A Targeted “Next-Generation” Sequencing-Informatic Approach to Define Genetic Diversity in Theileria orientalis Populations within Individual Cattle: Proof-of-Principle


Abstract
:1. Introduction
2. Results
2.1. Characteristics of Sequence Datasets, and Clustering of Reads to Define Genotypes
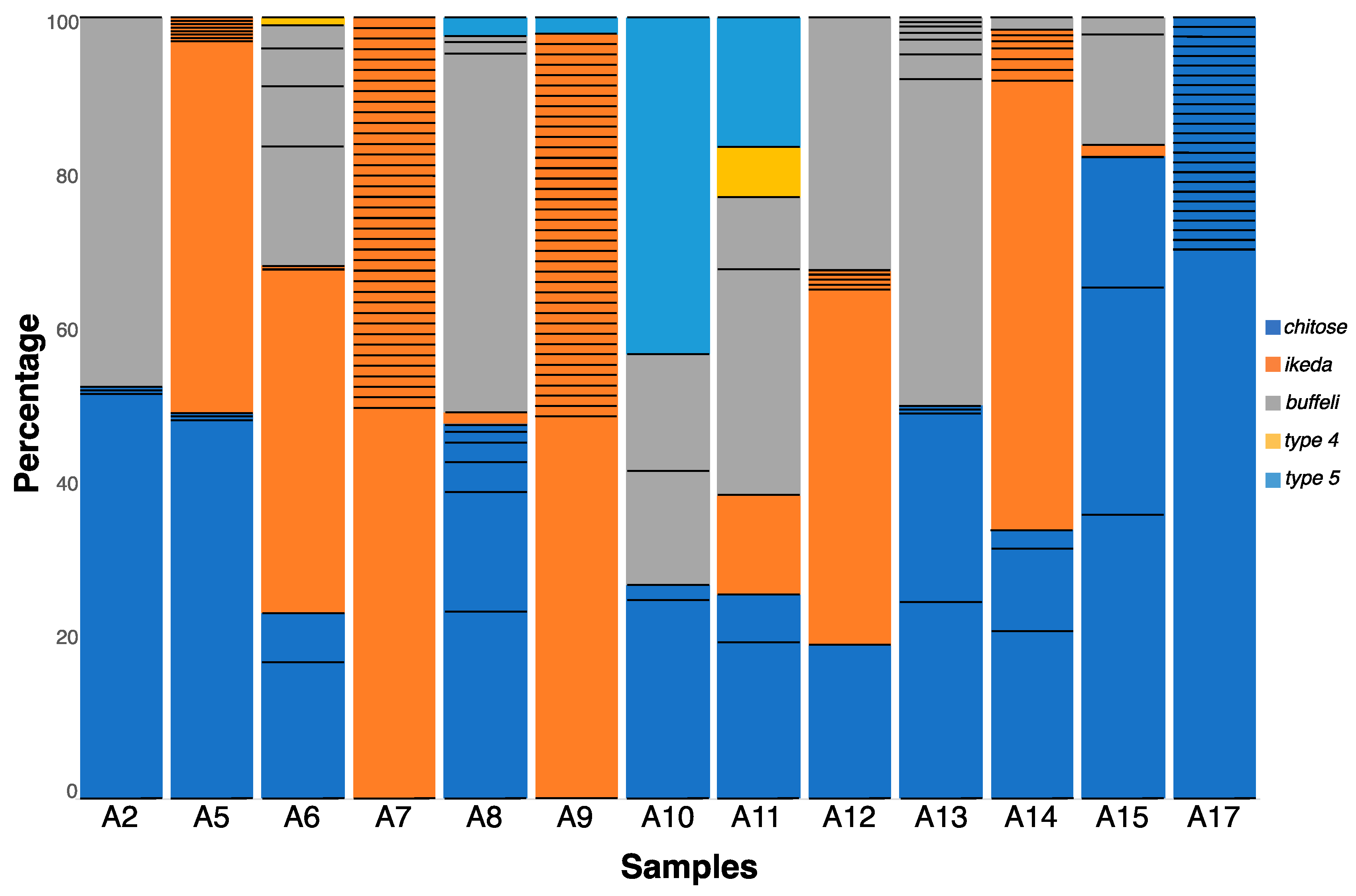
2.2. Extensive MPSP Sequence Diversity within and among Samples
2.3. Genotypes within Samples Usually Represented by a Unique, Dominant Nucleotide Sequence
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Makala, L.H.; Mangani, P.; Fujisaki, K.; Nagasawa, H. The current status of major tick-borne diseases in Zambia. Vet. Res. 2003, 34, 27–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbar, A.; Abbas, T.; Sandhu, Z.U.; Saddiqi, H.A.; Qamar, M.F.; Gasser, R.B. Tick-borne diseases of bovines in Pakistan: Major scope for future research and improved control. Parasit Vectors 2015, 8, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamau, J.; de Vos, A.J.; Playford, M.; Salim, B.; Kinyanjui, P.; Sugimoto, C. Emergence of new types of Theileria orientalis in Australian cattle and possible cause of theileriosis outbreaks. Parasit Vectors 2011, 4, 22. [Google Scholar] [CrossRef]
- Perera, P.K.; Gasser, R.B.; Anderson, G.A.; Jeffers, M.; Bell, C.M.; Jabbar, A. Epidemiological survey following oriental theileriosis outbreaks in Victoria, Australia, on selected cattle farms. Vet. Parasitol. 2013, 197, 509–521. [Google Scholar] [CrossRef]
- Perera, P.K.; Gasser, R.B.; Firestone, S.M.; Anderson, G.A.; Malmo, J.; Davis, G.; Beggs, D.S.; Jabbar, A. Oriental theileriosis in dairy cows causes a significant milk production loss. Parasit Vectors 2014, 7, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.G.; Playford, M.C.; Hickey, K.L. Theileria orientalis: A review. N. Z. Vet. J. 2016, 64, 3–9. [Google Scholar] [CrossRef]
- Gebrekidan, H.; Nelson, L.; Smith, G.; Gasser, R.B.; Jabbar, A. An outbreak of oriental theileriosis in dairy cattle imported to Vietnam from Australia. Parasitology 2017, 144, 738–746. [Google Scholar] [CrossRef]
- Gebrekidan, H.; Perera, P.K.; Ghafar, A.; Abbas, T.; Gasser, R.B.; Jabbar, A. An appraisal of oriental theileriosis and the Theileria orientalis complex, with an emphasis on diagnosis and genetic characterisation. Parasitol. Res. 2020, 119, 11–22. [Google Scholar] [CrossRef]
- Sivakumar, T.; Hayashida, K.; Sugimoto, C.; Yokoyama, N. Evolution and genetic diversity of Theileria. Infect. Genet. Evol. 2014, 27, 250–263. [Google Scholar] [CrossRef] [Green Version]
- Mans, B.J.; Pienaar, R.; Latif, A.A. A review of Theileria diagnostics and epidemiology. Int. J. Parasitol. Parasit Wildl. 2015, 4, 104–118. [Google Scholar] [CrossRef] [Green Version]
