
Naturally Occurring 8ß,13ß-kaur-15-en-17-al and Anti-Malarial Activity from Podocarpus polystachyus Leaves

 ,
,  , ,
, ,
Abstract
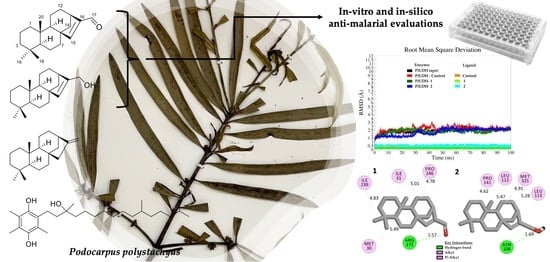
:
1. Introduction
2. Results and Discussion
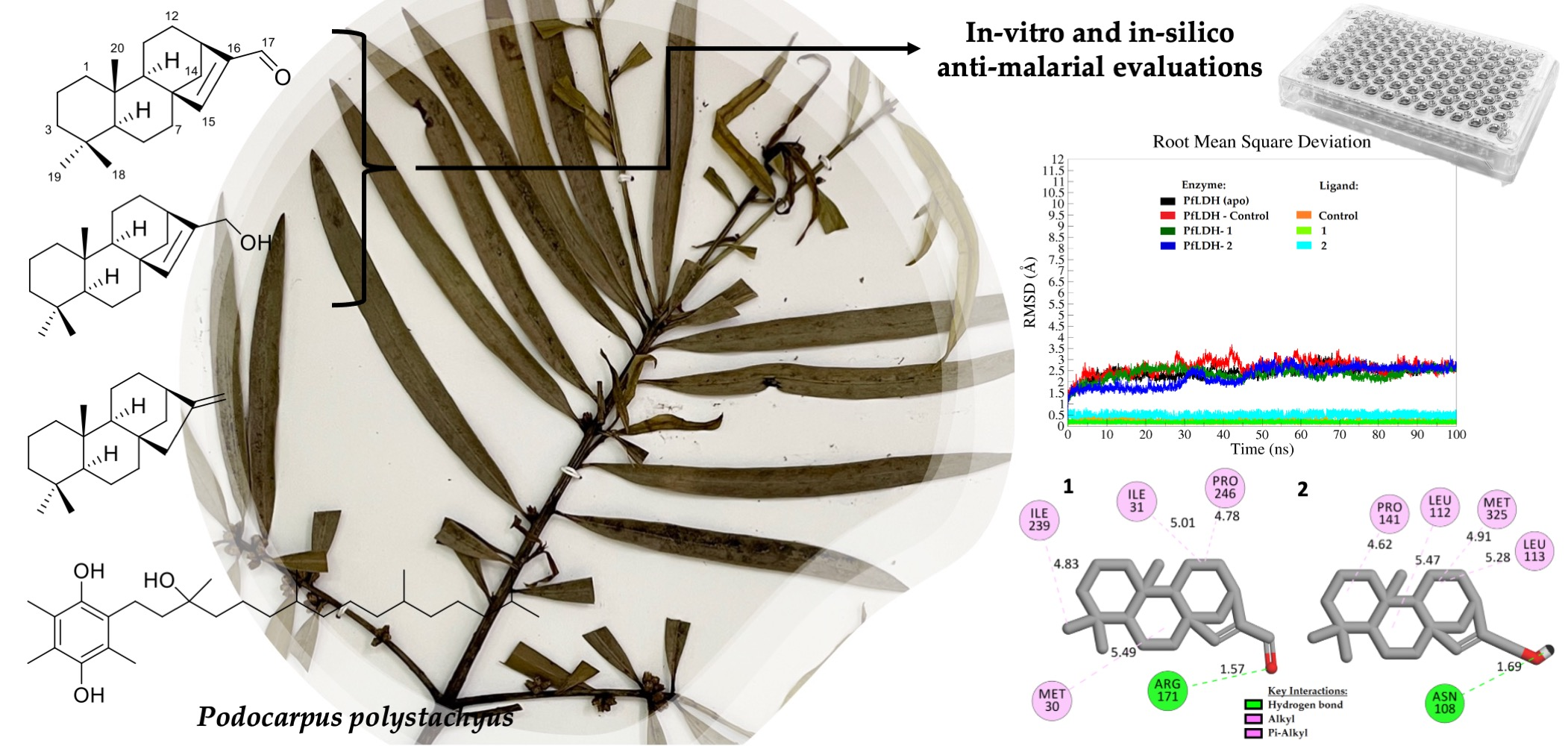
2.1. Isolation and Identification of Chemical Constituents
2.2. In Vitro Anti-Malarial Activity
2.3. In Silico Anti-Malarial Activity
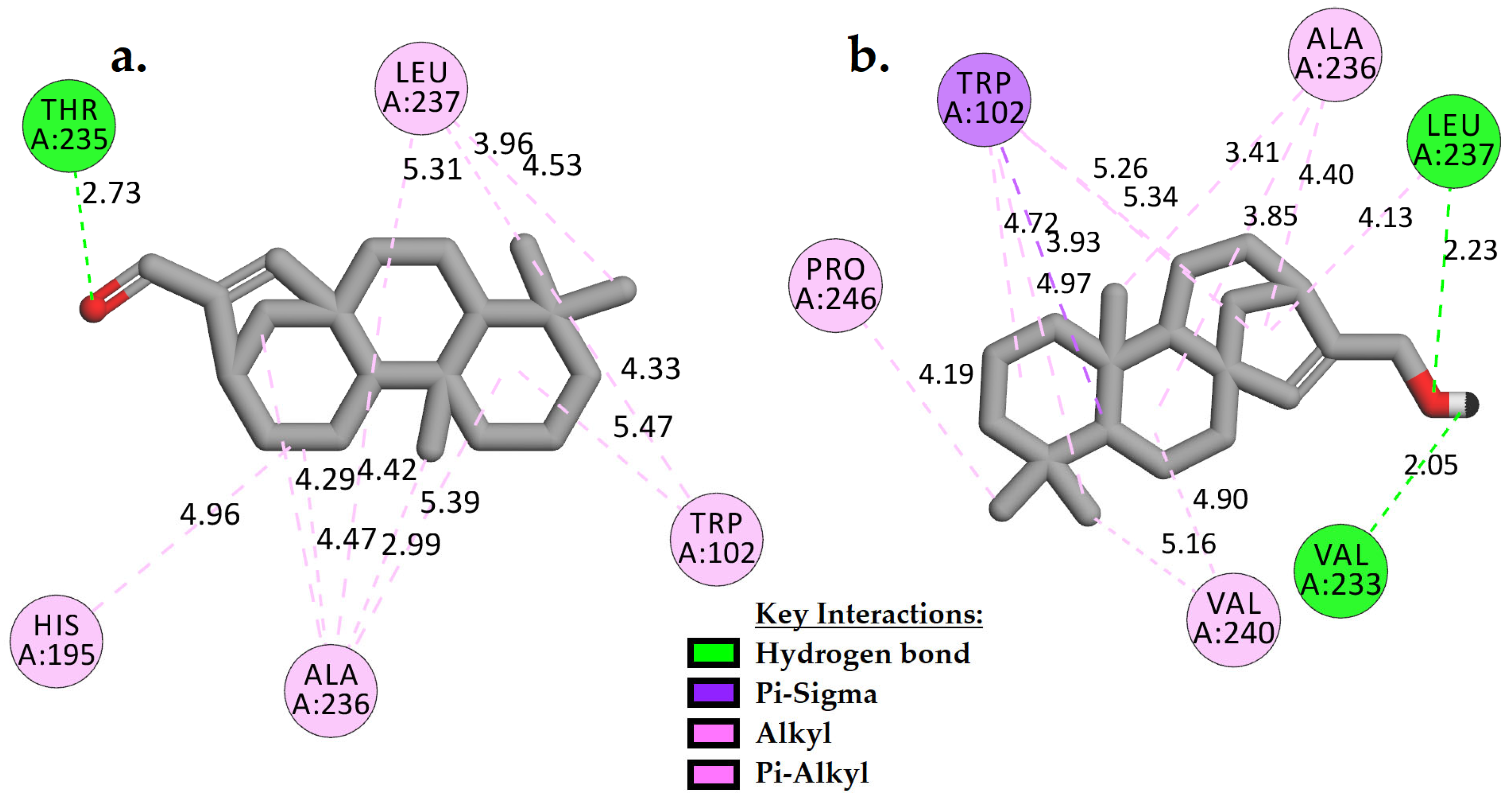
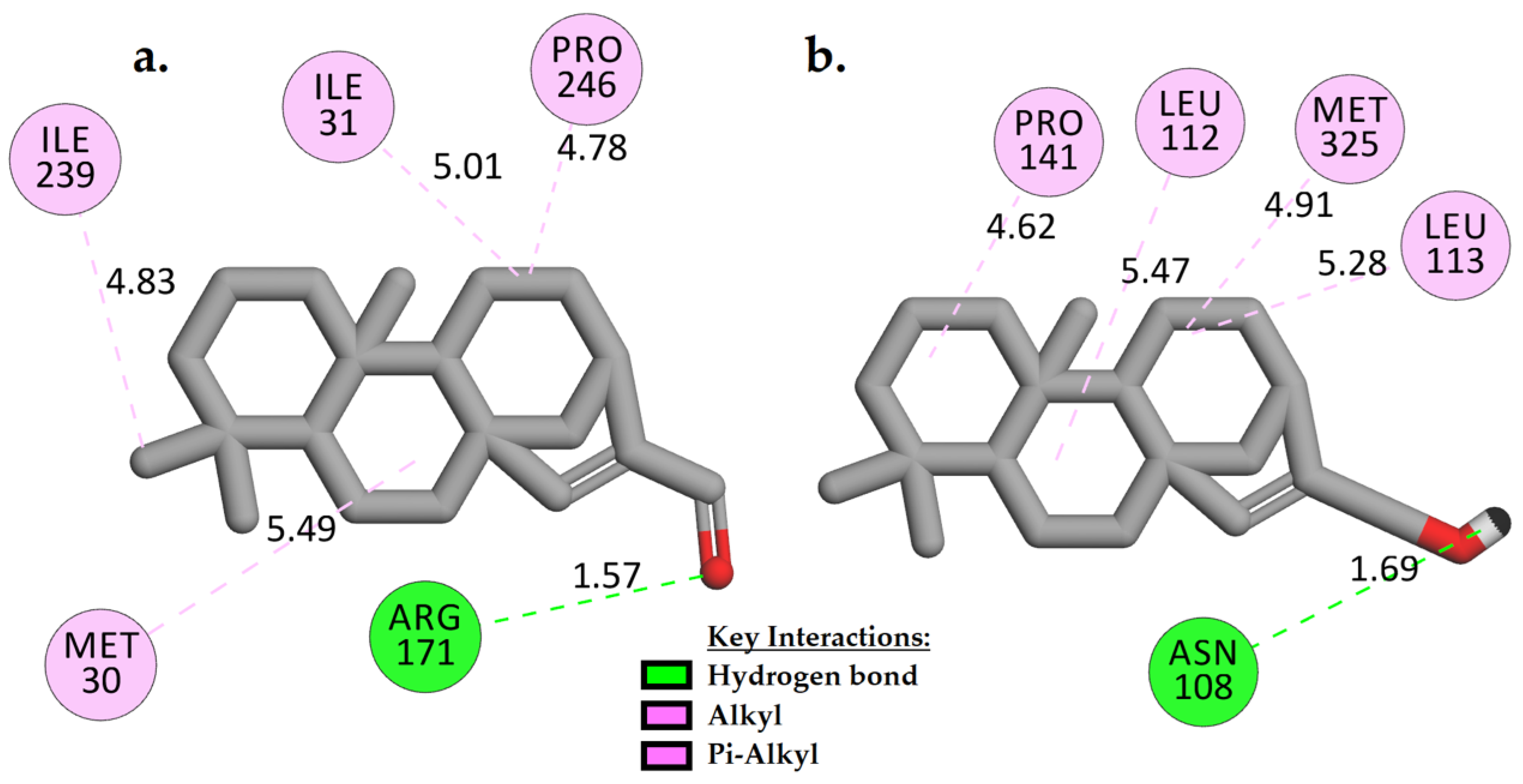
2.3.1. Molecular Docking Analysis
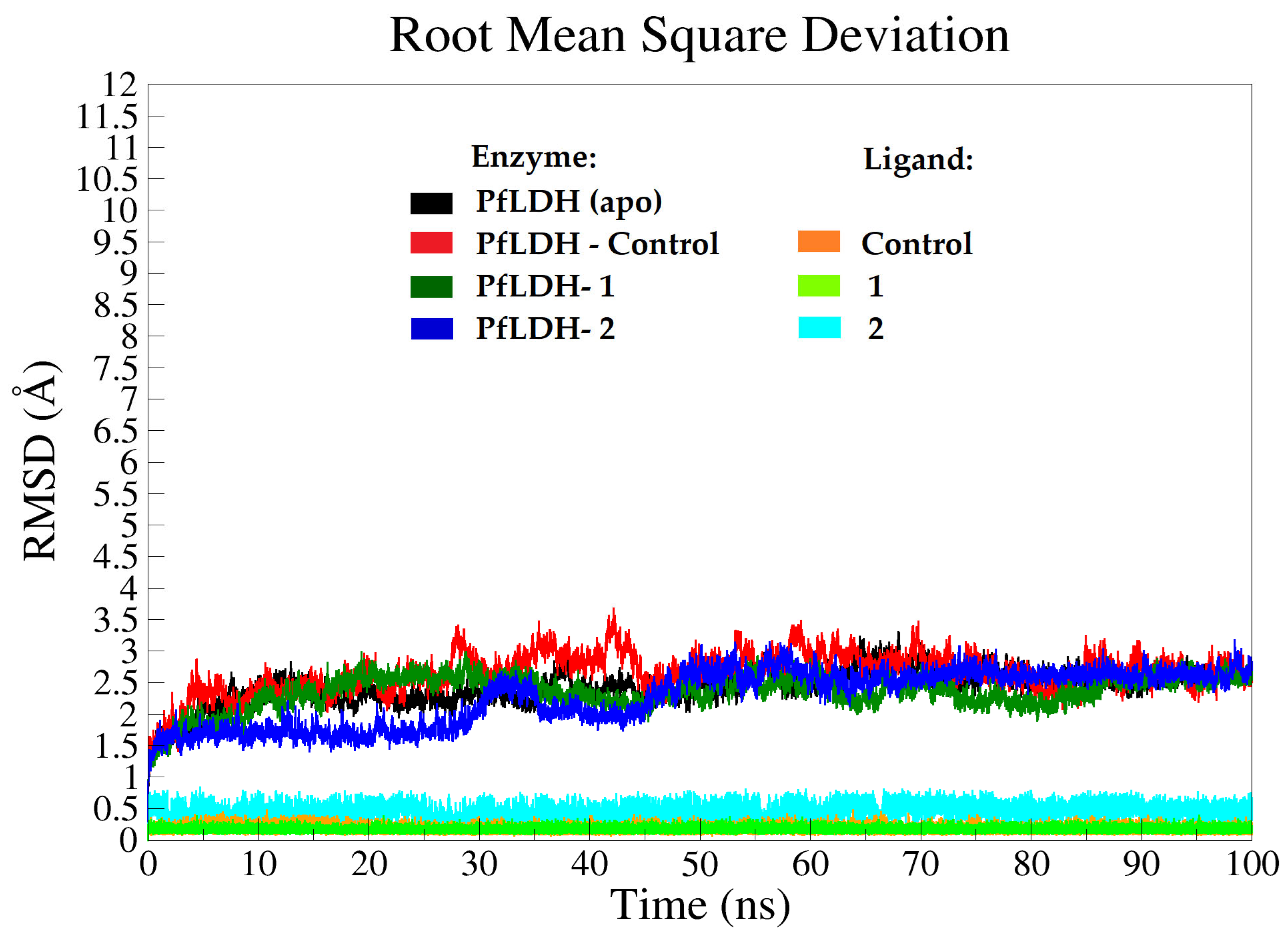
