2.2. Construction and Evaluation of Pharmacophore Models
Pharmacophore models are considered the most successful approach in the development of new therapeutic agents, especially since the last two decades [
17]. One of the advantages is that this technique allows to prioritize quickly and inexpensively molecules with high potential for interaction with the therapeutic target in large databases [
18]. Ten pharmacophore models for MtKasA inhibitors were generated using six molecules of the training set (see
Table 1).
From the models generated, only the third model was excluded from the analysis, because they presented an energy value higher than 100.00 kcal/mol, which reflects the difficulty of the molecules to align with the model [
19]. Discrepancies between energies between the remaining models can be explained mainly to steric hindrances [
20]. The values for Hbond (pharmacophoric concordance) varied from 478.10 to 650.30, while Mol_QRY (fitting of each inhibitor to the model) showed a lower variation from 112.40 to 127.7 kcal/mol.
For the pharmacophore models generated for MtKasA, it was possible to identify that the KasA_002 model (see
Table 1) presents the best values for the GALAHAD™ parameters: an energy value equal to 7.53 kcal/mol, Hbond equal to 650.30 and Mol_QRY equal to 112.40.
Small values for energy and high for Hbond and Mol_QRY are the ideal ones to select the best model [
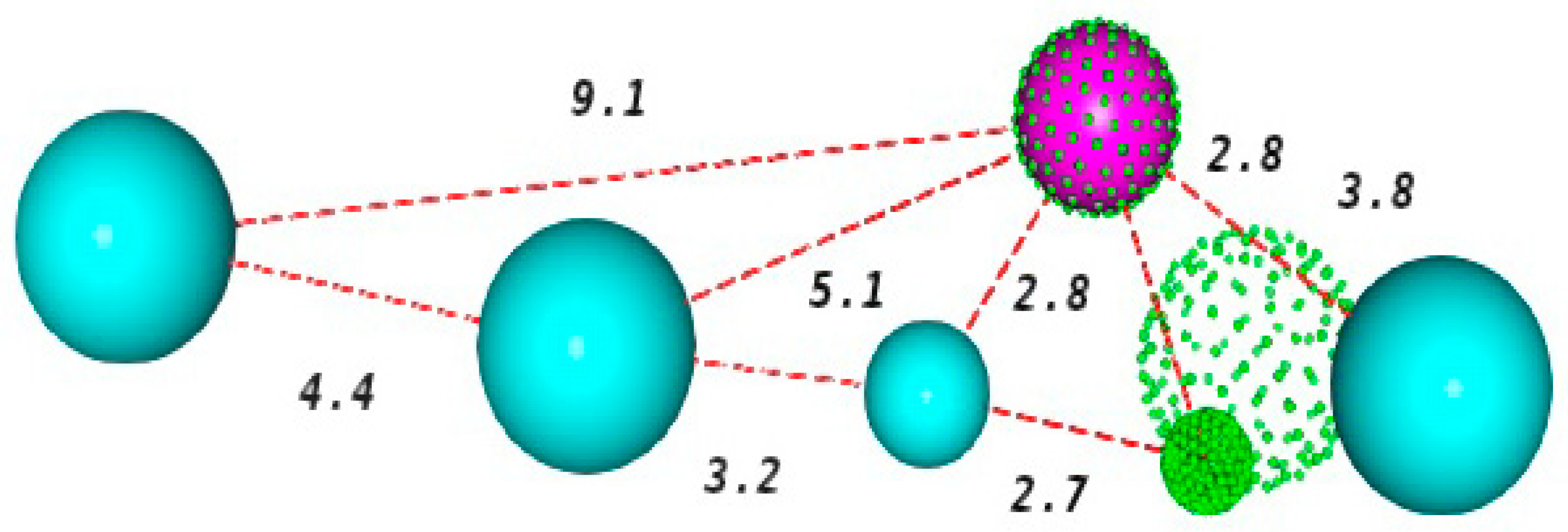
19], because reflects a greater ease of the molecules to mold when aligned with the model, a better pharmacophore agreement and a good fit of each inhibitor to the model. Consequently, such facts have led to the choice of the KasA_002 model. The schematic representation of this model, and the pharmacophore characteristics are shown in
Figure 2.
The spheres describe the area that should be occupied by a certain functional group with identical characteristics to those presented by that of the pharmacophore point [
21]. The selected pharmacophore model has six characteristics, being two hydrogen acceptor centers (green), one hydrogen donor center (magenta) and four hydrophobic centers (blue).
Pharmacophore-Based Virtual Screening
The pharmacophore model displays the key features involved in the interactions within the ligand-target complex. Thereby, this technique can be applied in the quest of compounds, which meet the main molecular requirements for the inhibition of a given target [
22].
In order to select candidate molecules for the biological assays against MtKasA, virtual screening was performed based on the pharmacophore characteristics of the selected model. With the total number of compounds present in the ZINC natural-product database (142,788 molecules), 3896 were aligned with the characteristics of the pharmacophore model KasA_002.
The UNITY platform provides a QFIT value as a scoring form for alignment in the model. The value of QFIT can vary from 0 to 100, in which 100 is the best value and presents a greater complementarity with the pharmacophore [
23]. The QFIT values for the molecules submitted to screening ranged from 1.47 to 66.76. The five molecules with the best QFIT value are shown in
Table 2. ZINC35465970 gives the higher QFIT value for the alignment in the best pharmacophore model of KasA (QFIT = 66.76).
However, the obtained results do not allow the identification of the mode of interaction, molecular volume, as well as do not quantify the affinity energy between the molecule and the receptor, which makes necessary the accomplishment of the docking. Thus, docking assays were performed. The molecules selected for the docking followed the evaluation criterion by the mean value + 2 × standard deviation (34.73). Thereby, 186 molecules were submitted to the docking calculation.
