
A Density Functional Theory and Information-Theoretic Approach Study of Interaction Energy and Polarizability for Base Pairs and Peptides


1
Institute of Biomedical Research, Yunnan University, Kunming 650500, China
2
Research Computing Center, University of North Carolina, Chapel Hill, NC 27599-3420, USA
3
Department of Chemistry, University of Nortrefh Carolina, Chapel Hill, NC 27599-3290, USA
*
Authors to whom correspondence should be addressed.
Pharmaceuticals 2022, 15(8), 938; https://doi.org/10.3390/ph15080938
Submission received: 29 June 2022
/
Revised: 21 July 2022
/
Accepted: 26 July 2022
/
Published: 28 July 2022
(This article belongs to the Special Issue Applications of Conceptual Density Functional Theory to the Chemistry and Discovery of Bioactive Compounds)
Abstract
:Using density functional theory (DFT) and the information-theoretic approach (ITA) quantities to appreciate the energetics and properties of biopolymers is still an unaccomplished and ongoing task. To this end, we studied the building blocks of nucleic acid base pairs and small peptides. For base pairs, we have dissected the relative importance of energetic components by using two energy partition schemes in DFT. Our results convincingly show that the exchange-correlation effect predominantly governs the molecular stability of base pairs while the electrostatic potential plays a minor but indispensable role, and the steric effect is trivial. Furthermore, we have revealed that simple density-based ITA functions are in good relationships with molecular polarizabilities for a series of 30 hydrogen-bonded base pairs and all 20 natural α-amino acids, 400 dipeptides, and 8000 tripeptides. Based on these lines, one can easily predict the molecular polarizabilities of larger peptides, even proteins as long as the total molecular wavefunction is available, rather than solving the computationally demanding coupled-perturbed Hartree–Fock (CPHF) equation or its DFT counterpart coupled-perturbed Kohn–Sham (CPKS) equation.
1. Introduction
Nucleic acids and proteins are important biological polymers in living organisms. Proteins are responsible for the catalysis of biological processes, while nucleic acids or base pairs play a role as carriers of the genetic information. However, ab initio calculations of entire nucleic acids or proteins are still a tough nut to crack or even computationally intractable. Rather, one can easily delve into their building blocks, base pairs of nucleic acids and amino acids of proteins or peptides, respectively.
For base pairs, there exists a type of important noncovalent interaction: hydrogen-bonding interactions, which could stabilize the two strands of nucleic acids (DNA and RNA). To figure out the origin and nature of hydrogen-bonding interactions in base pairs is still of heated discussion and an ongoing task in the literature [1,2,3,4,5]. Normally, the role of electrostatic potential is greatly emphasized [2], and in most cases, this is the dominantly important energetic component of the interaction energy. However, this is not the whole picture of H-bonded interactions! Parthasarathi et al. [4] have shown that the presence of critical points between H-bonded DNA base pairs is indicative of the closed-shell kind of interactions. With Bader’s quantum theory of atoms in molecules (QTAIM), Toczyłowski et al. [5] have revealed that the electrostatic and exchange energies are found to be the most important components of the overall interaction energy, although the dispersion and the induction energies also play important roles. Of note, the exchange term used is not the same quantity as that in DFT [6]. In this work, we also resort to the supermolecule interaction model as that of Toczyłowski et al. We employed the energetic components in DFT to dictate which is responsible for the molecular stability. From two energy decomposition schemes [6,7], we have clearly shown that the exchange-correlation effect in DFT predominantly governs the molecular stability of base pairs while the electrostatic potential plays a minor but indispensable role, and the steric effect is trivial.
Besides energetics, we have also explored the molecular polarizabilities of 30 base pairs and 20 natural α-amino acids, 400 dipeptides and 8000 tripeptides. A few simple density-based ITA functions, such as Shannon entropy [8], Fisher information [9], Ghosh−Berkowitz−Parr entropy [10], Onicescu information energy [11], relative Rényi entropy [11], information gain [12], and relative Fisher information [13,14] are utilized to correlate with molecular polarizabilities. In addition, molecular volumes are also employed. We have found that there exist strong linear correlations between molecular polarizabilities and ITA quantities. The implication of these lines is straightforward, that one can easily predict the molecular polarizabilities of larger peptides, or even an entire protein when the molecular wavefunction can be obtained. In other words, one can bypass the time-consuming CPHF/CPKS equation [15,16,17] by numerically integrating some density-based functions. Moreover, our work on molecular polarizability is physics-based (locality of electron density) and is markedly different from other mathematics-based predictions, such as machine learning (ML) [18,19,20] and regression model [21].
2. Results
2.1. Validation
Hait et al. [22] have verified that hybrid density functionals are satisfactory in calculating molecular polarizabilities. A few popular density functionals, such as M06-2X [23], B3LYP [24,25], CAM-B3LYP [26], PBE0 [27] and ωB97XD [28] were chosen together with a series of atomic-centered Gaussian-type basis sets 6-311G(d,p) [29], Def2-SVP [30], Def2-TZVP [30] and aug-cc-pVTZ [31]. We calculated the molecular polarizabilities for a total of 20 natural α-amino acids (in its neutral form in the gas phase) at different theoretical levels as shown in Table 1. The MP2 results from the literature are also collected for comparison. Two statistical parameters, mean unsigned error (MUE) and mean signed error (MSE), are employed to gauge the quality of the method. The experimental values [32] of αiso are taken as reference to obtain MUE and MSE values. It is clearly shown that the combination of 6-311G(d,p) with 5 functionals from this work or MP2 (MSE = ~16 Bohr3) from the literature [33] leads to large deviations compared with the experiment. This is also the case for a small double-zeta basis set Def2-SVP, when combined with M06-2X, B3LYP, PBE0, and ωB97XD, with an exception of CAM-B3LYP. The reason behind is unknown. When it comes to a relatively larger triple-zeta basis set Def2-TZVP, the MSE dramatically decreases, for example from 18.4 (Def2-SVP) to 8.9 Bohr3 for M06-2X. If an even better basis set aug-cc-pVTZ is used, the MSE is only 3.8 Bohr3. Of note, the relative deviation of 5 density functionals should be comparable when combined with, for example, Def2-TZVP. In addition, though B3LYP performs well in predicting molecular polarizabilities here, its failure in delineating dispersion will undoubtedly hinder the wide applicability of ITA quantities for various kinds of inorganic, organic and biological systems. The results will be presented in our forthcoming publication. Collectively, with both accuracy and efficiency taken into consideration, we selected M06-2X/Def2-TZVP for predicting molecular polarizabilities.