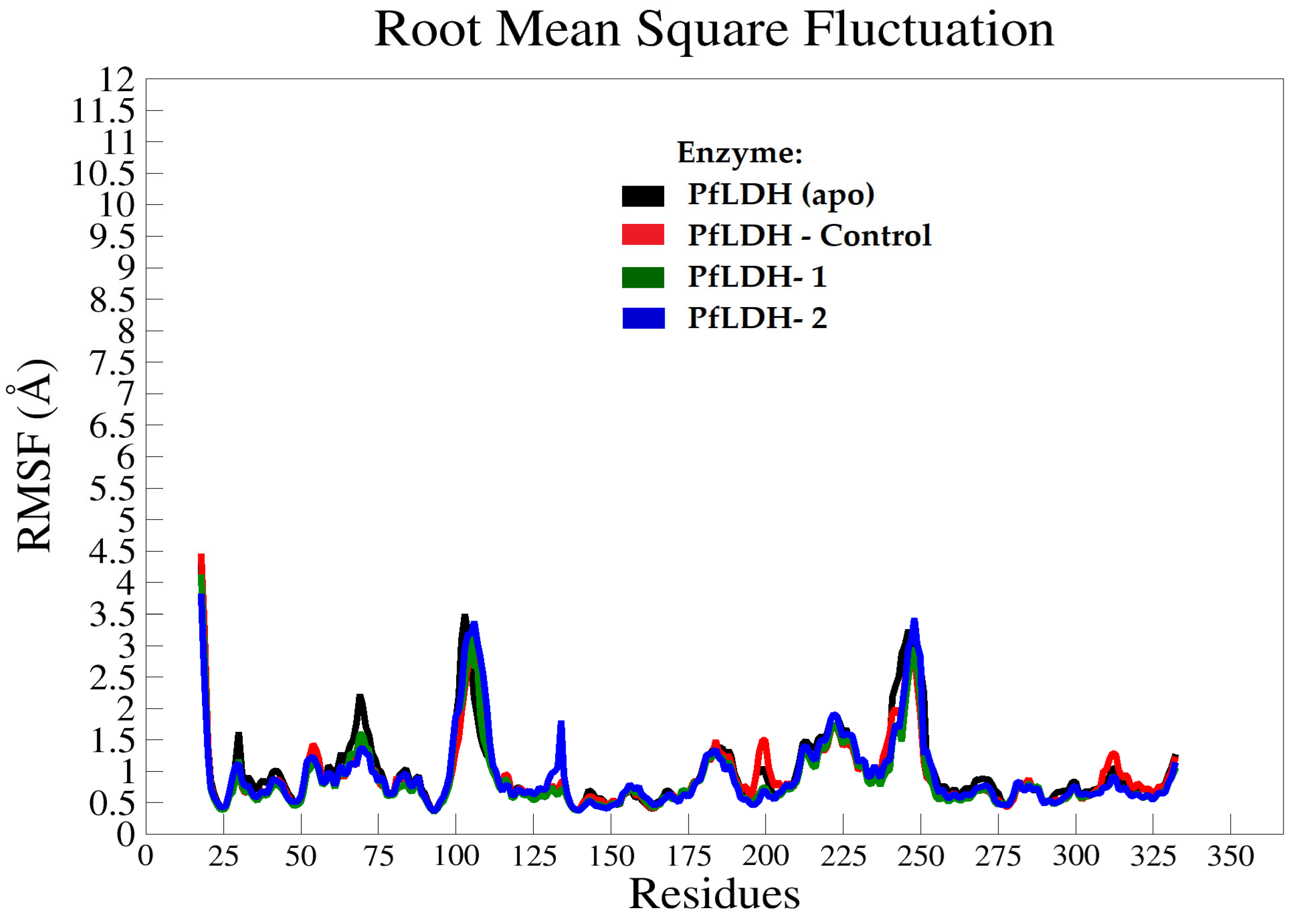
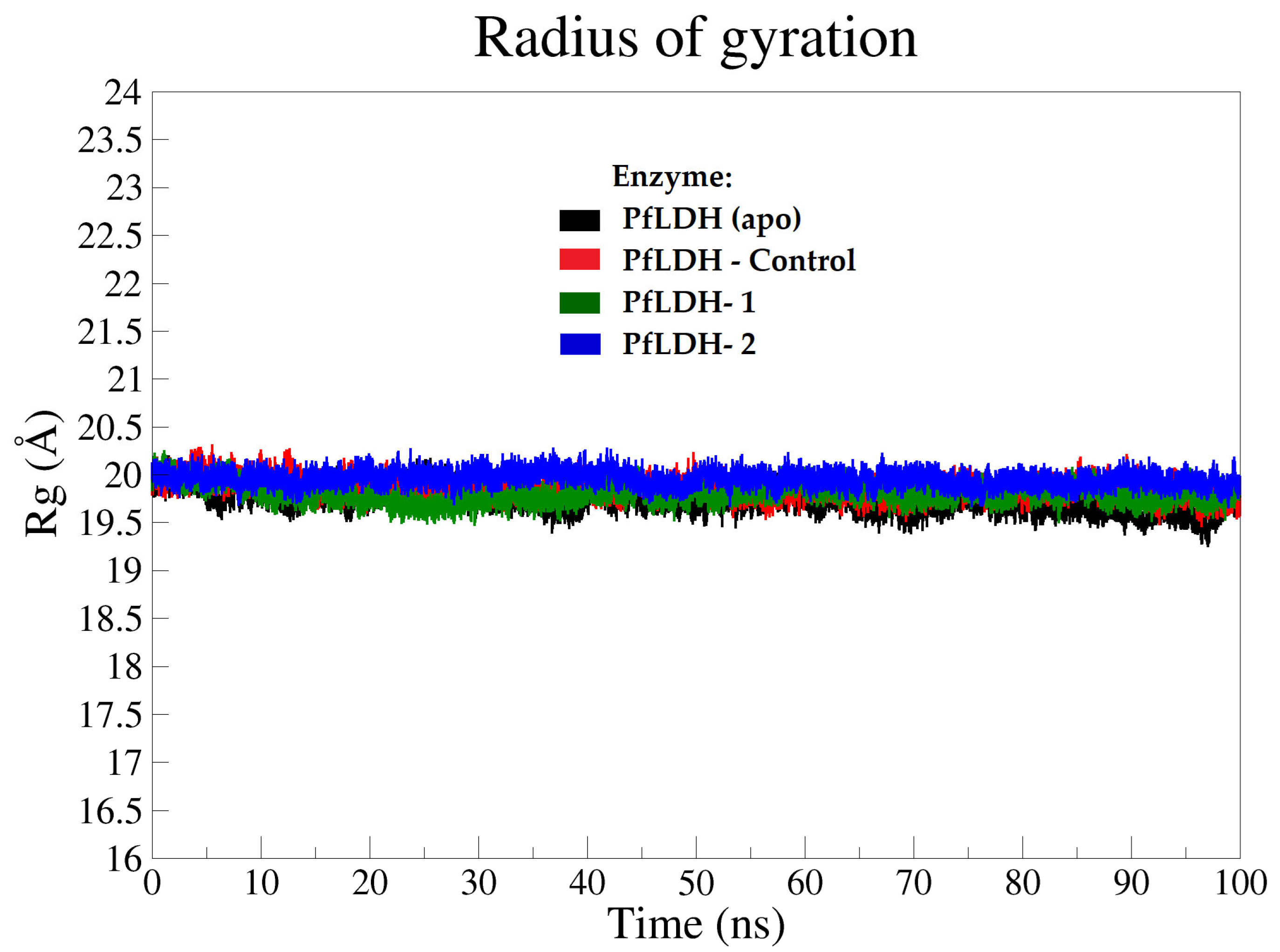
2.3.2. Molecular Dynamics (MD) Analysis
3. Materials and Methods
3.1. Instrument
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Computer Calculation of 13C NMR Chemical Shifts
3.5. Computer Calculation of ECD Spectra
3.6. In Vitro Anti-Malarial Activity
3.7. In silico Anti-Malarial Activity
3.7.1. Molecular Docking Simulation
3.7.2. Molecular Dynamic Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Little, D.P.; Knopf, P.; Schulz, C. DNA