2.3. Docking-Based Virtual Screening
Initially, the scoring function was selected. For this, we used some evaluation metrics such as molecular redocking, KISS score calculation, ROC curve analysis and the enrichment factor. The evaluation of the positioning of the ligand in the orthosteric site from the DOCK’s search algorithm was performed through the values of RMSDs, RMSDh and RMSDm. The RMSDs evaluate the deviation between different heavy atom pairs of hydrogen from the reference conformation to the pose after redocking. In turn, the RMSDm is an implementation of RMSD used in AutoDock Vina [
24], which does not explicitly consider a mapping atom by atom; the deviation is estimated from the minimum distance between any atom of the same element in reference conformations and recoupled. The RMSDh performs a correction of the symmetry between the heavy atoms [
25]. A value of RMSD < 2.0 Å is considered acceptable [
26].
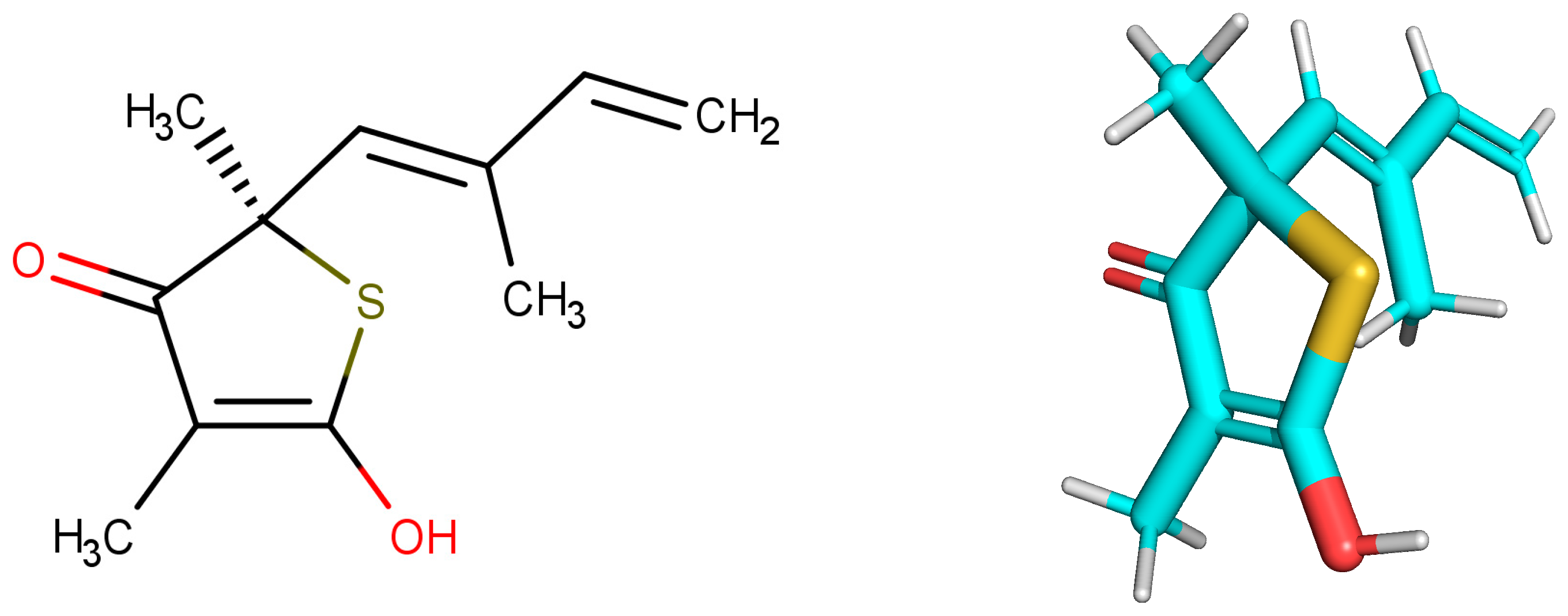
The redocking showed RMSDs = 0.26Å, RMSDh = 0.26Å and RMSDm = 0.14Å (see
Figure 3). The methods of calculation of RMSD indicated excellent results, and the DOCK program similarly reproduced the conformation of the crystallographic ligand in the active site of the protein, since the values found were ideal as recommended by literature [
11,
26]. The second stage of evaluation for the methodology of docking refers to the analysis of the intermolecular interactions after redocking. KISS score is the result of the ratio between the number of hydrogen bonds in the redocked ligand and the number of hydrogen bonds in the crystallographic ligand (PDB = 4C6X). KISS score = 1 was obtained for the target. The interaction of the hydrogen observed with the crystallographic ligand was reproduced after redocking, and no interaction of the same nature was formed. Variations occurred only in the hydrophobic interactions, with the His275 and Phe236 residues.