- Perera, P.K.; Gasser, R.B.; Firestone, S.M.; Smith, L.; Roeber, F.; Jabbar, A. Semiquantitative multiplexed tandem PCR for detection and differentiation of four Theileria orientalis genotypes in cattle. J. Clin. Microbiol. 2015, 53, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasser, R.B. Molecular tools—Advances, opportunities and prospects. Vet. Parasitol. 2006, 136, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Hemmink, J.D.; Sitt, T.; Pelle, R.; de Klerk-Lorist, L.M.; Shiels, B.; Toye, P.G.; Morrison, W.I.; Weir, W. Ancient diversity and geographical sub-structuring in African buffalo Theileria parva populations revealed through metagenetic analysis of antigen-encoding loci. Int. J. Parasitol. 2018, 48, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, U.; Ali, Q.; Rashid, I.; Shabbir, M.Z.; Ijaz, M.; Abbas, M.; Evans, M.; Ashraf, K.; Morrison, I.; Morrison, L.; et al. Development of a deep amplicon sequencing method to determine the species composition of piroplasm haemoprotozoa. Ticks Tick Borne Dis. 2019, 10, 101276. [Google Scholar] [CrossRef] [PubMed]
- Huggins, L.G.; Koehler, A.V.; Ng-Nguyen, D.; Wilcox, S.; Schunack, B.; Inpankaew, T.; Traub, R.J. A novel metabarcoding diagnostic tool to explore protozoan haemoparasite diversity in mammals: A proof-of-concept study using canines from the tropics. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Combrink, L.; Glidden, C.K.; Beechler, B.R.; Charleston, B.; Koehler, A.V.; Sisson, D.; Gasser, R.B.; Jabbar, A.; Jolles, A.E. Age of first infection across a range of parasite taxa in a wild mammalian population. Biol. Lett. 2020, 16, 20190811. [Google Scholar] [CrossRef] [Green Version]
- Glidden, C.K.; Koehler, A.V.; Hall, R.S.; Saeed, M.A.; Coppo, M.; Beechler, B.R.; Charleston, B.; Gasser, R.B.; Jolles, A.E.; Jabbar, A. Elucidating cryptic dynamics of Theileria communities in African buffalo using a high-throughput sequencing informatics approach. Ecol. Evol. 2019, 10, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.H.; Hathaway, N.J.; Kharabora, O.; Mwandagalirwa, K.; Tshefu, A.; Meshnick, S.R.; Taylor, S.M.; Juliano, J.J.; Stewart, V.A.; Bailey, J.A. A deep sequencing approach to estimate Plasmodium falciparum complexity of infection (COI) and explore apical membrane antigen 1 diversity. Malar. J. 2017, 16, 490. [Google Scholar] [CrossRef] [Green Version]
- Zhong, D.; Lo, E.; Wang, X.; Yewhalaw, D.; Zhou, G.; Atieli, H.E.; Githeko, A.; Hemming-Schroeder, E.; Lee, M.C.; Afrane, Y.; et al. Multiplicity and molecular epidemiology of Plasmodium vivax and Plasmodium falciparum infections in east Africa. Malar. J. 2018, 17, 185. [Google Scholar] [CrossRef] [Green Version]
- Metoh, T.N.; Chen, J.H.; Fon-Gah, P.; Zhou, X.; Moyou-Somo, R.; Zhou, X.N. Genetic diversity of Plasmodium falciparum and genetic profile in children affected by uncomplicated malaria in Cameroon. Malar. J. 2020, 19, 115. [Google Scholar] [CrossRef] [Green Version]