Barcode Identification of Podocarpaceae—The Second Largest Conifer Family. PLoS ONE 2013, 8, e81008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mill, R.R. Towards a biogeography of the podocarpaceae. In Proceedings of the Acta Horticulturae: International Society for Horticultural Science (ISHS), Leuven, Belgium, 30 September 2003; pp. 137–147. [Google Scholar]
- Abdillahi, H.S.; Stafford, G.I.; Finnie, J.F.; Van Staden, J. Ethnobotany, Phytochemistry and Pharmacology of Podocarpus sensu Latissimo (Sl). S. Afr. J. Bot. 2010, 76, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Su, J.; Zhang, Z.; Li, L.; Fan, M.; Zhu, Y.; Wu, X.; Zhao, Q. Two New Anti-Proliferative C18-Norditerpenes from the Roots of Podocarpus macrophyllus. Chem. Biodivers. 2018, 15, e1800043. [Google Scholar] [CrossRef]
- Zhou, B.; Ren, Y.-H.; Han, Y.-S.; Mesplède, T.; Yue, J.-M. Diverse Types of Diterpenoids with an Aromatized C Ring from the Twigs of Podocarpus imbricatus. J. Nat. Prod. 2020, 83, 2416–2424. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Le, D.; Lee, M. Diterpenoids Isolated from Podocarpus macrophyllus Inhibited the Inflammatory Mediators in LPS-Induced HT-29 and RAW 264.7 Cells. Molecules 2021, 26, 4326. [Google Scholar] [CrossRef]
- Meharie, B.G.; Tunta, T.A. Evaluation of Diuretic Activity and Phytochemical Contents of Aqueous Extract of the Shoot Apex of Podocarpus falcactus. J. Exp. Pharmacol. 2020, 12, 629–641. [Google Scholar] [CrossRef]
- Nparks. Podocarpus polystachyus R.Br. Ex Endl. Available online: https://www.nparks.gov.sg/florafaunaweb/flora/3/0/3077 (accessed on 23 September 2021).