The third stage for the analysis of the scoring functions consisted in the evaluation of the Receiver Operating Characteristic (ROC) curve. This step is one of the best ways to compare the performance of scoring functions and classifiers [
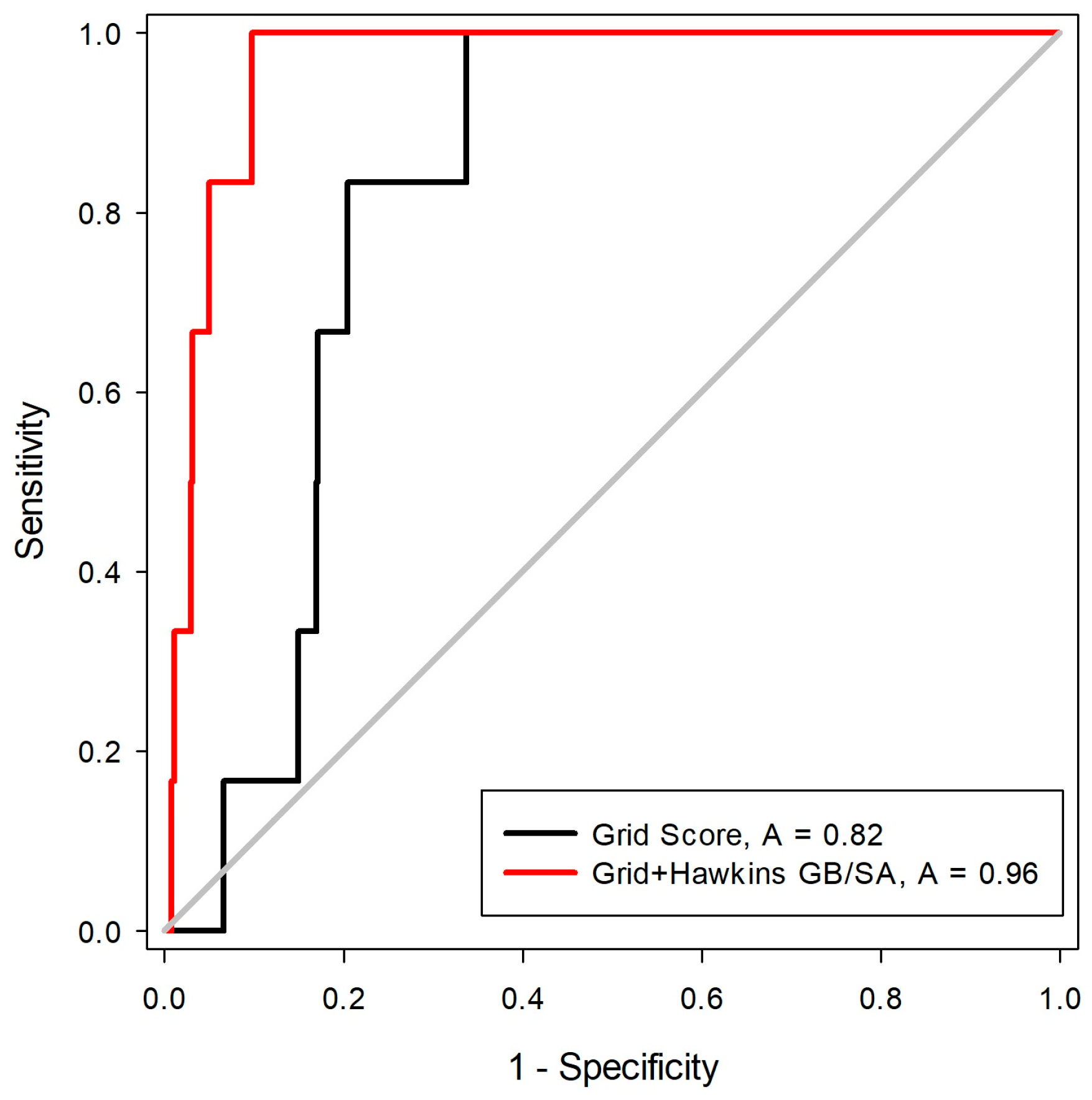
27]. For this, a library was constructed with 550 decoys and six inhibitors, five of these with two states of protonation. After the construction of the molecule bank (ligands + decoys), the docking tests were carried out with two scoring functions (Grid-Hawkins GB/SA and Grid Score). The graph with the ROC curve with the two scoring functions applied can be visualized in
Figure 4.
From the ROC curve graph, it was possible to evaluate the performance of scalar measures of classification, as specificity and sensitivity. The ROC curve is a graphical representation of the sensitivity (proportion of true positives) as a function of specificity (proportion of false positives). The value of the area on the curve (AUC) provides an objective measure of the overall performance of a classifier. An AUC value equal to 1 (or 100%) indicates that the active and inactive compounds are perfectly discriminated, while a value of 0.5 (or 50%) is understood as a random performance [
28]. Matsubara’s studies [
29] show that, in general, the accuracy of the classification method can be evaluated with the following scale: 0.9–1: excellent; 0.8–0.89: good; 0.6–0.79: reasonable; 0.5–0.59: poor; and below 0.49 corresponds to a complete failure.
