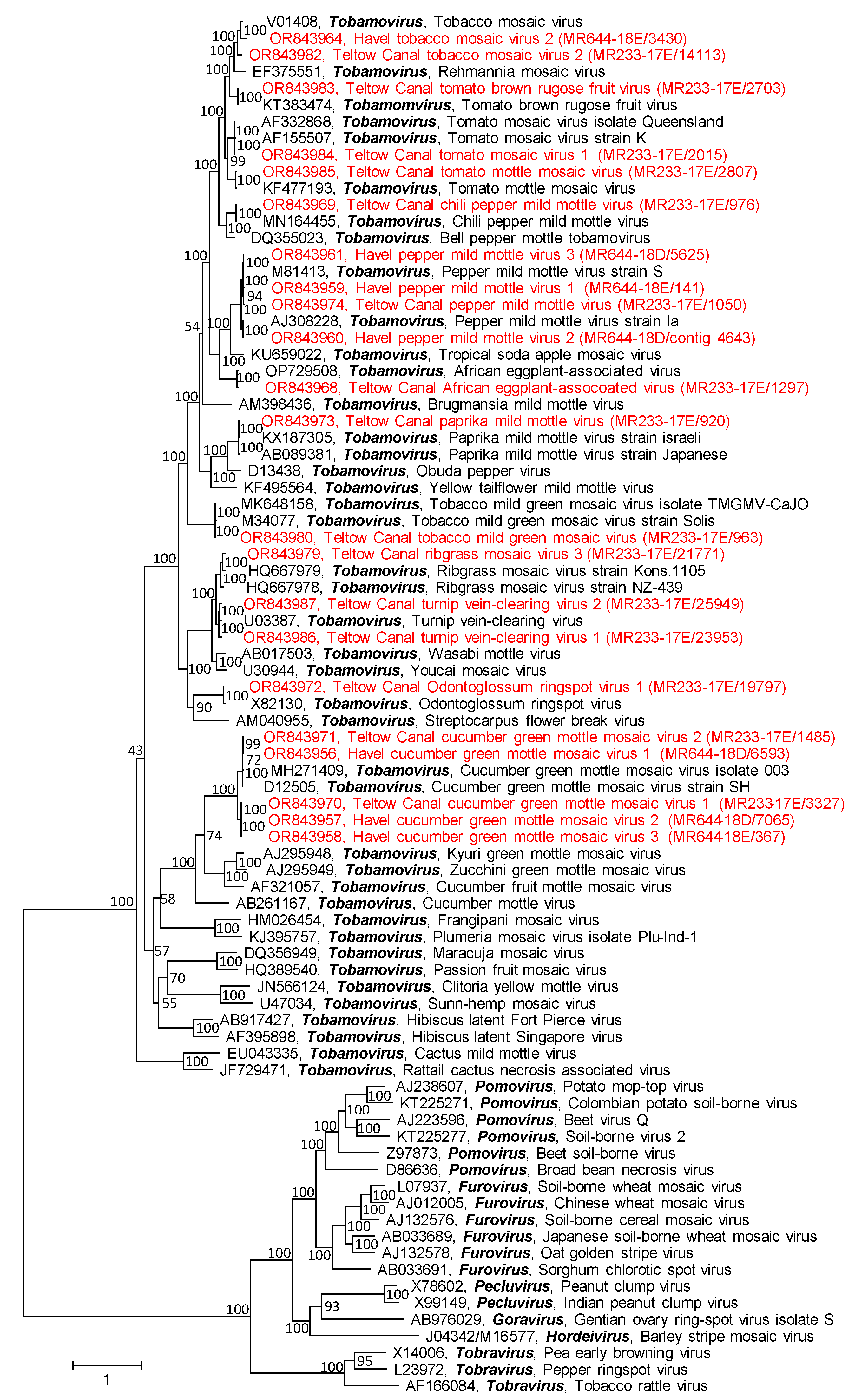
3.2.3. Virgaviridae
3.2.4. Endornaviridae
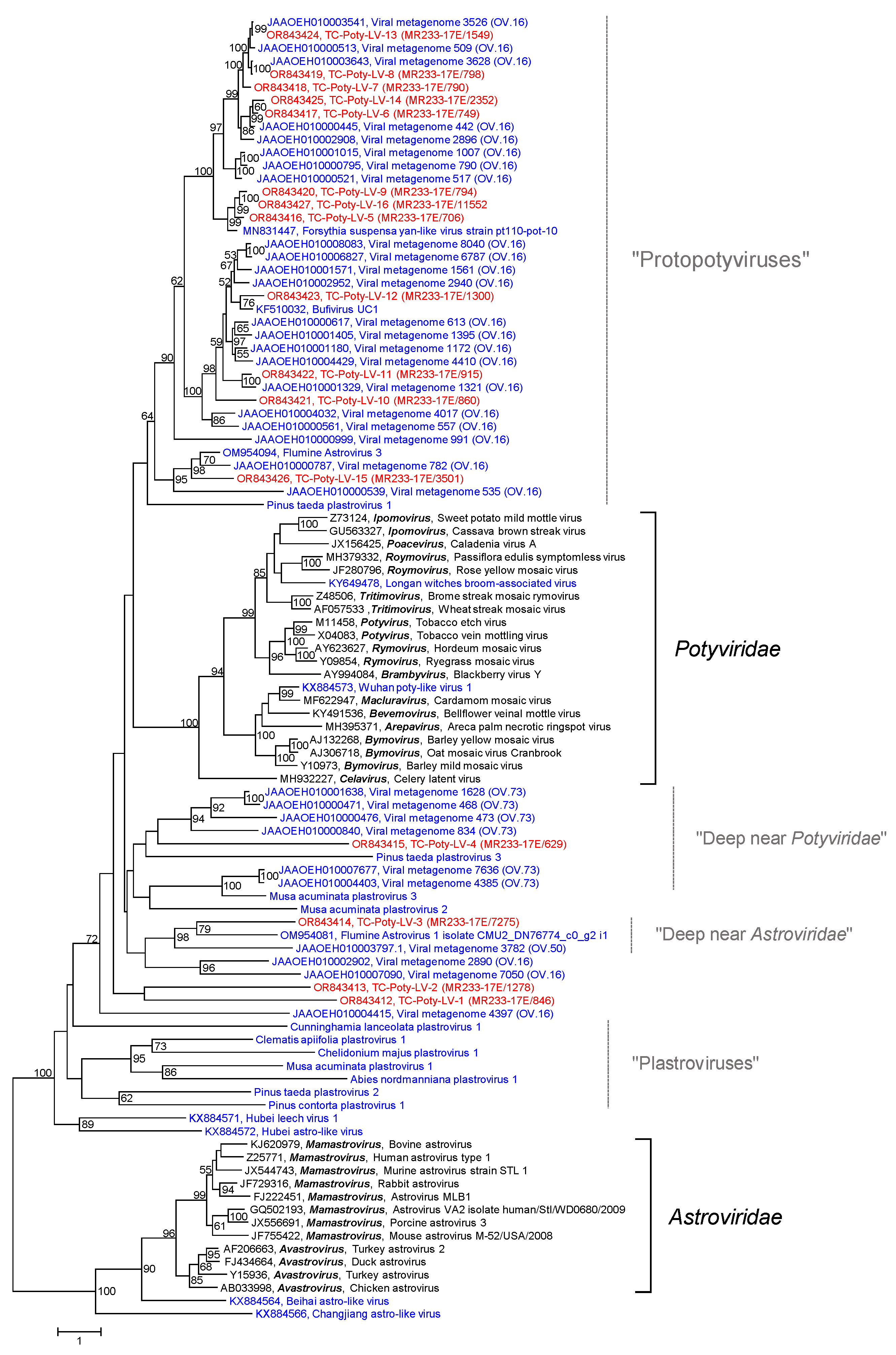
3.2.5. Poty-Like Viruses
3.2.6. Partitiviridae
3.2.7. Albeto-Like Viruses
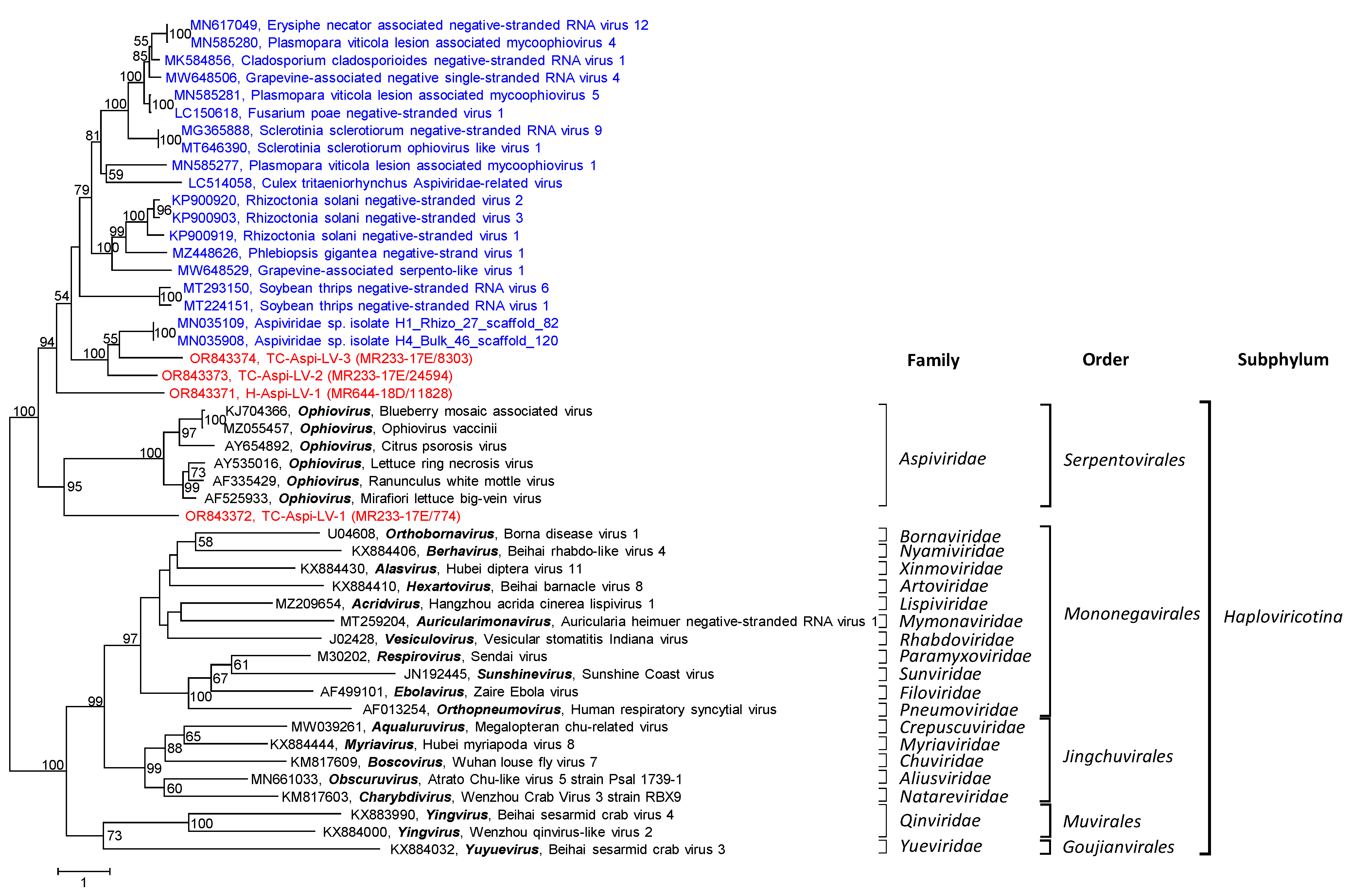
3.2.8. Aspiviridae
3.2.9. Alphaflexiviridae
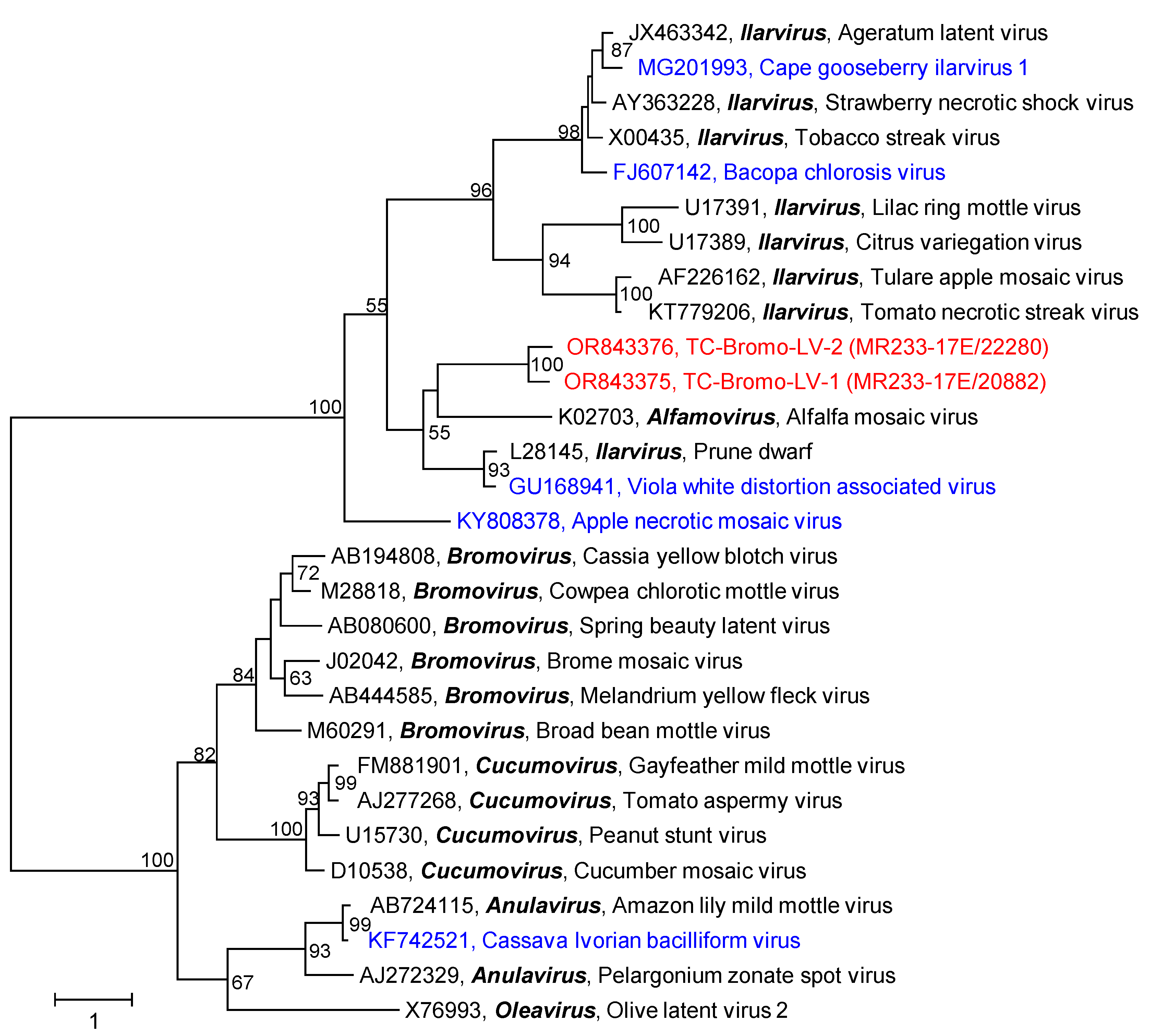
3.2.10. Bromoviridae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edwards, R.A.; Rohwer, F. 2005. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, P.G.; Pipas, J.M. Detecting viral sequences in NGS data. Curr. Opin. Virol. 2019, 39, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Chen, Y.M.; Wang, W.; Qin, X.C.; Holmes, E.C. Expanding the RNA virosphere by unbiased metagenomics. Annu. Rev. Virol. 2019, 6, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benko, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Krupovic, M.; Mushegian, A.; Kropinski, A.M.; Siddell, S.G.; Varsani, A.; Adams, M.J.; Davison, A.J.; Dutilh, B.E.; Harrach, B.; et al. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar]
- Dutilh, B.E.; Varsani, A.; Tong, Y.; Simmonds, P.; Sabandzovic, S.; Rubino, L.; Roux, S.; Munoz, A.R.; Lood, C.; Lefkowitz, E.J.; et al. Perspective on taxonomic classification of uncultivated viruses. Curr. Opin. Virol. 2021, 51, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and evolution of the global RNA virome. mBio 2018, 9, e02329-18. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef]
- Fuhrman, J.A. Marine viruses and their biochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Suttle, C.A. Viruses and nutrient cycles in the sea. BioScience 1999, 49, 781–788. [Google Scholar] [CrossRef]