- Dadrasnia, A.; Agamuthu, P. Bioremediation of Diesel Fuel Contaminated Soil by Podocarpus polystachyus Enhanced with Organic Wastes. Malays. J. Sci. 2013, 32, 9–14. [Google Scholar]
- Hussain, I.; Puschenreiter, M.; Gerhard, S.; Schöftner, P.; Yousaf, S.; Wang, A.; Syed, J.H.; Reichenauer, T.G. Rhizoremediation of Petroleum Hydrocarbon-Contaminated Soils: Improvement Opportunities and Field Applications. Environ. Exp. Bot. 2018, 147, 202–219. [Google Scholar] [CrossRef]
- Cambie, R.C.; Cox, R.E.; David Sidwell, K.D.C. Phenolic Diterpenoids of Some Podocarps. Phytochemistry 1983, 22, 1163–1166. [Google Scholar] [CrossRef]
- World Health Organization. World Maria Report 2020; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Neafsey, D.E.; Taylor, A.R.; MacInnis, B.L. Advances and Opportunities in Malaria Population Genomics. Nat. Rev. Genet. 2021, 22, 502–517. [Google Scholar] [CrossRef]
- Bhatt, S.; Weiss, D.J.; Cameron, E.; Bisanzio, D.; Mappin, B.; Dalrymple, U.; Battle, K.E.; Moyes, C.L.; Henry, A.; Eckhoff, P.A.; et al. The Effect of Malaria Control on Plasmodium falciparum in Africa between 2000 and 2015. Nature 2015, 526, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, S.; Durgam, L.; Guruprasad, L. Multiple E-Pharmacophore Modelling Pooled with High-Throughput Virtual Screening, Docking and Molecular Dynamics Simulations to Discover Potential Inhibitors of Plasmodium falciparum Lactate Dehydrogenase (PfLDH). J. Biomol. Struct. Dyn. 2019, 37, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Bhardwaj, V.; Purohit, R. Identification of a Novel Binding Mechanism of Quinoline Based Molecules with Lactate Dehydrogenase of Plasmodium falciparum. J. Biomol. Struct. Dyn. 2021, 39, 348–356. [Google Scholar] [CrossRef]
- Van Niekerk, D.D.; Penkler, G.P.; du Toit, F.; Snoep, J.L. Targeting Glycolysis in the Malaria Parasite Plasmodium falciparum. FEBS J. 2016, 283, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, M.A.; Clark, J.; Connelly, M.; Rivas, F. Antimalarial Activity of Abietane Ferruginol Analogues Possessing a Phthalimide Group. Bioorg. Med. Chem. Lett. 2014, 24, 5234–5237. [Google Scholar] [CrossRef] [PubMed]
- Briggs, L.H.; Cambie, R.C.; Rutledge, P.S.; Stanton, D.W. 1158. Diterpenes. Part IX. Bromo-Derivatives in the (+)-Phyllocladene and (–)-Isokaurene Series. J. Chem. Soc. 1965, 6212–6221. [Google Scholar] [CrossRef]
- Duc, D.K.M.; Fetizon, M.; Lazare, S.; Grant, P.K.; Nicholls, M.J.; Liau, H.T.L.; Francis, M.J.; Poisson, J.; Bernassau, J.-M.; Roque, N.F.; et al. 13C Nmr Spectroscopy of Tetracarbocyclic Diterpenes and Related Substances. Tetrahedron 1981, 37, 2371–2374. [Google Scholar] [CrossRef]
- Jutiviboonsuk, A.; Zhang, H.-J.; Kondratyuk, T.P.; Herunsalee, A.; Chaukul, W.; Pezzuto, J.M.; Fong, H.H.S.; Bunyapraphatsara, N. Isolation and Characterization of Cancer Chemopreventive Compounds from Barringtonia maunwongyathiae. Pharm. Biol. 2007, 45, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Su, W.-C.; Fang, J.-M.; Cheng, Y.-S. Abietanes and Kauranes from Leaves of Cryptomeria japonica. Phytochemistry 1994, 35, 1279–1284. [Google Scholar] [CrossRef]
- Henderson, R.; Hodges, R. Ketones Derived from Phyllocladene. Tetrahedron 1960, 11, 226–230. [Google Scholar] [CrossRef]
- Fujita, E.; Ochiai, M. Terpenoids. XLII. A Convenient Stereoselective Transformation of 16-Exocyclic Methylene Group into Carboxy Group in Ent-Kaurene and Its 19-Oic Acid. Chem. Pharm. Bull. 1977, 25, 3013–3017. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, Y.; Hashimoto, N.; Kusumoto, N.; Saijo, H.; Goto, I.; Kobayashi, H.; Kurihara, Y.; Takahashi, K.; Ashitani, T. Acaricidal Activity of Components of Cryptomeria japonica against Spider Mites. J. Wood Sci. 2015, 61, 60–64. [Google Scholar] [CrossRef]
- Fujita, E.; Fuji, K.; Nagao, Y.; Node, M.; Ochiai, M. The Chemistry on Diterpenoids in 1975 Part-I. Bull. Inst. Chem. Res. Kyoto Univ. 1976, 54, 197–227. [Google Scholar]