2.2. Total Energy Decomposition of Base Pairs
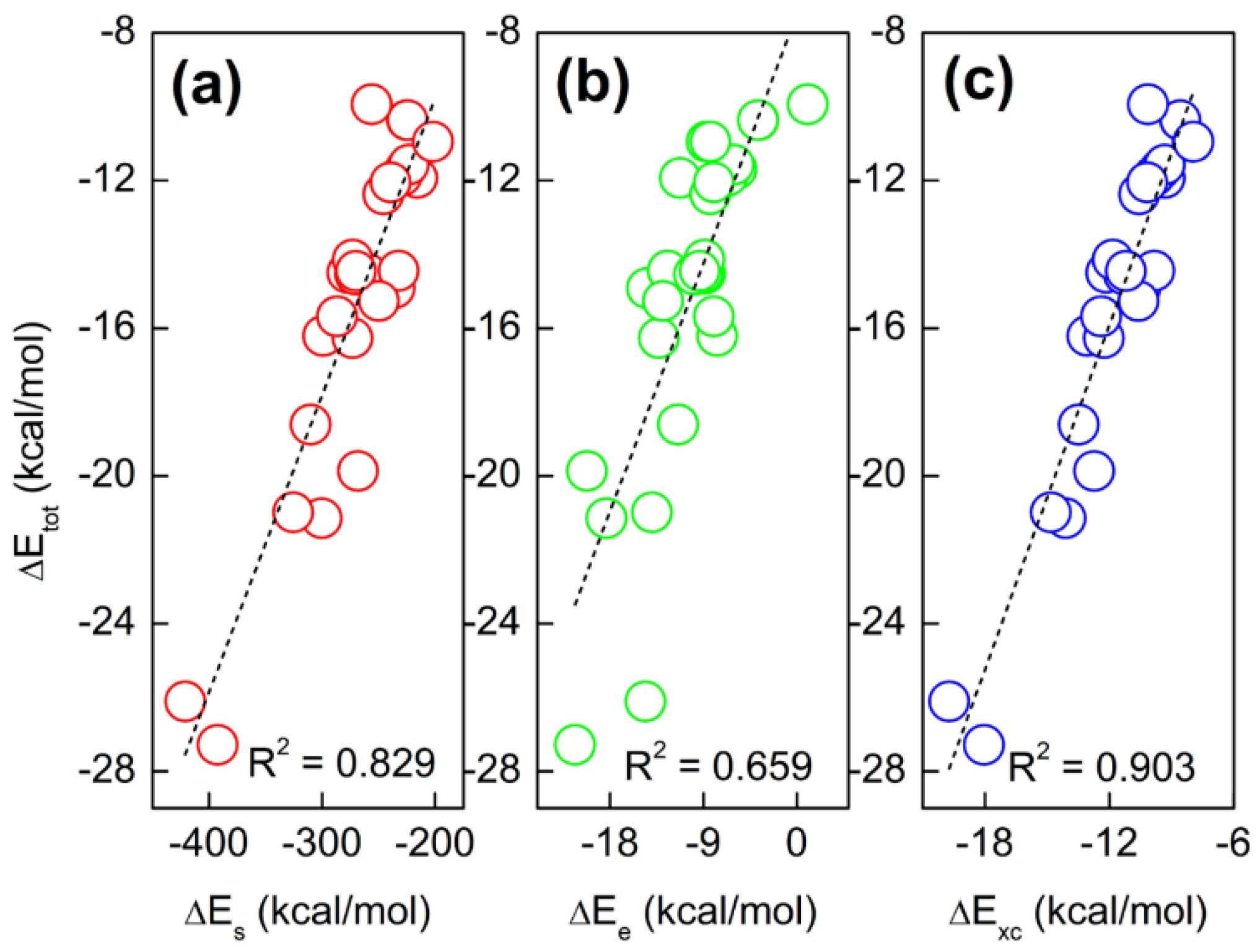
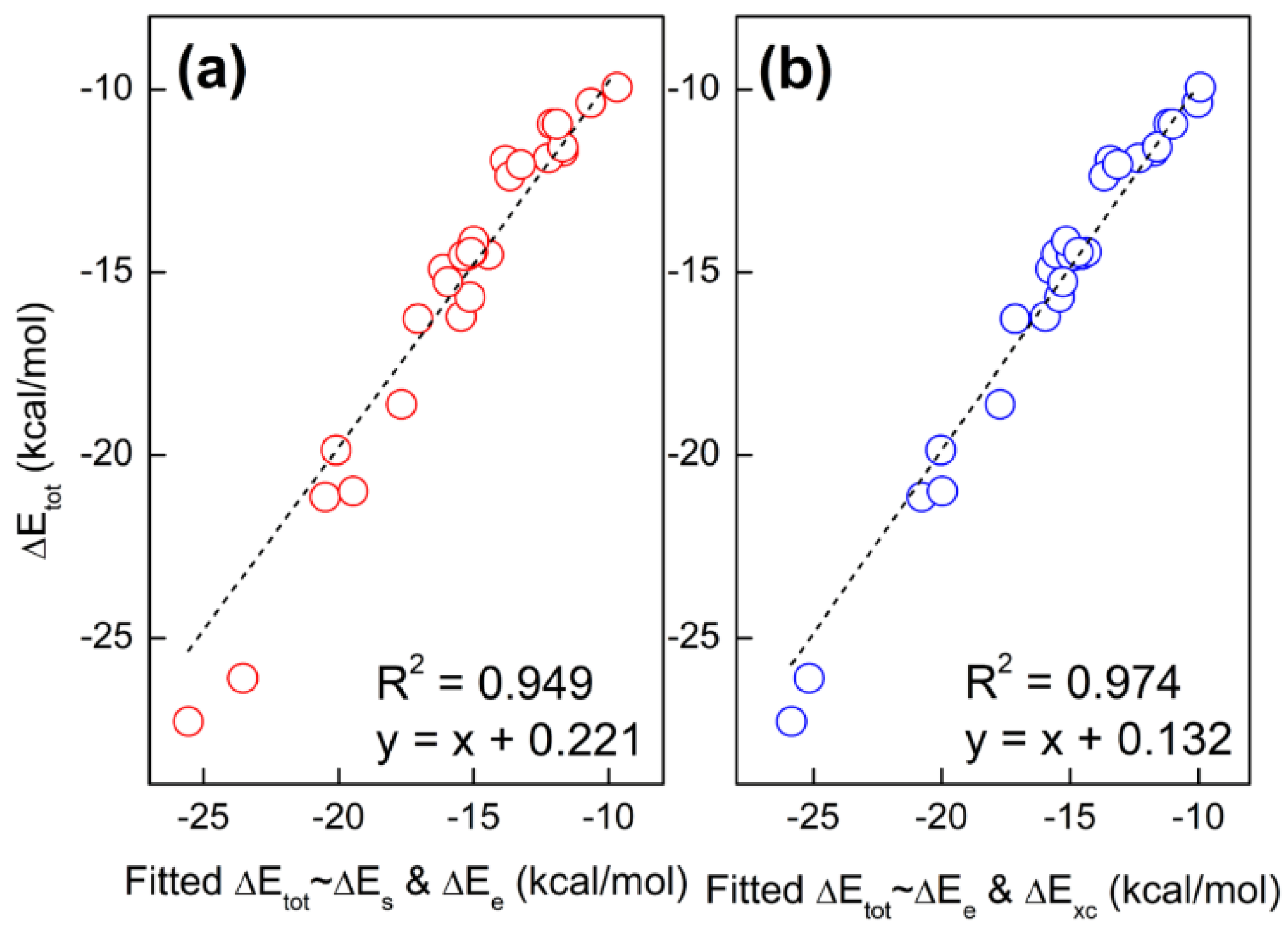
To determine the nature of hydrogen bonding interactions in base pairs is of heated discussion in the literature. In this work, we aim to use two energy decomposition schemes as mentioned above to dictate which energetic component is responsible for the hydrogen bonding interactions. Using the supramolecular approach, the interaction energy (ΔE) and its components, including kinetic (ΔTs), exchange-correlation (ΔExc), electrostatic potential (ΔEe), steric hindrance (ΔEs), and quantum effect (ΔEq), are obtained at the M06-2X/Def2-TZVP level, as collected in Table 2. Here, we only consider the electronic energy, with thermal contributions and BSSE (basis set superposition error) [34,35] corrections neglected. The total energy difference is always negative, indicating that hydrogen-bonding interactions are energetically favorable. In addition, one can see that exchange-correlation (ΔExc), electrostatic potential (ΔEe), and steric hindrance (ΔEs) all make positive contributions to the total energy difference. However, the large negative values of steric hindrance are compensated by the positive Fermionic quantum effect in the new scheme. In the conventional scheme, the positive kinetic energy also cancels out part of the exchange-correlation effect and electrostatic potential. Next, we will figure out the relative importance of these three components. Shown in Figure 1 are the linear correlations between the total energy difference (ΔE) and ΔEs, ΔEe, and ΔExc. These lines indicate that single-variable linear regression cannot distinguish the relative importance of these three energetic components. To resolve this issue, two-variable fits are used to make that happen. Shown in Figure 2 are the strong linear correlations between the fitted and predicted total energy difference. The fitted regression equations for Figure 2a,b are
and
ΔE = 0.040ΔEs + 0.467ΔEe
ΔE = 1.017ΔExc+ 0.350ΔEe
It is lucidly shown that it is the exchange-correlation effect that dominates the hydrogen-bonding interactions, while the electrostatic potential also plays an important role and the steric hindrance is trivial.