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Thurber, R.V. Viruses manipulate the marine environment. Nature 2009, 459, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Ignacio-Espinoza, J.C.; Roux, S.; Doulcier, G.; Acinas, S.G.; Alberti, A.; Chaffron, S.; Cruaud, C.; de Vargas, C.; Gasol, J.M.; et al. Patterns and ecological drivers of ocean viral communities. Science 2015, 348, 1261498. [Google Scholar] [CrossRef] [PubMed]
- Rossinck, M.J. Viruses in the phytobiome. Curr. Opin. Virol. 2019, 37, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Martin, D.P.; Elena, S.F.; Shepherd, D.N.; Roumagnac, P.; Varsani, A. Evolution and ecology of plant viruses. Nat. Rev. Microbiol. 2019, 17, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Rossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant virus metagenomics: Advances in virus discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef]
- Schoelz, J.E.; Stewart, L.R. The role of viruses in the phytobiome. Annu. Rev. Virol. 2018, 5, 93–111. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Matteson, A.R. Freshwater and marine virioplankton: A brief overview of commonalities and differences. Freshw. Biol. 2008, 53, 1076–1089. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef]
- Peduzzi, P. Virus ecology of fluvial systems: A blank spot on the map? Biol. Rev. 2016, 91, 937–949. [Google Scholar] [CrossRef]
- Williamson, K.E.; Fuhrmann, J.J.; Wommack, K.E.; Radosevich, M. Viruses in soil ecosystems: An unknown quantity within an unexplored territory. Annu. Rev. Virol. 2017, 4, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Culley, A. New insight into the RNA aquatic virosphere via viromics. Virus Res. 2018, 244, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L. A decade of RNA virus metagenomics is (not) enough. Virus Res. 2018, 244, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Silas, S.; Wang, Y.; Wu, S.; Bocek, M.; Kazlauskas, D.; Krupovic, M.; Fire, A.; Dolja, V.V.; Koonin, E.V. Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat. Microbiol. 2020, 5, 1262–1270. [Google Scholar] [CrossRef]