- Villasmil, T.; Rojas, J.; Aparicio, R.; Gamboa, N.; Acosta, M.E.; Rodrigues, J.; Usubillaga, A. Antimalarial Activity of Some Kaurenes. Nat. Prod. Commun. 2017, 12, 1934578X1701200219. [Google Scholar] [CrossRef] [Green Version]
- Batista, R.; García, P.A.; Castro, M.A.; Miguel del Corral, J.M.; Speziali, N.L.; de Pilla Varotti, F.; de Paula, R.C.; García-Fernández, L.F.; Francesch, A.; San Feliciano, A.; et al. Synthesis, Cytotoxicity and Antiplasmodial Activity of Novel Ent-Kaurane Derivatives. Eur. J. Med. Chem. 2013, 62, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.; Hada, R.; Gupta, S.; Khan, J.; Dobrowolski, J.; Dhar, P.K.; Kumar, N.; Singh, S. Highly Potent Anti-Malarial Activity of Benzopyrano(4,3-b)Benzopyran Derivatives and in Silico Interaction Analysis with Putative Target Plasmodium falciparum Lactate Dehydrogenase. J. Biomol. Struct. Dyn. 2021, 40, 5159–5174. [Google Scholar] [CrossRef]
- Tegar, M.; Purnomo, H. Tea Leaves Extracted as Anti-Malaria Based on Molecular Docking PLANTS. Procedia Environ. Sci. 2013, 17, 188–194. [Google Scholar] [CrossRef] [Green Version]
- Kalani, K.; Agarwal, J.; Alam, S.; Khan, F.; Pal, A.; Srivastava, S.K. In Silico and In Vivo Anti-Malarial Studies of 18β Glycyrrhetinic Acid from Glycyrrhiza glabra. PLoS ONE 2013, 8, e74761. [Google Scholar] [CrossRef] [Green Version]
- Conners, R.; Schambach, F.; Read, J.; Cameron, A.; Sessions, R.B.; Vivas, L.; Easton, A.; Croft, S.L.; Brady, R.L. Mapping the Binding Site for Gossypol-like Inhibitors of Plasmodium falciparum Lactate Dehydrogenase. Mol. Biochem. Parasitol. 2005, 142, 137–148. [Google Scholar] [CrossRef]
- Shamsuddin, M.A.; Ali, A.H.; Zakaria, N.H.; Mohammat, M.F.; Hamzah, A.S.; Shaameri, Z.; Lam, K.W.; Mark-Lee, W.F.; Agustar, H.K.; Mohd Abd Razak, M.R.; et al. Synthesis, Molecular Docking, and Antimalarial Activity of Hybrid 4-Aminoquinoline-Pyrano[2,3-c]Pyrazole Derivatives. Pharmaceuticals 2021, 14, 1174. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O. V Radius of Gyration as an Indicator of Protein Structure Compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Petukh, M.; Li, M.; Alexov, E. Predicting Binding Free Energy Change Caused by Point Mutations with Knowledge-Modified MM/PBSA Method. PLoS Comput. Biol. 2015, 11, e1004276. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and Accurate Predictions of Binding Free Energies Using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the Molecular Mechanics/Poisson Boltzmann Surface Area and Molecular Mechanics/Generalized Born Surface Area Methods. II. The Accuracy of Ranking Poses Generated from Docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, C.N.; Fu, H.; Lee Fryar, K.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K. Contribution of Hydrogen Bonds to Protein Stability. Protein Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef]
- Amir Rawa, M.S.; Hassan, Z.; Murugaiyah, V.; Nogawa, T.; Wahab, H.A. Anti-Cholinesterase Potential of Diverse Botanical Families from Malaysia: Evaluation of Crude Extracts and Fractions from Liquid-Liquid Extraction and Acid-Base Fractionation. J. Ethnopharmacol. 2019, 245, 112160. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in Molecular Quantum Chemistry Contained in the Q-Chem 4 Program Package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Otaka, J.; Shimizu, T.; Futamura, Y.; Hashizume, D.; Osada, H. Structures and Synthesis of Hitoyopodins: Bioactive Aromatic Sesquiterpenoids Produced by the Mushroom Coprinopsis cinerea. Org. Lett. 2018, 20, 6294–6297. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the Worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- BIOVIA. Discovery Studio Visualizer; BIOVIA: San Diego, CA, USA, 2017; Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 14 March 2022).