2.3. Molecular Polarizabilities of Base Pairs
In Table 3, we have collected the molecular polarizabilities, molecular volumes, and ITA quantities for 30 base pairs, which are obtained at the M06-2X/Def2-TZVP level. Also included in Table 3 are the correlation coefficients (R2) between the molecular polarizabilities and molecular volumes and ITA quantities. One can observe that Shannon entropy (SS), Fisher information (IF), 2nd and 3rd relative Reényi entropy (rR2 and rR3), and G3 are in strong linear relationships with molecular polarizabilities, with R2 > 0.8. It is intriguing to note that the absolute values of G3 are very close to molecular polarizabilities. However, the reason is unknown at present. In Table 4, we compare the polarizability results from conventional calculations, G3 data, and those predicted on top of the molecular wavefunctions obtained at the M06-2X/Def2-TZVP level. One can easily observe that the G3 data are very accurate in comparison to conventional polarizability data with MSE(%) to 1.5. However, the two sets of predicted data employing the original Tkatchenko–Scheffler (TS) formula [36] on top of Becke [37] or Hirshfeld [38] partitions are either strongly underestimated or overestimated, with MUE(%) up to –23.4 and 11.6, respectively. It is found that a mean value can greatly reduce the MSE(%) to 5.9. Though a rational theoretical explanation is lacking at the moment, we can take a careful look of the TS formula, where the sum runs over all atoms in a molecule. The weights measuring the volume ratio for atom A in a molecule to the free atom A in vacuum, can be obtained by the Becke or Hirshfeld partitioning of the electron density. Of note, denotes the atomic polarizabilities and they have been obtained accurately [39]. Thus, the main problem should come from the weights. Very recently, a revised formula has been proposed for small molecules with improved performance [40]. Yet, the performance of both original TS method and its variant for macromolecules requires more extensive work.