- Chen, Y.M.; Sadiq, S.; Tian, J.H.; Chen, X.; Lin, X.D.; Shen, J.J.; Chen, H.; Hao, Z.Y.; Wille, M.; Zhou, Z.C.; et al. RNA viromes from terrestrial sites across China expang environmental viral diversity. Nat. Microbiol. 2022, 7, 1312–1323. [Google Scholar] [CrossRef]
- Zell, R.; Groth, M.; Selinka, L.; Selinka, H.C. Picorna-like viruses of the Havel River, Germany. Front. Microbiol. 2022, 13, 865287. [Google Scholar] [CrossRef]
- Zell, R.; Groth, M.; Selinka, L.; Selinka, H.C. Hepeliviruses in two waterbodies in Berlin, Germany. Arch. Virol. 2023, 168, 9. [Google Scholar] [CrossRef]
- Wyn-Jones, A.P.; Carducci, A.; Cook, N.; D’Agostino, M.D.; Divizia, M.; Fleischer, J.; Gantzer, A.; Girones, R.; Höller, C.; de Roda Husman, A.M.; et al. Surveillance of adenoviruses and noroviruses in European recreational waters. Water Res. 2011, 45, 1025–1038. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 2011, 17, 200. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQTREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Rochon, D.; Lommel, S.; Martelli, G.P.; Rubino, L.; Russo, M. Chapter: Family Tombusviridae. In Virus Taxonomy: Ninth Report of the International Committe on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2012; pp. 1111–1138. [Google Scholar]
- Firth, A.E.; Brierley, I. Non-canonical translation in RNA viruses. J. Gen. Virol. 2012, 93, 1385–1409. [Google Scholar] [CrossRef]
- Greninger, A.L.; DeRisi, J.L. Draft genome sequence of tombunodavirus UC1. GenomeA 2015, 3, e00655-15. [Google Scholar] [CrossRef]