From the analysis of the ROC curve, it was possible to observe that the scoring function proved to be the most efficient in the recovery of the bioactive compounds for MtKasA was Grid + Hawkins GB/SA, with an AUC = 0.96, which can be considered as an excellent value for the classification.
One of the problems pointed out to the AUC value is the fact that it is a global measure, not clearly presenting information about the early recognition of active compounds [
30,
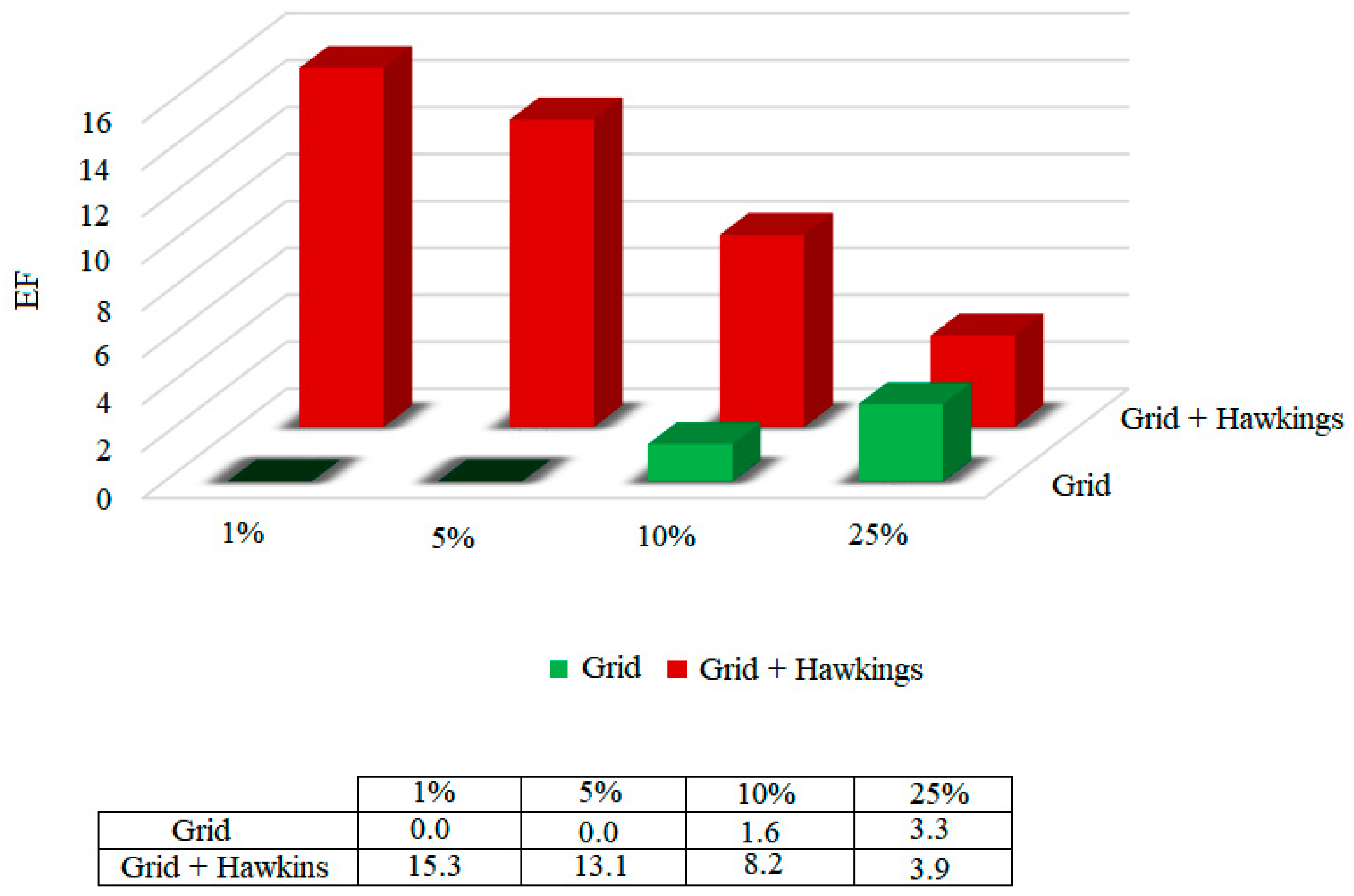
31]. On the other hand, the enrichment factor (EF) quantifies the proportion of active compounds identified when analyzed in a given proportion (nX%) of the total set of ordered compounds. The enrichment factors were calculated for the functions, considering 1%, 5%, 10% and 25% of the bank that was coupled to. The results are shown in
Figure 5.
According to the results presented in
Figure 5, it was possible to highlight that, among the two scoring functions tested, the Grid + Hawkins GB/SA function presents a 15.3 times greater chance of identifying the active compounds in only 1% of the database.
Considering the two analyzes, the area on the curve and the enrichment factor, the Grid + Hawkins GB/SA scoring function was presented as the methodology that best classifies, and therefore presents the greatest accuracy.
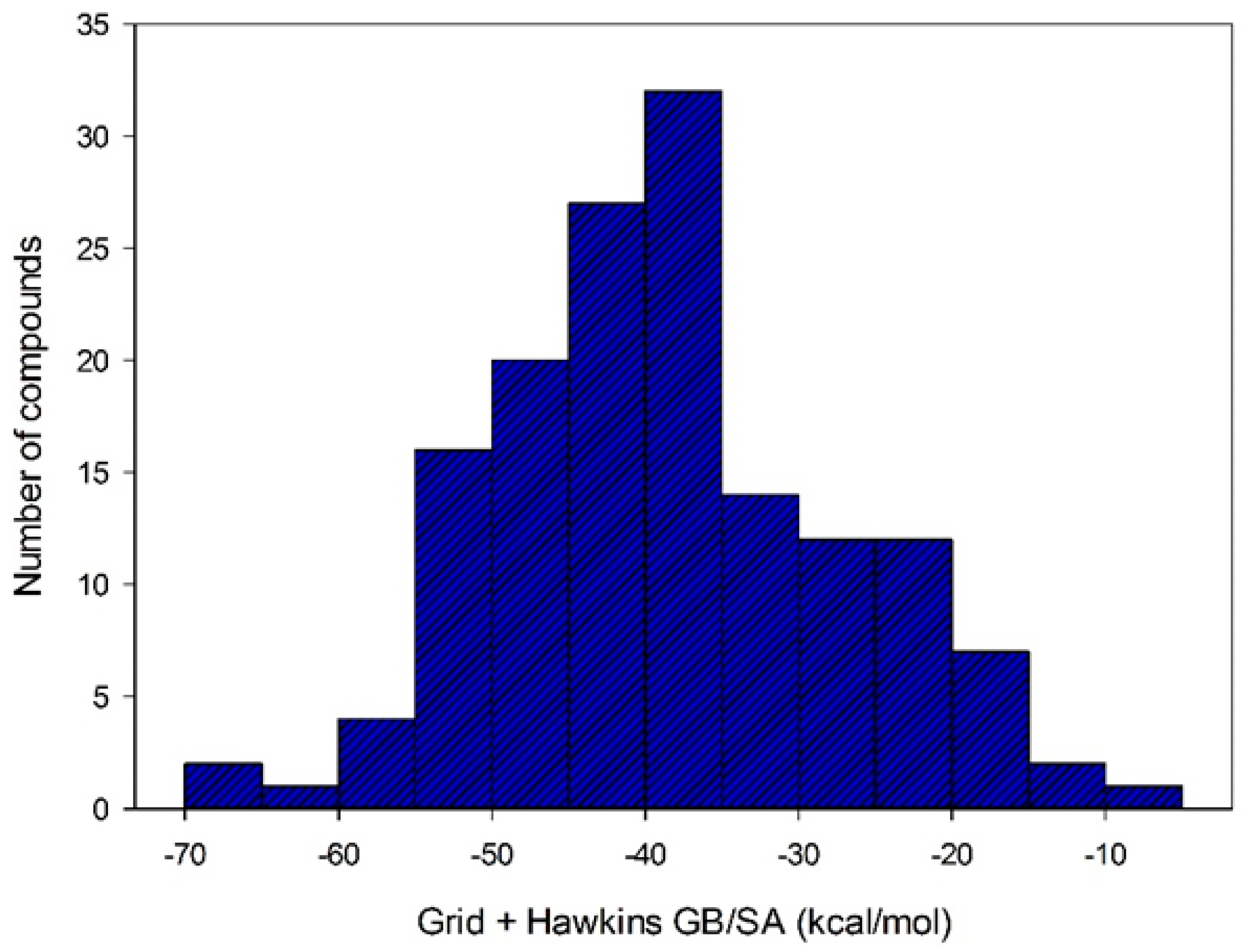
After selecting the scoring function with greater accuracy for each target, the virtual screening was performed by docking using structures with a QFIT value equal to or greater than 34.73. Out the 186 molecules subjected to the docking, only 152 achieved some affinity to MtKasA. The affinity energy values for these molecules ranged from −4.87 to −67.70 kcal/mol (see
Figure 6).