2.4. Molecular Polarizabilities of Amino Acids, Dipeptides and Tripeptides
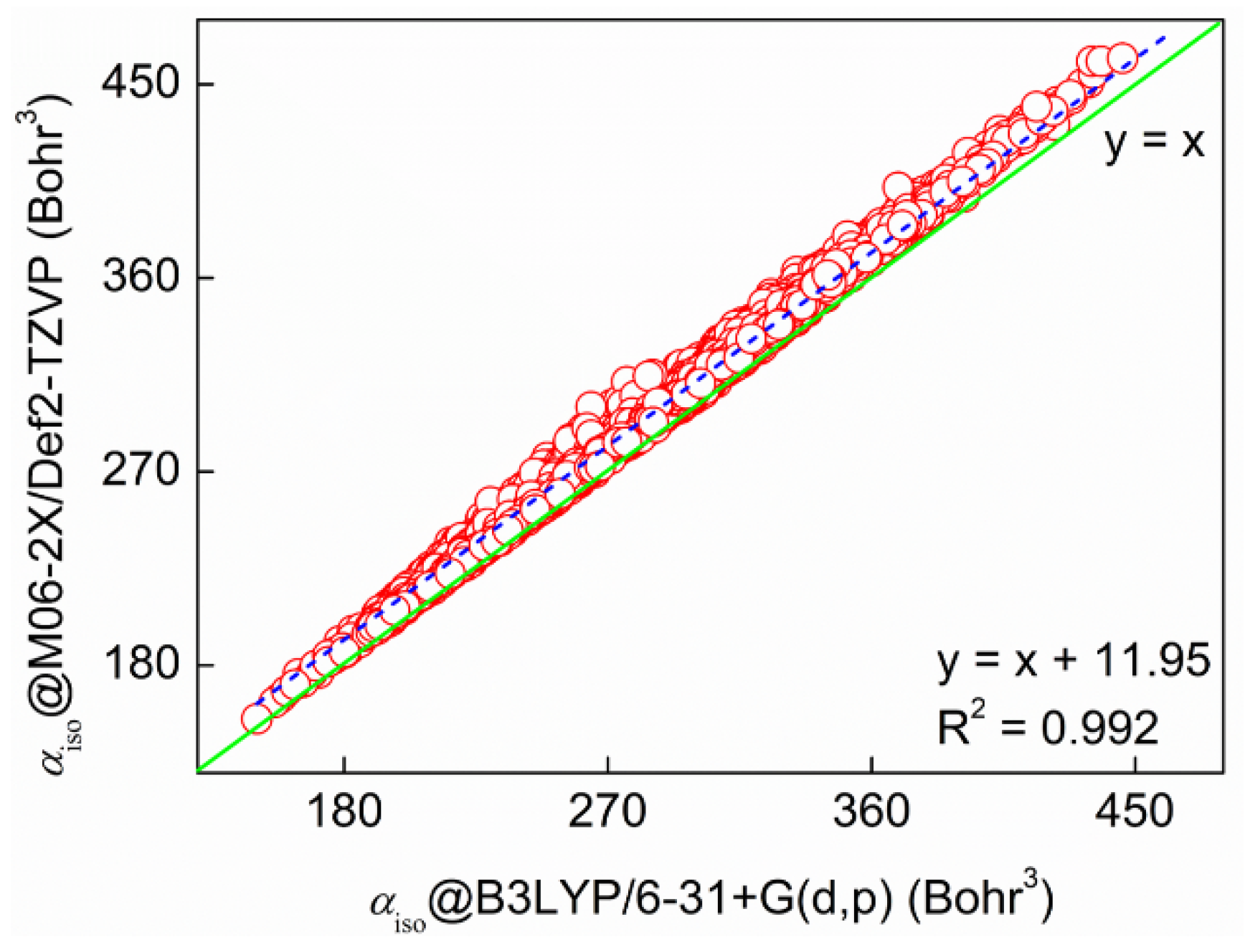
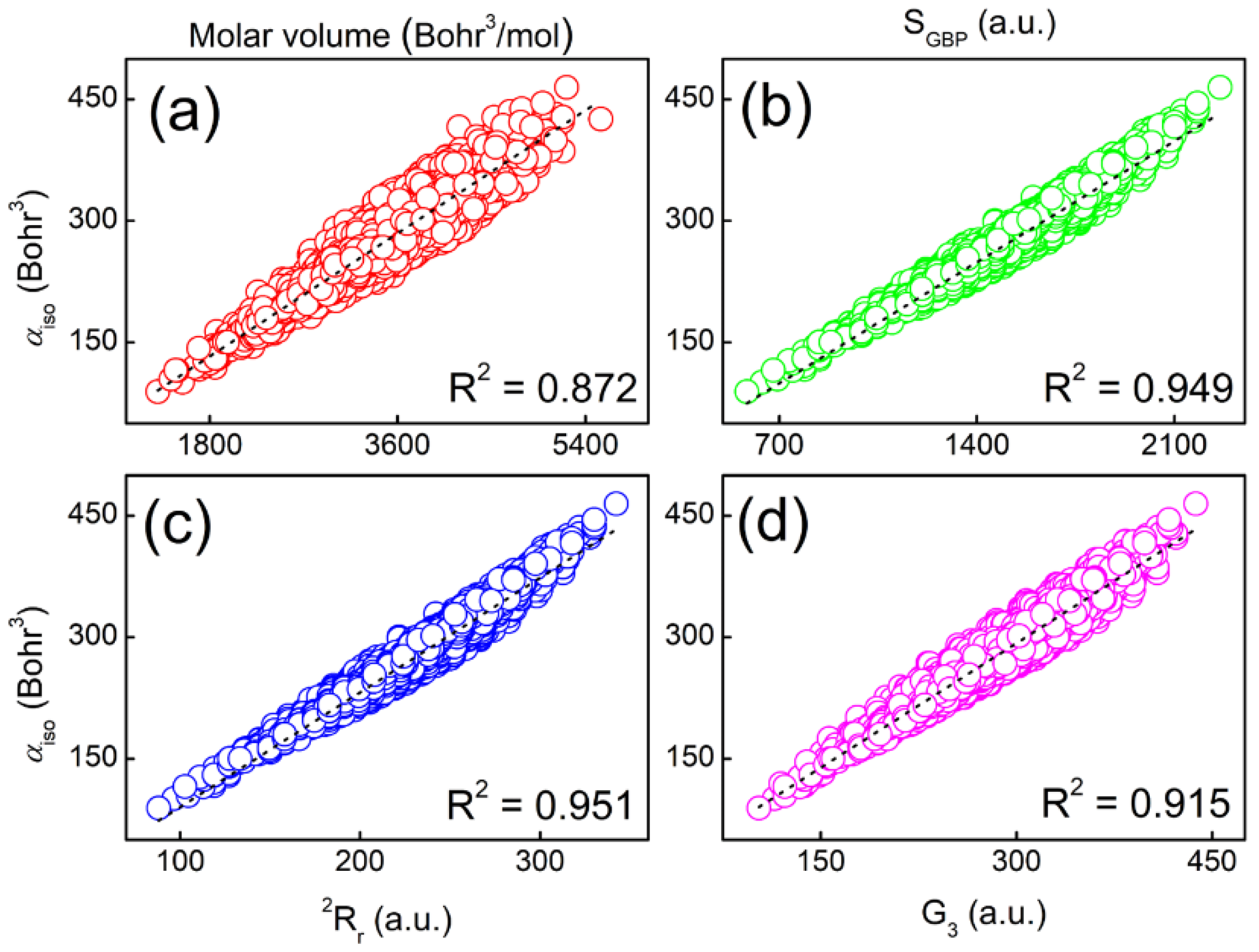
We first cross-validate the accuracy of the molecular polarizabilities of 8000 tripeptides. The two theoretical levels are M06-2X/Def2-TZVP//M06-2X/Def2-SVP and B3LYP/6-31+G(d,p)//B3LYP/6-31G level, respectively. Shown in Figure 3 is the strong correlation coefficient (R2 = 0.992) between the two sets of calculated molecular polarizabilities. The gap between the two methods is only 11.95 Bohr3, indicating that one can use a relatively cheaper theoretical method to obtain accurate results. In addition, we have found some strong correlations for a total of 20 amino acids, 400 dipeptides, and 8000 tripeptides between molecular polarizabilities and molecular volumes, GBP entropy (SGBP), 2nd relative Rényi entropy (rR2), and G3 as shown in Figure 4. The correlation coefficients are in the range of 0.87 to 0.95. We have to point out that molecular polarizabilities can be linearly correlated with molecular volumes at both atomic and molecular levels [41,42,43,44,45,46,47,48]. Here, we have verified that ITA quantities can serve as good indicators of molecular properties, in our case, molecular polarizability. This means that one can directly predict molecular polarizabilities of larger peptides, or even proteins. It is well-documented that it is time-consuming to solve the CPHF/CPKS equations [15,16,17]. When the molecular system becomes larger, it may be even intractable. For ITA quantities, as long as the total molecular wavefunction is obtained in a single-point calculation, numerical integration of ITA quantities normally requires much less time than iteratively solving the CPHF/CPKS equations. It is anticipated when it comes to real systems, like proteins, one can combine the linear-scaling fragment-based methods, such as GEBF (generalized-energy based fragmentation) [49,50,51,52], and ITA quantities to accurately and efficiently predict the molecular polarizabilities.
3. Discussion
We have systematically investigated the energetics of 30 base pairs and molecular polarizabilities of 30 base pairs and 20 amino acids, 400 dipeptides, and 8000 tripeptides. It is the first time that ITA quantities have been applied to correlate with molecular polarizabilities. Though at present, in theory we have not verified the existence of such a linear correlation