- Somera, M.; Fargette, D.; Hébrard, E.; Sarmiento, C.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Solemoviridae 2021. J. Gen. Virol. 2021, 102, 001707. [Google Scholar] [CrossRef]
- Ling, R.; Pate, A.E.; Carr, J.P.; Firth, A.E. An essential fifth coding ORF in the sobemoviruses. Virology 2013, 446, 397–408. [Google Scholar] [CrossRef]
- Schneemann, A.; Zhong, W.; Gallagher, T.M.; Rueckert, R.R. Maturation cleavage required for infectivity of a nodavirus. J. Virol. 1992, 66, 6728–6734. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Adkins, S.; Bragard, C.; Gilmer, D.; Li, D.; MacFarlane, S.A.; Wong, S.M.; Melcher, U.; Ratti, C.; Ryu, K.H.; et al. ICTV Virus Taxonomy Profile: Virgaviridae. J. Gen. Virol. 2017, 98, 1999–2000. [Google Scholar] [CrossRef] [PubMed]
- Valverde, R.A.; Khalifa, M.E.; Okada, R.; Fukuhara, T.; Sabanadzovic, S.; ICTV Report Consortium. Virus Taxonomy Profile: Endornaviridae. J. Gen. Virol. 2019, 100, 1204–1205. [Google Scholar] [CrossRef] [PubMed]
- Inoue-Nagata, A.K.; Jordan, R.; Kreuze, J.; Li, F.; López-Moya, J.J.; Mäkinen, K.; Ohshima, K.; Wylie, S.J.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Potyviridae 2022. J. Gen. Virol. 2022, 103, 001738. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Seifert, M.; Bartenschlager, R.; Seitz, S. Discovery of highly divergent lineages of plant-associated astro-like viruses sheds light on the emergence of potyviruses. Virus Res. 2019, 260, 38–48. [Google Scholar] [CrossRef]
- French, R.; Charon, J.; Le Lay, C.; Muller, C.; Holmes, E.C. Human land use impacts viral diversity and abundance in a New Zealand river. Virus Evol. 2022, 8, veac032. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Shan, T.; Wang, Y.; Yang, J.; Chen, X.; Xiao, Y.; You, Z.; He, Y.; Zhao, M.; Lu, J.; et al. Virome of riverside phytocommunity ecosystem of an ancient canal. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Dodds, J.A. Satellite tobacco mosaic virus. Annu. Rev. Phytopathol. 1998, 36, 295–310. [Google Scholar] [CrossRef]