In an attempt to relate the QFIT values obtained from similarity screening and the affinity energy values resulting from docking calculations, a consensual analysis was performed using these two variables (see
Table 3).
The structure with the best score for MtbKasA was ZINC35465970, which has 332.5 g/mol of molecular mass, three Hbond acceptors, three Hbond donors, nine rotatable bonds and partition coefficient (logP) of 6.44. Information extracted from ZINC [
32] PubChem [
33] and ChemSpyder [
34] indicate that such a molecule has not been subjected to biological evaluation. ZINC31170017 presented the second best value of consensus, and the analysis of the physical-chemical properties indicates that this molecule has 462.5 g/mol of molecular mass, nine Hbond acceptors, six Hbond donors, ten rotatable bonds and 1.02 xlogP. It was not possible to identify experiments related with the biological activity for ZINC31170017. Therefore, from the analysis of the scoring function and the similarity alignment (QFIT), molecules ZINC35465970 and ZINC31170017 were submitted for analysis of the intermolecular interactions.
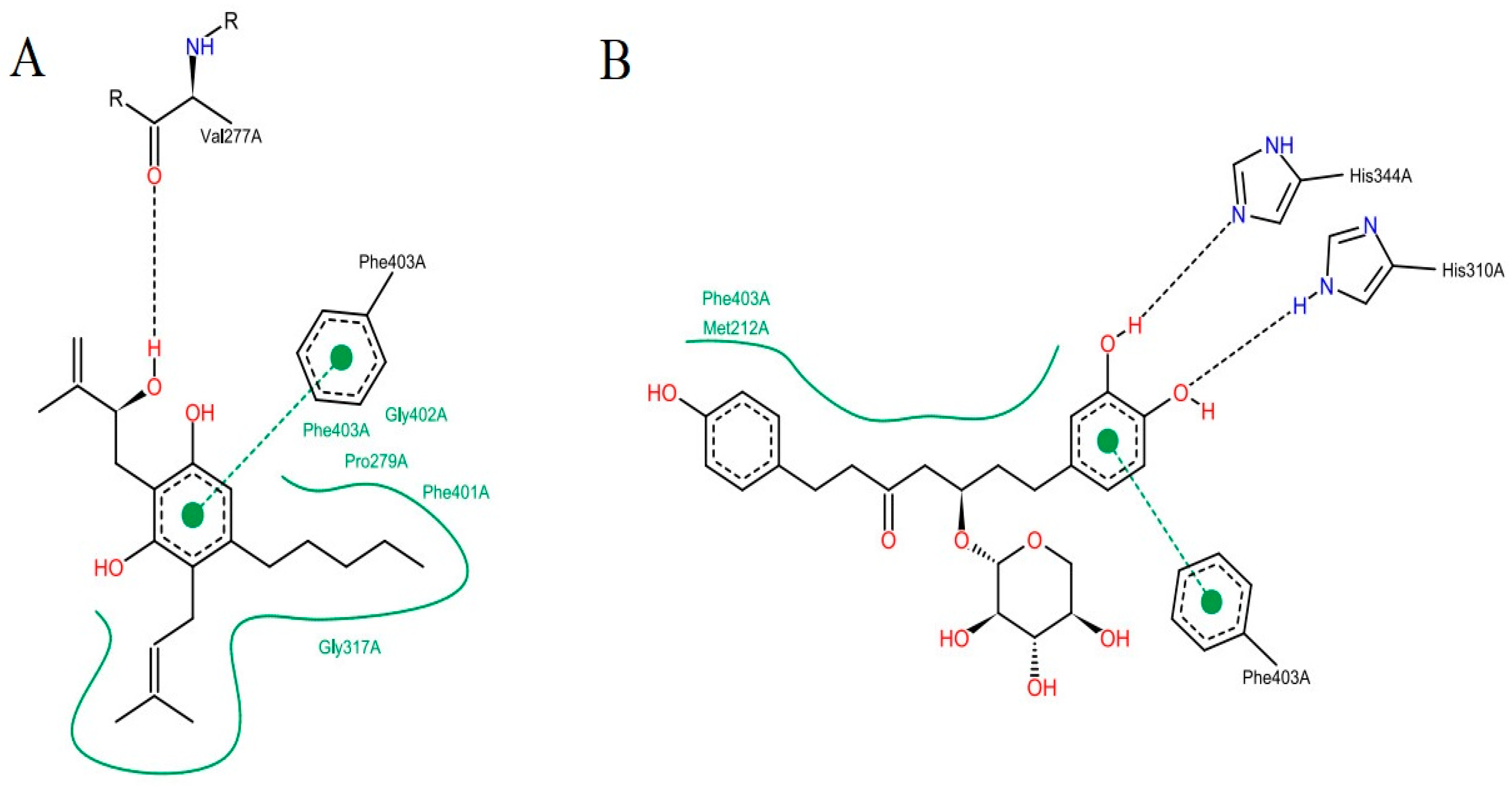
The analysis of the intermolecular interactions is useful for identification and optimization of contacts between ligands and target [
35]. The results for the two molecules selected are shown in
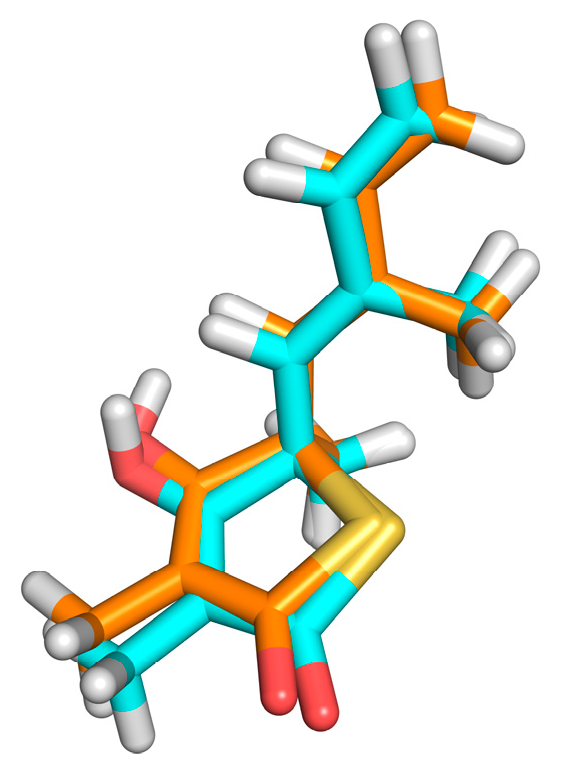
Figure 7: It is possible to point out a greater contribution of the hydrophobic interactions in both molecules. ZINC35465970 (
Figure 7A) performs hydrophobic interactions of its aliphatic chains attached to the dihydroxybenzene ring with the Phe403, Pro 279, Phe401, Gly402 and Gly317 residues. In addition to these interactions, this molecule forms aromatic interaction π-π, between the dihydroxybenzene ring and the aromatic ring of the side chain of Phe403. It is also possible to observe a hydrogen interaction between a hydroxyl of the molecule with the Val277 ketone.