between ITA quantities and molecular polarizabilities, they are both related to electron density and its derivatives. Additionally, our results presented here demonstrate that one can determine the molecular polarizability by numerically integrating the simple density-based functions when the molecular wavefunction of a given system is obtained from single-point calculations, rather than resorting to iteratively obtaining the molecular orbital derivatives. The implication of our work is straightforward, that one can use the linear relationships as shown in the text to predict molecular polarizabilities of relatively larger peptides, or even larger proteins. Can the ITA quantities be widely applied to various inorganic, organic and biological systems? More work along this line is required.
We have to point out that we are not the first to predict the molecular polarizabilities just as employing the electron density as the input. Jayatilaka et al. [53] has cast the molecular polarizability in terms of moments of the ground-state electron density matrix and the results are reasonably good against CPHF results. The corresponding performance for hyperpolarizability is far from satisfactory. Since hyperpolarizability is not considered in this work, we will figure out if the abovementioned density-based ITA functions can predict the molecular hyperpolarizability well.
4. Materials and Methods
4.1. Energy Decomposition Schemes in DFT
In Kohn–Shan DFT, the total energy difference (ΔE) can be decomposed into its components via [3,4]
and
where Ts, Ee, and Exc are the noninteracting kinetic, electrostatic, and exchange-correlation terms, respectively. The electrostatic potential Ee has three components: the nuclear−electron attraction, (Vne), the classical Coulombic repulsion, (J), and the nuclear−nuclear repulsion (Vnn). The last term, Exc, consists of exchange (Ex) and correlation (Ec) components. In Equation (2), Es stands for the steric hindrance, and Eq signifies the contribution from Fermionic quantum effect. The steric effect Es has been proved to be simply the Weizsäcker kinetic energy,
Combining Equations (1)–(3), one can simply define Eq, which reads
This new formulation has its own distinct physical meaning with a corresponding physical state. It has been applied to a number of molecular systems and phenomena, whose results are consistent with our chemical intuition and conventional wisdom [54].