- García, M.L.; Bó, E.D.; da Graca, J.V.; Gago-Zachert, S.; Hammond, J.; Moreno, P.; Natsuaki, T.; Pallás, V.; Navarro, J.A.; Reyes, C.A.; et al. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Ophioviridae. J. Gen. Virol. 2017, 98, 1161–1162. [Google Scholar] [CrossRef]
- Kreuze, J.F.; Vaira, A.M.; Menzel, W.; Candresse, T.; Zavriev, S.K.; Hammond, J.; Ryu, K.H.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Alphaflexiviridae. J. Gen. Virol. 2020, 101, 699–700. [Google Scholar] [CrossRef]
- Bujarski, J.; Gallitelli, D.; García-Arenal, F.; Pallás, V.B.; Palukaitis, P.; Reddy, M.K.; Wang, A.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Bromoviridae. J. Gen. Virol. 2019, 100, 1206–1207. [Google Scholar] [CrossRef] [PubMed]
- Beyer, S.; Szewzyk, R.; Gnirss, R.; Johne, R.; Selinka, H.C. Detection and characterization of hepatitis-E virus genotype 3 in wastewater and urban surface waters in Germany. Food Environ. Virol. 2020, 12, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.; Guinot-Moreno, F.J.J.; Sanz-Lopez, C.; Galipienso, L. Discovery and diagnosis of a new sobemovirus infecting Cyperus esculentus showing leaf yellow mosaic and dwarfism using small-RNA high throughput sequencing. Plants 2022, 11, 2002. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA viroshere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Zhang, T.; Breitbart, M.; Lee, W.H.; Run, J.Q.; Wei, C.L.; Soh, S.W.I.; Hibberd, M.L.; Liu, E.T.; Rohwer, F.; Ruan, Y.J. RNA viral community in human feces: Prevalence of plant pathogenic viruses. PLoS Biol. 2006, 4, e3. [Google Scholar] [CrossRef]