Considering the complex formed between compound ZINC31170017 (
Figure 7B) and MtbKasA, hydrophobic interactions with Phe403 and Met212 residues can be observed. In addition, a hydrophobic π-π interaction with Phe403 and the dihydroxy benzene ring of the molecule can be observed, also present in the molecule with the best score, indicating that this residue may be involved in the molecular recognition process. The third type of binding present is a hydrogen interaction, occurring between the two hydroxyls of the dihydroxy benzene ring with the imidazole rings of His344 and His310. Hydrogen interactions with the same residues (His310 and His344) have already been verified in other studies [
36].
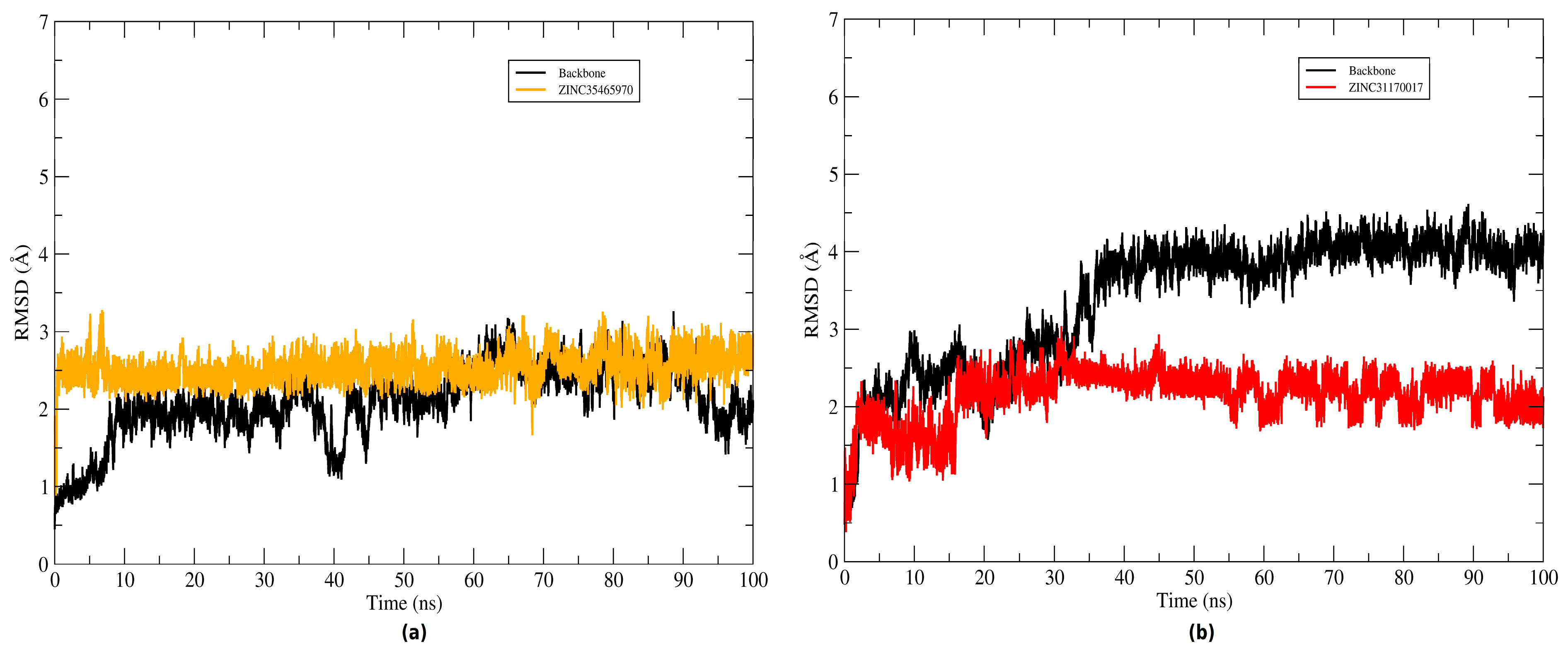
2.4. Structural Analysis of Systems
The root mean square deviation (RMSD) of the atomic positions of protein was plotted to evaluate the structural stability of the complexes (KasA-ZINC31170017 and KasA-ZINC35465970) and the binder backbone along the molecular dynamics trajectory. To plot the backbone and RMSD of the complex graphs the Cα atoms and heavy atoms were used, respectively. The graphs plotted of systems can be seen in
Figure 8. Throughout the simulation time, the inhibitors remained bound to the active site of the protein. The ZINC35465970 ligand remained in equilibrium exhibiting slight structural divergences. During approximately the 20 ns of the initial simulation, ZINC31170017 underwent several conformational changes, but along the trajectory reached a balance and started to exhibit small changes in KasA conformation. Thus, the complexes were considered stable and not discarded for a further analysis.