4.2. Information-Theoretic Approach Quantities
Shannon entropy SS [8] is a measure of the spatial delocalization of the electron density, and Fisher information IF [9] measures the sharpness or localization of the same. They are defined as Equations (5) and (6), respectively.
Additionally, for atoms and molecules, IF has an equivalent expression [55] in terms of the Laplacian of the electron density, ∇2ρ(r).
Ghosh−Berkowitz−Parr (GBP) entropy SGBP [10]
where t(r; ρ) is the kinetic energy density, which is related to the total kinetic energy TS via
tTF(r; ρ) is the Thomas−Fermi (TF) kinetic energy density given by
with K as the Boltzmann constant (set to be unity for convenience in this work), c = (5/3) + ln(4πcK/3), and cK = (3/10)(3π2)2/3, the specific form of the local kinetic energy
More recently, additional ITA quantities have been introduced as new reactivity descriptors in conceptual density functional theory (CDFT) [11]. One example is Onicescu information energy [11] of order n
relative Rényi entropy [8] of order n
and information gain [12] (also called Kullback−Leibler divergence) IG is given in Equation (14)
where ρ0(r) is the reference-state density satisfying the same normalization condition as ρ(r).
Very recently [13,14], we have proposed another three functions G1, G2, and G3, whose analytical forms as shown below:
The G2 function involves Laplacian contribution and can be regarded as the relative Laplacian contribution to the steric potential. The quantifications and applications of Equations (15)–(17) can be found in [14]. Suffice to note, during the past decade, we have attempted to seamlessly glue the density functional theory and information theory together, as electron density can be used as a linker of these two theories. The progress and applications can be found in our recent review [54]. For example, very recently we have applied the information-theoretic approach to appreciate homochirality [56,57], which is another very fundamental problem in biology and we will look into it in the near future.
4.3. Computational Details
4.3.1. Base Pairs
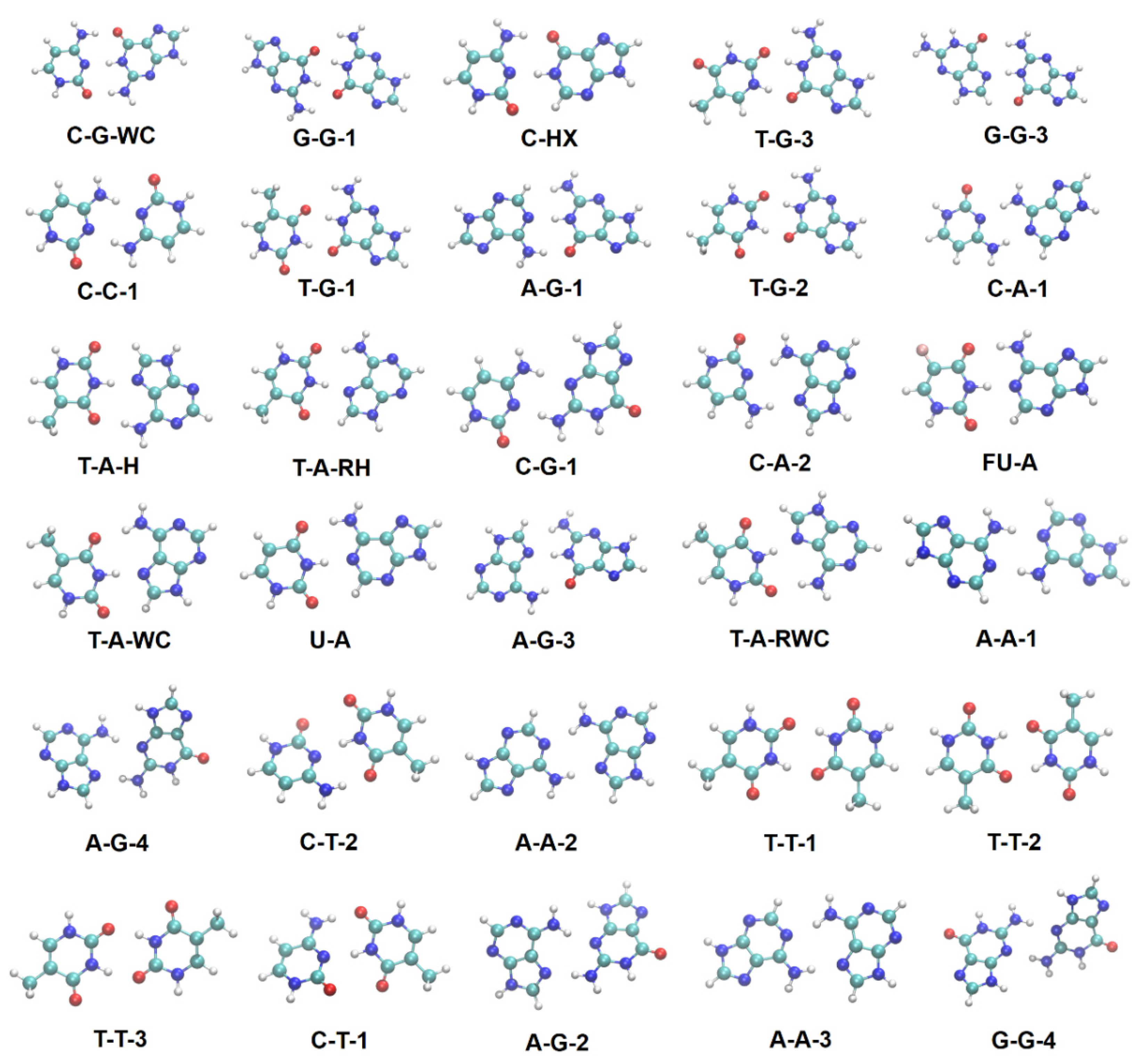
Following the notation of [2], we build a total of 30 hydrogen-bonded nucleic acid base pairs as shown in Scheme 1. For each complex, the nucleic acid bases are denoted by one or two letter abbreviations, among which are the four DNA bases: adenine (A), cytosine (C), guanine (G), and thymine (T), as well as fluorouracil (FU), hypoxanthine (HX), and uracil (U). For the most common configurations, we use the usual abbreviations: Watson–Crick (WC), reversed Watson–Crick (RWC), Hoogsteen (H), and reversed Hoogsteen (RH). We also use numbers to distinguish between different configurations of the same complex and not to introduce any kind of ordering.