- Rosario, K.; Symonds, E.M.; Sinigalliano, C.; Stewart, J.; Breitbart, M. Pepper mild mottle virus as an indicator of fecal pollution. Appl. Environ. Microbiol. 2009, 75, 7261–7267. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assignment by DIAMOND to | Teltow Canal (Sample MR233-17) | Havel River (Sample MR644-18) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Order | Family or Genus | No. of Scaffolds | No. of Contigs | No. of Scaffolds | No. of Contigs | ||||
| Total No. | >1 kb | Total No. | >1 kb | Total No. | >1 kb | Total No. | >1 kb | ||
| - | Albetovirus | 30 | 8 | 19 | 4 | 8 | 3 | 8 | 1 |
| Tymovirales | Alphaflexiviridae | 26 | 2 | 15 | 2 | 2 | - | - | - |
| Durnavirales | Amalgaviridae | 2 | 2 * | - | - | - | - | - | - |
| Serpentovirales | Aspiviridae | 4 | 3 | 4 | 2 | 2 | 1 | 2 | 1 |
| - | Aumaivirus | - | - | - | - | - | - | - | - |
| Hepelivirales | Benyviridae | 2 | - | - | - | - | - | - | - |
| Tymovirales | Betaflexiviridae | 9 | - | - | - | - | - | - | - |
| Ourlivirales | Botourmiaviridae | 224 | 142 * | - | - | - | - | - | - |
| Martellivirales | Bromoviridae | 19 | 2 | 16 | 1 | 2 | 1 | 2 | - |
| Martellivirales | Closteroviridae | 25 | - | - | - | - | - | - | - |
| Tymovirales | Deltaflexiviridae | - | - | - | - | - | - | - | - |
| Martellivirales | Endornaviridae | 119 | 42 | 106 | 31 | 12 | - | 8 | - |
| Bunyavirales | Fimoviridae | - | - | - | - | - | - | - | - |
| Tymovirales | Gammaflexiviridae | - | - | - | - | - | - | - | - |
| Martellivirales | Kitaviridae | 1 | 1 * | - | - | - | - | - | - |
| Martellivirales | Mayoviridae | - | - | - | - | - | - | - | - |
| Mononegavirales | Mymonaviridae | 1 | - | - | - | - | - | - | - |
| - | Papanivirus | - | - | - | - | - | - | - | - |