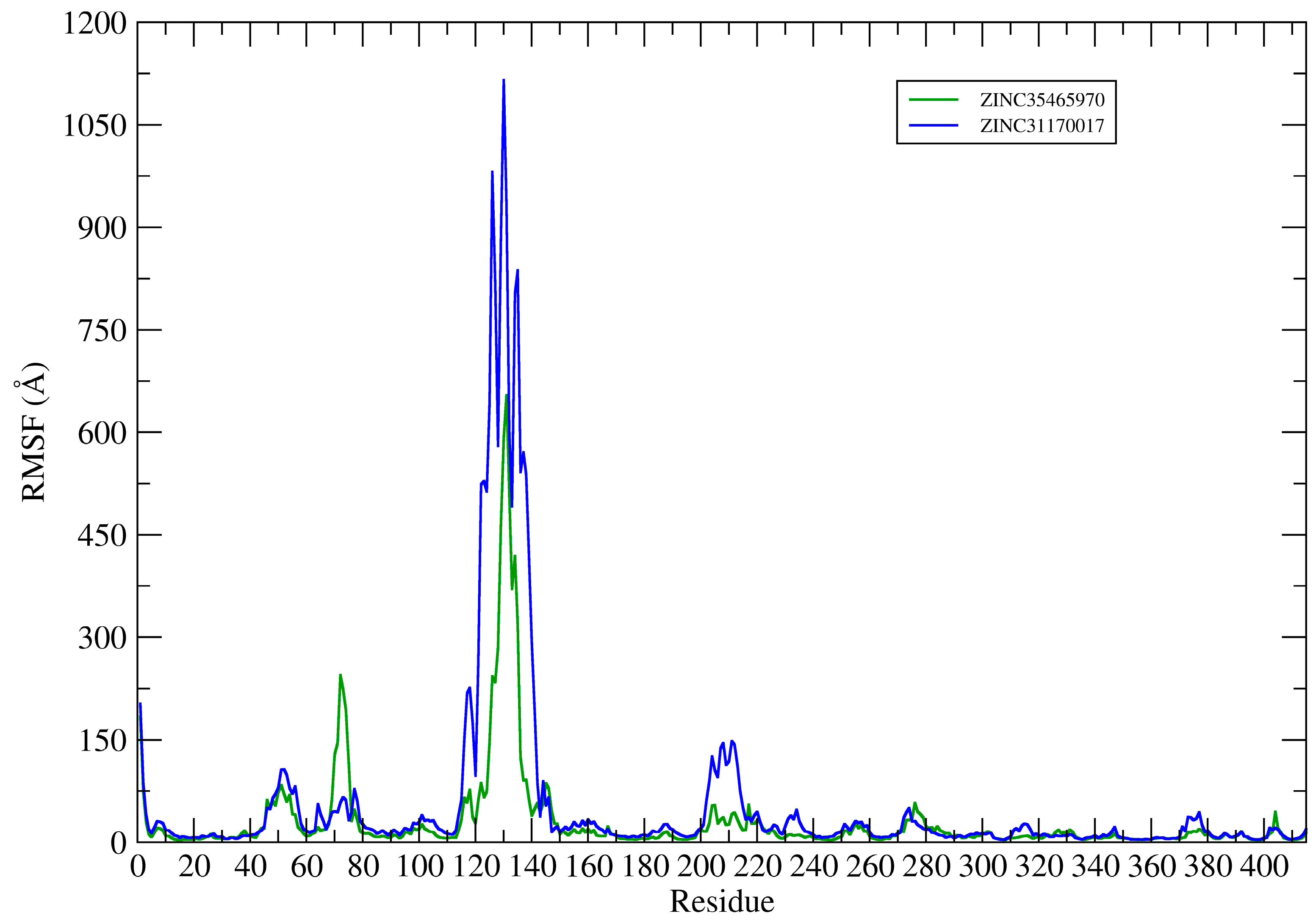
This way, the root mean square fluctuation (RMSF) of the complexes residues (KasA-ZINC31170017 and KasA-ZINC35465970) have been plotted using the Cα atoms to analyze the protein backbone (see
Figure 9).