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and Upgrading Automated Preparation of Biomolecular Structures for Molecular Simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical PKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Al-Thiabat, M.G.; Gazzali, A.M.; Mohtar, N.; Murugaiyah, V.; Kamarulzaman, E.E.; Yap, B.K.; Rahman, N.A.; Othman, R.; Wahab, H.A. Conjugated β-Cyclodextrin Enhances the Affinity of Folic Acid towards FRα: Molecular Dynamics Study. Molecules 2021, 26, 5304. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B. MolProbity: More and Better Reference Data for Improved All-atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Al-Thiabat, M.G.; Saqallah, F.G.; Gazzali, A.M.; Mohtar, N.; Yap, B.K.; Choong, Y.S.; Wahab, H.A. Heterocyclic Substitutions Greatly Improve Affinity and Stability of Folic Acid towards FRα. an In Silico Insight. Molecules 2021, 26, 1079. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.-P.A.; Katzmann, D.J.; Sosa, C.P. Multilevel Parallelization of AutoDock 4.2. J. Cheminform. 2011, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018: San Francisco 2018. Available online: https://ambermd.org/doc12/Amber18.pdf (accessed on 19 June 2022).
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An Accessory Software Package for Molecular Mechanical Calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Tan, C.; Tan, Y.-H.; Luo, R. Implicit Nonpolar Solvent Models. J. Phys. Chem. B 2007, 111, 12263–12274. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 13C | 1 | ent-Kaur-15-en-17-al [22] | Calculated NMR | ||
|---|---|---|---|---|---|
| δ13C | δ1H (J) | δ13C | δ1H (J) | δ13C | |
| 1 | 38.8 | 40.4 | 38.5 | ||
| 2 | 18.8 | 1.38, m 1.47, m | 18.5 | 19.2 | |
| 3 | 42.2 | 42.0 | 40.6 | ||
| 4 | 33.4 | 33.3 | 33.0 | ||
| 5 | 56.0 | 0.88, m | 55.9 | 0.80, dd (12,2) | 55.7 |
| 6 | 20.2 | 1.40, m 1.61, m | 18.7 | 21.2 | |
| 7 | 36.3 | 1.40, m 1.71, m | 38.2 | 36.3 | |
| 8 | 50.0 | 51.0 | 49.4 | ||
| 9 | 54.20 | 1.20, m | 46.8 | 1.06, bd (12) | 53.6 |
| 10 | 37.7 | 39.8 | 38.1 | ||
| 11 | 19.1 | 1.10, m 1.58, m | 18.4 | 19.7 | |
| 12 | 24.9 | 1.44, m 1.58, m | 25.3 | 24.5 | |
| 13 | 36.4 | 2.85, m | 38.0 | 3.01, bd (5) | 36.4 |
| 14 | 54.17 | 1.25, m 1.82, ddd (8.0, 5.7, 2.3) | 43.0 | 1.36, dd (10.5,5) 2.17, dd (10.5,1) | 54.3 |
| 15 | 157.5 | 6.85, s | 162.3 | 6.55, s | 156.8 |
| 16 | 147.6 | 148.5 | 147.9 | ||
| 17 | 190.1 | 9.71, s | 189.5 | 9.70, s | 191.4 |
| 18 | 22.1 | 0.86, s | 33.5 | 0.85, s | 22.7 |
| 19 | 33.8 | 0.82, s | 21.5 | 0.79, s | 34.0 |
| 20 | 15.7 | 0.73, s | 17.7 | 1.04, s | 17.1 |
| Compound | *ΔGbind (kcal/mol) | Experimental IC50 (µM) |
|---|---|---|
| 1 | −8.03 | 12 |
| 2 | −7.97 | 52 |
| Control | −5.94 | --- |
| Complex with PfLDH | ΔGbind kcal/mol | VDW kcal/mol | EEL kcal/mol | Gpolar kcal/mol | Gnon-polar kcal/mol | AutoDock 4.2 kcal/mol |
|---|---|---|---|---|---|---|
| 1 | −30.87 ± 0.15 | −30.33 ± 0.09 | −7.06 ± 0.81 | 9.87 ± 0.14 | −3.96 ± 0.01 | −8.03 |
| 2 | −24.70 ± 0.15 | −35.85 ± 0.09 | −7.37 ± 0.14 | 22.92 ± 0.11 | −4.40 ± 0.07 | −7.97 |
| Control | −16.64 ± 0.12 | −15.07 ± 0.14 | −116.83 ± 0.51 | 117.49 ± 0.47 | −2.23 ± 0.01 | −5.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amir Rawa, M.S.; Al-Thiabat, M.G.; Nogawa, T.; Futamura, Y.; Okano, A.; Wahab, H.A. Naturally Occurring 8ß,13ß-kaur-15-en-17-al and Anti-Malarial Activity from Podocarpus polystachyus Leaves. Pharmaceuticals 2022, 15, 902. https://doi.org/10.3390/ph15070902
Amir Rawa MS, Al-Thiabat MG, Nogawa T, Futamura Y, Okano A, Wahab HA. Naturally Occurring 8ß,13ß-kaur-15-en-17-al and Anti-Malarial Activity from Podocarpus polystachyus Leaves. Pharmaceuticals. 2022; 15(7):902. https://doi.org/10.3390/ph15070902
Chicago/Turabian StyleAmir Rawa, Mira Syahfriena, Mohammad G. Al-Thiabat, Toshihiko Nogawa, Yushi Futamura, Akiko Okano, and Habibah A. Wahab. 2022. "Naturally Occurring 8ß,13ß-kaur-15-en-17-al and Anti-Malarial Activity from Podocarpus polystachyus Leaves" Pharmaceuticals 15, no. 7: 902. https://doi.org/10.3390/ph15070902