4.3.2. Amino Acids, Dipeptides, and Tripeptides
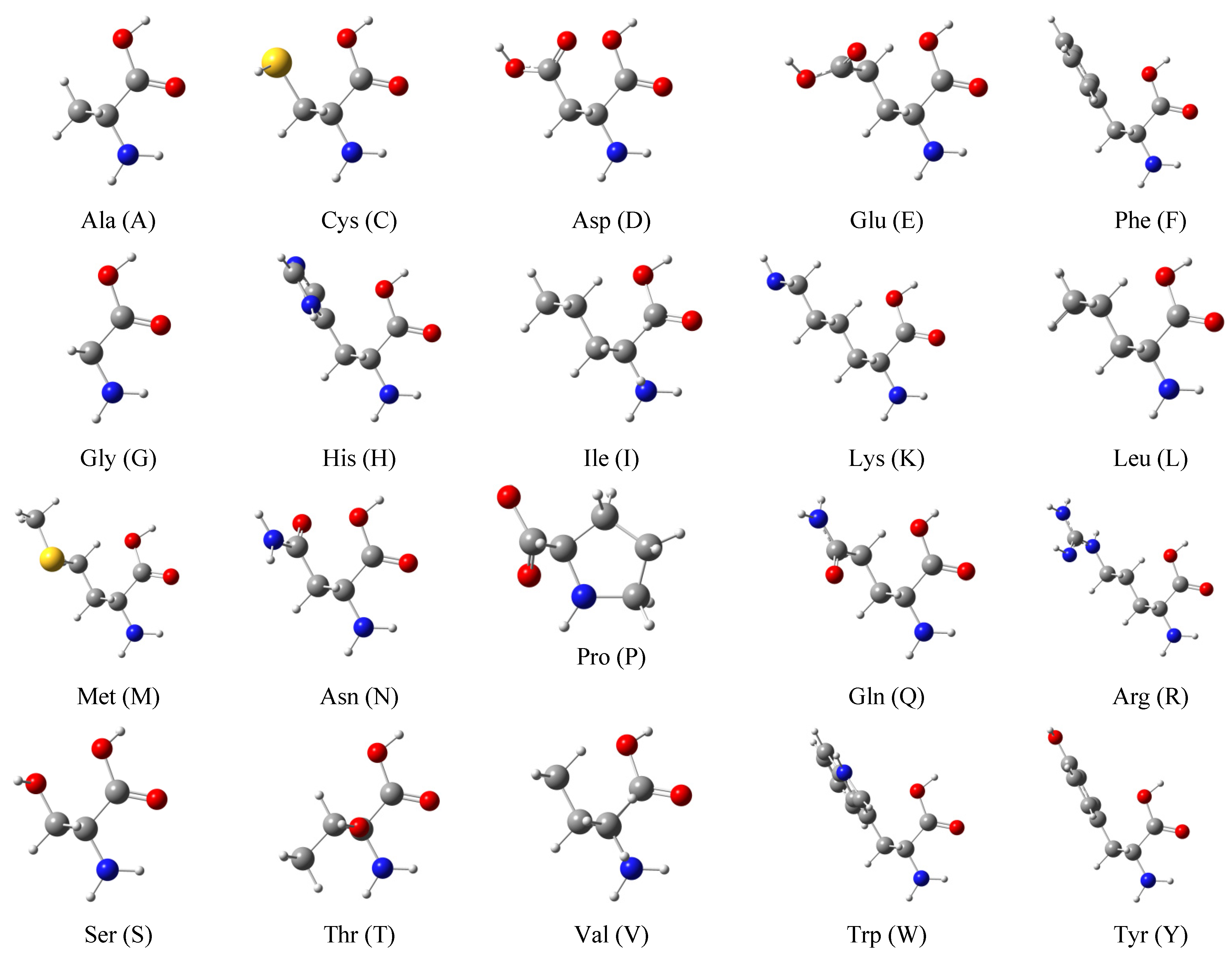
Shown in Scheme 2 are the 20 natural amino acids, directly taken from the template of the GaussView [58] program. We used the tleap module in the AmberTools package to generate a series of capped (ACE and NME) dipeptides (400) and tripeptides (8000). For each of the peptides, a simulation box was built with water molecules and Na+ and Cl− ions. We performed a total of 1 ns MD simulations with the ff19SB [59] force field for the peptide and TIP3P [60] for the water molecules. An NPT (1 atm, 300 K) ensemble was used with a time step of 2 fs. The temperature was controlled with underdamped Langevin simulations of the “virtual” solvent with the damping coefficient γ = 5 ps−1 [61]. The pressure was held constant by applying the Langevin piston method [62,63]. Periodic long-range electrostatic interactions were computed using the particle mesh Ewald summation [64]. Covalent bonds associated with hydrogen atoms were constrained by the SHAKE algorithm [65]. All classical MD simulations were carried out with the Amber20 [66,67] CUDA version. A total of 10 MD snapshot structures of each peptide were evenly extracted for further quantum chemical calculations. Since we were not going to locate the global minimum conformers, we first performed single-point calculations of the 10 structures at the semi-empirical PM7 [68] level, and selected the structure with the lowest energy for subsequent analysis.