- Hathaway, N.J.; Parobek, C.M.; Juliano, J.J.; Bailey, J.A. SeekDeep: Single-base resolution de novo clustering for amplicon deep sequencing. Nucleic Acids Res. 2017, 46, e21. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, C.; Micallef, M.; Alex, S.M.; Collins, D.; Djordjevic, S.P.; Bogema, D.R. Temporal dynamics and subpopulation analysis of Theileria orientalis genotypes in cattle. Infect. Genet. Evol. 2015, 32, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Bogema, D.R.; Micallef, M.L.; Liu, M.; Padula, M.P.; Djordjevic, S.P.; Darling, A.E.; Jenkins, C. Analysis of Theileria orientalis draft genome sequences reveals potential species-level divergence of the ikeda, chitose and buffeli genotypes. BMC Genom. 2018, 19, 298. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, A.; Hekimoglu, O.; Dincer, E.; Hagedorn, P.; Nitsche, A.; Ergunay, K. A cross-sectional screening by next-generation sequencing reveals Rickettsia, Coxiella, Francisella, Borrelia, Babesia, Theileria and Hemolivia species in ticks from Anatolia. Parasit Vectors 2019, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Pienaar, R.; Josemans, A.; Latif, A.A.; Mans, B.J. The host-specificity of Theileria sp. (sable) and Theileria sp. (sable-like) in African bovidae and detection of novel Theileria in antelope and giraffe. Parasitology 2020, 147, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Boyce, R.M.; Hathaway, N.; Fulton, T.; Reyes, R.; Matte, M.; Ntaro, M.; Mulogo, E.; Waltmann, A.; Bailey, J.A.; Siedner, M.J.; et al. Reuse of malaria rapid diagnostic tests for amplicon deep sequencing to estimate Plasmodium falciparum transmission intensity in western Uganda. Sci. Rep. 2018, 8, 10159. [Google Scholar] [CrossRef] [PubMed]
- Koepfli, C.; Mueller, I. Malaria epidemiology at the clone level. Trends Parasitol. 2017, 33, 974–985. [Google Scholar] [CrossRef]
- Kobayashi, N.; Onuma, M.; Kirisawa, R.; Ohgitani, T.; Takahashi, K.; Sasaki, N.; Kawakami, Y. Monoclonal antibodies against intraerythrocytic merozoites (piroplasms) of Theileria sergenti. Nihon Juigaku Zasshi 1987, 49, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Gubbels, M.J.; Katzer, F.; Hide, G.; Jongejan, F.; Shiels, B.R. Generation of a mosaic pattern of diversity in the major merozoite-piroplasm surface antigen of Theileria annulata. Mol. Biochem. Parasitol. 2000, 110, 23–32. [Google Scholar] [CrossRef]
- Freitas-Junior, L.H.; Bottius, E.; Pirrit, L.A.; Deitsch, K.W.; Scheidig, C.; Guinet, F.; Nehrbass, U.; Wellems, T.E.; Scherf, A. Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature 2000, 407, 1018–1022. [Google Scholar] [CrossRef]
- Duffy, M.F.; Byrne, T.J.; Carret, C.; Ivens, A.; Brown, G.V. Ectopic recombination of a malaria var gene during mitosis associated with an altered var switch rate. J. Mol. Biol. 2009, 389, 453–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claessens, A.; Hamilton, W.L.; Kekre, M.; Otto, T.D.; Faizullabhoy, A.; Rayner, J.C.; Kwiatkowski, D. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of var genes during mitosis. PLoS Genet. 2014, 10, e1004812. [Google Scholar] [CrossRef]