| Durnavirales | Partitiviridae | 95 | 14 | 61 | 9 | 23 | 1 | 11 | 1 |
| Patatavirales | Potyviridae | 34 | 17 | 29 | 22 | 4 | - | 2 | - |
| Mononegavirales | Rhabdoviridae | 3 * | 1 * | - | - | - | - | - | - |
| Picornavirales | Secoviridae | 25 | 5 * | - | - | - | - | - | - |
| Reovirales | Sedoreoviridae | - | - | - | - | - | - | - | - |
| Sobelivirales | Solemoviridae | 190 | 59 | 175 | 68 | 58 | 23 | 47 | 27 |
| Reovirales | Spinareoviridae | - | - | - | - | - | - | - | - |
| Tolivirales | Tombusviridae | 1710 | 356 | 1174 | 295 | 603 | 98 | 395 | 100 |
| Bunyavirales | Tospoviridae | - | - | - | - | - | - | - | - |
| Tymovirales | Tymoviridae | 7 | - | - | - | - | - | - | - |
| Martellivirales | Virgaviridae | 498 | 30 | 232 | 23 | 108 | 10 | 78 | 7 |
| - | Virtovirus | - | - | - | - | - | - | - | - |
| ∑ 2725 | ∑ 533 | ∑ 1831 | ∑ 457 | ∑ 822 | ∑ 137 | ∑ 553 | ∑ 137 | ||
| Reference Viruses of this Study | Teltow Canal Samples | Havel River Sample | ||||
|---|---|---|---|---|---|---|
| MR137-16 | 20161295 | MR233-17 | MR644-18 | |||
| CLC Contigs | CLC Contigs | CLC Contigs | metaSPAdes Scaffolds | CLC Contigs | metaSPAdes Scaffolds | |
| Havel albeto-like viruses (n = 4) | - | - | 2 | 6 | 5 | 4 |
| Havel aspi-like virus (n = 1) | - | - | - | - | 1 | 1 |
| Havel partiti-like virus (n = 1) | - | - | - | - | 1 | 2 |
| Havel solemo-like viruses (n = 25) | 13 | 14 | 64 | 68 | 52 | 35 |
| Havel tombus-like viruses (n = 106) | 37 | 55 | 181 | 324 | 206 | 132 |
| Havel virga-like viruses (n = 13) | 33 | 36 | 27 | 233 | 30 | 25 |
| Teltow Canal albeto-like viruses (n = 23) | - | 1 | 28 | 27 | 6 | 8 |
| Teltow Canal alphaflexi-like viruses (n = 2) | - | 1 | 4 | 2 | - | - |
| Teltow Canal aspi-like viruses (n = 3) | - | - | 5 | 3 | - | - |
| Teltow Canal bromo-like viruses (n = 2) | - | 1 | 3 | 2 | - | 1 |