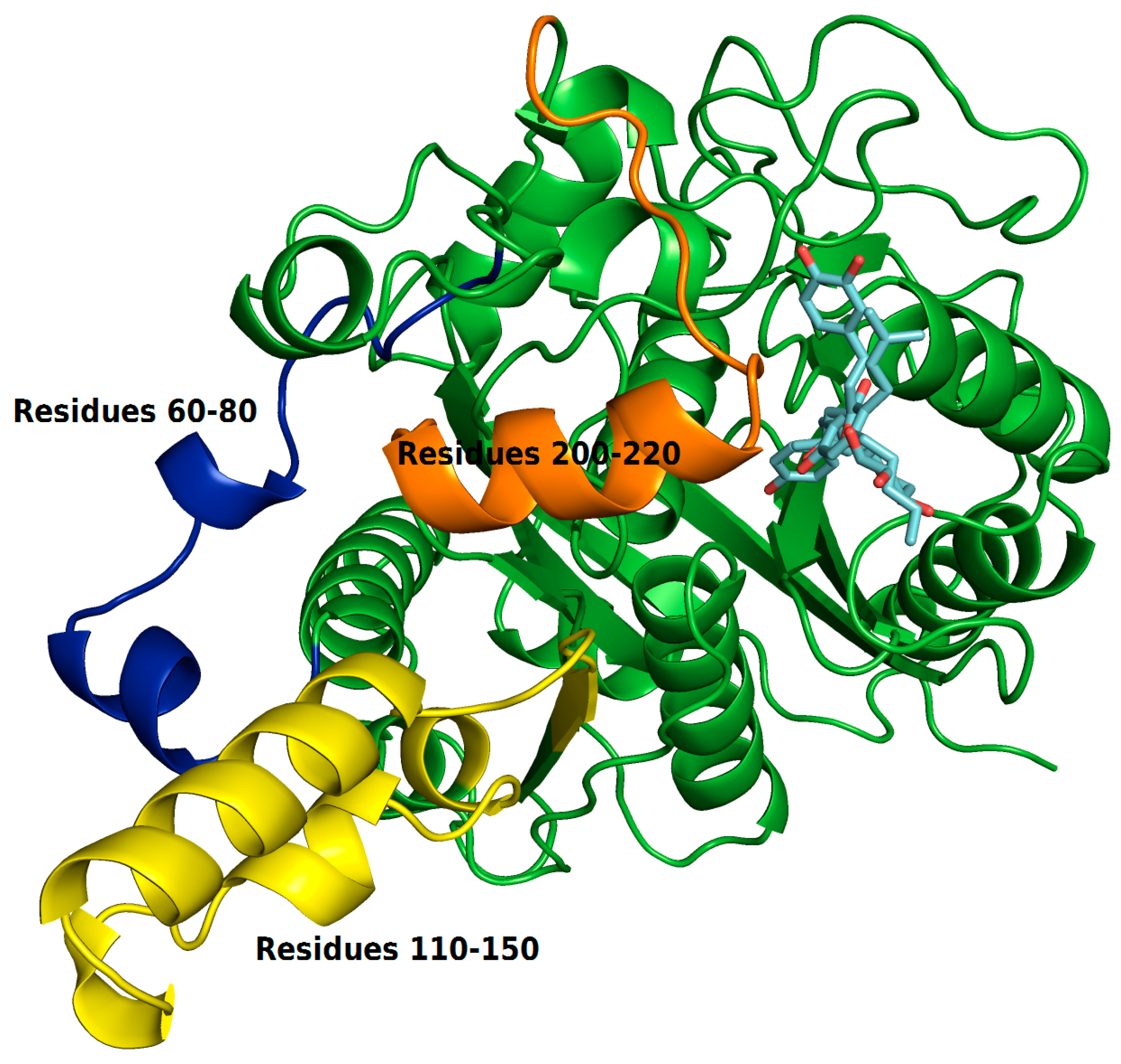
The RMSF plots revealed differences in protein flexibility throughout the trajectory. The largest differences in residue fluctuations occurred in the range of residues 60 to 80, 110 to 150 and 200 to 220. In
Figure 10 these protein segments were colored and identified.
The amino acids 60 to 80 form a loop region and two small alpha helices that are exposed to the solvent. The fluctuation of this backbone region may be related to structural features of loop regions that naturally have a degree of flexibility.
ZINC35465970 compound shifted from its molecular docking position further into the bonding cavity during the simulation. In the molecular docking pose, this ligand did not show many interactions with the alpha helix residues and the loop comprised between residues 200–220. However, after MD simulation and with the balance of ligand in the binding pocket, two hydrophobic chains of the complex underwent conformational reorientation, thus establishing greater hydrophobic interactions with the residues in the range of 200 to 220. ZINC31170017 showed interactions with few residues present at the beginning of alpha helix formed by the residues in the range that we have analyzed. In this way, the alpha helix region remained freer to move, and consequently demonstrating a greater fluctuation.
The region of the protein that showed the greatest fluctuation is composed by the residues 110 to 150. In the system established with ZINC31170017 this region presented a more open conformation, more exposed to the solvent, whereas the same region of the protein in the system formed with ZINC35465970 remained in a tightest conformation, in comparison to each other.
2.4.1. Hydrogen Bonds Established between Receptor-Ligands
Taking the RMSD and RMSF results are a visual analysis of representative MD trajectory, the formation of hydrogen bonds during the entire computational simulation time was carried out to investigate the interaction profile of complexes. The main interactions established are shown in
Table 4.
There was a difference between the number of hydrogen bonds established by compounds in the binding pocket. ZINC35465970 showed the highest number of hydrogen bonds formed, which can justify its greater structural stability verified in the RMSD graph and also its greater capacity to connect the enzyme, and consequently its greater power of inhibition verified in binding free energy value (ΔGbind = −30.90 kcal/mol). However, ZINC31170017 established only two hydrogen bonds with the receptor, with this presented lower conformational stability throughout the simulation and less capacity of interaction with the protein.
2.4.2. Bind Free Energy KasA-Ligands
The free energy values and their energy components are summarized in
Table 5. The results obtained by the MM/GBSA method suggest that ZINC35465970 has a greater capacity to inhibit the KasA enzyme, since we obtained the value of ΔG
bind = −30.90 kcal/mol, whereas the ZINC31170017 reached the value of ΔG
bind = −27.49 kcal/mol. The van der Waals interactions (ΔE
vdW) were the main responsible for maintaining the enzyme-inhibitor complexes, in the systems formed, ΔE
vdW presented values of −45.21 kcal/mol and −35.86 kcal/mol, to ZINC35465970 and ZINC31170017, respectively. The electrostatic (ΔE
ele) and non-polar (ΔG
NP) contributions also favored the established systems. For the interaction with ZINC35465970, ΔE
ele = −11.93 kcal/mol and ΔG
NP = −6.06 kcal/mol for ZINC31170017 ΔE
ele = −11.48 kcal/mol and ΔGNP = −5.18 kcal/mol were obtained.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