- Montgomery, E.; Charlesworth, B.; Langley, C.H. A test for the role of natural selection in the stabilization of transposable element copy number in a population of Drosophila melanogaster. Genet. Res. 1987, 49, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Gebrekidan, H.; Abbas, T.; Wajid, M.; Ali, A.; Gasser, R.B.; Jabbar, A. Molecular characterisation of Theileria orientalis in imported and native bovines from Pakistan. Infect. Genet. Evol. 2017, 47, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Gebrekidan, H.; Gasser, R.B.; Baneth, G.; Yasur-Landau, D.; Nachum-Biala, Y.; Hailu, A.; Jabbar, A. Molecular characterization of Theileria orientalis from cattle in Ethiopia. Ticks Tick Borne Dis. 2016, 7, 742–747. [Google Scholar] [CrossRef] [Green Version]
- Cufos, N.; Jabbar, A.; de Carvalho, L.M.; Gasser, R.B. Mutation scanning-based analysis of Theileria orientalis populations in cattle following an outbreak. Electrophoresis 2012, 33, 2036–2040. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Sample Code | Total No. of Reads (MOI) | Genotype | |||||
|---|---|---|---|---|---|---|---|
| Chitose A | Chitose B | Ikeda | Buffeli | Type 4 | Type 5 | ||
| A2 | 18,573 (4) | MT428436 | MT428452 | ||||
| A5 | 19,215 (11) | MT428436 | MT428433 | ||||
| A6 | 20,523 (9) | MT428435 | MT428433 | MT428453 | MT428454 | ||
| A7 | 8167 (38) | MT428433 | |||||
| A8 | 14,038 (11) | MT428436 | MT428435 | MT428495 | MT428434 | MT428479 | |
| A9 | 8957 (39) | MT428433 | MT428439 | ||||
| A10 | 26,800 (5) | MT428435 | MT428434 | MT428439 | |||
| A11 | 27,608 (7) | MT428436 | MT428435 | MT428433 | MT428434 | MT428454 | MT428439 |
| A12 | 14,171 (7) | MT428435 | MT428433 | MT428434 | |||
| A13 | 10,032 (11) | MT428435 | MT428434 | ||||
| A14 | 8813 (11) | MT428440 | MT428433 | MT428452 | |||
| A15 | 13,332 (6) | MT428436 | MT428435 | MT428433 | MT428434 | ||
| A17 | 9570 (25) | MT428436 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koehler, A.V.; Jabbar, A.; Hall, R.S.; Gasser, R.B. A Targeted “Next-Generation” Sequencing-Informatic Approach to Define Genetic Diversity in Theileria orientalis Populations within Individual Cattle: Proof-of-Principle. Pathogens 2020, 9, 448. https://doi.org/10.3390/pathogens9060448
Koehler AV, Jabbar A, Hall RS, Gasser RB. A Targeted “Next-Generation” Sequencing-Informatic Approach to Define Genetic Diversity in Theileria orientalis Populations within Individual Cattle: Proof-of-Principle. Pathogens. 2020; 9(6):448. https://doi.org/10.3390/pathogens9060448
Chicago/Turabian StyleKoehler, Anson V., Abdul Jabbar, Ross S. Hall, and Robin B. Gasser. 2020. "A Targeted “Next-Generation” Sequencing-Informatic Approach to Define Genetic Diversity in Theileria orientalis Populations within Individual Cattle: Proof-of-Principle" Pathogens 9, no. 6: 448. https://doi.org/10.3390/pathogens9060448