| Teltow Canal endorna-like viruses (n = 20) | - | - | 57 | 20 | 19 | 31 |
| Teltow Canal partiti-like viruses (n = 14) | - | - | 28 | 15 | 4 | 9 |
| Teltow Canal poty-like viruses (n = 16) | 1 | 19 | 48 | 31 | 48 | 72 |
| Teltow Canal solemo-like viruses (n = 63) | 24 | 67 | 161 | 78 | 60 | 69 |
| Teltow Canal tombus-like viruses (n = 335) | 117 | 319 | 815 | 847 | 274 | 362 |
| Teltow Canal virga-like viruses (n = 21) | 36 | 64 | 78 | 105 | 40 | 70 |
| Virus | Coverage * |
|---|---|
| TC-Tombus-LV-172 | 68,815.7 |
| TC-Solemo-LV-9 | 29,928.8 |
| TC-Tombus-LV-57 | 20,918.6 |
| Teltow Canal cucumber green mottle mosaic virus 1 (Virgaviridae) | 12,382.4 |
| Teltow Canal cucumber green mottle mosaic virus 2 (Virgaviridae) | 9145.23 |
| TC-Tombus-LV-114 | 5535.93 |
| Teltow Canal pepper mild mottle virus (Virgaviridae) | 5325.65 |
| TC-Tombus-LV-40 | 5098.56 |
| TC-Tombus-LV-102 | 4143.25 |
| H-Tombus-LV-2 | 3620.59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zell, R.; Groth, M.; Selinka, L.; Selinka, H.-C. Exploring the Diversity of Plant-Associated Viruses and Related Viruses in Riverine Freshwater Samples Collected in Berlin, Germany. Pathogens 2023, 12, 1458. https://doi.org/10.3390/pathogens12121458
Zell R, Groth M, Selinka L, Selinka H-C. Exploring the Diversity of Plant-Associated Viruses and Related Viruses in Riverine Freshwater Samples Collected in Berlin, Germany. Pathogens. 2023; 12(12):1458. https://doi.org/10.3390/pathogens12121458
Chicago/Turabian StyleZell, Roland, Marco Groth, Lukas Selinka, and Hans-Christoph Selinka. 2023. "Exploring the Diversity of Plant-Associated Viruses and Related Viruses in Riverine Freshwater Samples Collected in Berlin, Germany" Pathogens 12, no. 12: 1458. https://doi.org/10.3390/pathogens12121458