All density functional theory (DFT) calculations were conducted with the Gaussian 16 [69] package with ultrafine integration grids and tight self-consistent field convergence.
A full geometric optimization at the M06-2X/Def2-SVP level without any symmetry constraint was conducted for 30 base pairs and 8420 peptides. The optimized Cartesian coordinates are supplied in the Supplementary Materials. Since it is computationally demanding to perform harmonic vibrational frequency analysis for all systems in this study, we chose 30 base pairs, 20 amino acids, 400 dipeptides, and 10 randomly selected tripeptides and no imaginary frequencies were observed. Molecular polarizabilities, volumes, and wavefunctions were obtained at the M06-2X/Def2-TZVP level. Note that only the isotropic molecular polarizability αiso = (αxx + αyy + αzz)/3 is reported in this work. Total energy components were obtained via the keyword iop(5/33 = 1). The Multiwfn 3.8 [70] program was utilized to calculate all ITA quantities introduced above by using the checkpoint or wavefunction file from the Gaussian calculations as the input. Molecular volumes were obtained at 0.001 e/Bohr3 contour surface of electronic density. The stockholder Hirshfeld partition scheme of atoms in molecules was employed when atomic contributions were concerned. The reference density was the neutral atom calculated at the same level of theory as molecules.
5. Conclusions
In this work, we have explored the interaction energy of base pairs and molecular properties of both base pairs and amino acids, dipeptides, and tripeptides. Using the total energy decomposition schemes, one can find that the exchange-correlation effect makes the predominant contribution to the molecular stability, followed by the electrostatic potential, and steric hindrance plays a trivial role. We further revealed that molecular polarizabilities can be linearly correlated with ITA quantities. These strong linear correlations can be used to predict the molecular polarizabilities of larger peptides and even proteins. We mention in passing that two directions merit further studies. (i) Verification of the wide applicability of ITA quantities to various systems needs intensive work. (ii) When a fragment-based method, such as the linear-scaling generalized energy-based fragmentation (GEBF) approach, is taken into consideration, one can directly predict the subsystem (fragment of a large system, usually with a few atoms within a distance threshold) polarizabilities rather than solving the derivatives of molecular orbitals and obtain the molecular polarizability of macromolecular systems via a linear combination of subsystem polarizabilities. More work along this line is ongoing, and results will be presented elsewhere in the very near future.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph15080938/s1, The optimized Cartesian coordinates of 30 base pairs and 20 amino acids, 400 peptides, and 8000 tripeptides.
Author Contributions
Conceptualization, S.L. and D.C.; data curation, D.Z.; formal analysis, D.Z.; funding acquisition, D.Z.; project administration, S.L. and D.C.; supervision, S.L. and D.C.; writing—original draft, D.Z.; writing—review and editing, S.L. and D.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Yunnan Fundamental Research Projects (grant NO. 202101AU070012).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Acknowledgments
Pratim K. Chattaraj is acknowledged for this invitation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Šponer, J.; Leszczynski, J.; Hobza, P. Structures and Energies of Hydrogen-Bonded DNA Base Pairs. A Nonempirical Study with Inclusion of Electron Correlation. J. Phys. Chem. 1996, 100, 1965–1974. [Google Scholar] [CrossRef]
- Hobza, P.; Šponer, J. Structure, Energetics, and Dynamics of the Nucleic Acid Base Pairs: Nonempirical Ab Initio Calculations. Chem. Rev. 1999, 99, 3247–3276. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.F.; Bickelhaupt, F.M.; Snijders, J.G.; Baerends, E.J. Hydrogen Bonding in DNA Base Pairs: Reconciliation of Theory and Experiment. J. Am. Chem. Soc. 2000, 122, 4117–4128. [Google Scholar] [CrossRef] [Green Version]
- Parthasarathi, R.; Amutha, R.; Subramanian, V.; Nair, B.U.; Ramasami, T. Bader’s and Reactivity Descriptors’ Analysis of DNA Base Pairs. J. Phys. Chem. A 2004, 108, 3817–3828. [Google Scholar] [CrossRef]
- Toczyłowski, R.R.; Cybulski, S.M. An Analysis of the Interactions between Nucleic Acid Bases: Hydrogen-Bonded Base Pairs. J. Phys. Chem. A 2003, 107, 418–426. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W.T. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Liu, S.B. Steric effect: A quantitative description from density functional theory. J. Chem. Phys. 2007, 126, 244103. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.A. Theory of statistical estimation. Math. Proc. Camb. Philos. Soc. 1925, 22, 700–725. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.K.; Berkowitz, M.; Parr, R.G. Transcription of ground-state density-functional theory into a local thermodynamics. Proc. Natl. Acad. Sci. USA 1984, 81, 8028–8031. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.B.; Rong, C.Y.; Wu, Z.M.; Lu, T. Rényi entropy, Tsallis entropy and Onicescu information energy in density functional reactivity theory. Acta Phys. Chim. Sin. 2015, 31, 2057–2063. [Google Scholar] [CrossRef]
- Kullback, S. Information Theory and Statistics; Dover Publications: Mineola, NY, USA, 1997. [Google Scholar]
- Liu, S.B. Identity for Kullback-Leibler divergence in density functional reactivity theory. J. Chem. Phys. 2019, 151, 141103. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, D.B.; Lu, T.; Liu, S.B.; Rong, C.Y. Quantifications and Applications of Relative Fisher Information in Density Functional Theory. J. Phys. Chem. A 2021, 125, 3802–3811. [Google Scholar] [CrossRef]
- McWeeny, R. Some recent advances in density matrix theory. Rev. Mod. Phys. 1960, 32, 335–369. [Google Scholar] [CrossRef]
- Langhoff, P.W.; Karplus, M.; Hurst, R.P. Approximations to Hartree—Fock Perturbation Theory. J. Chem. Phys. 1966, 44, 505–514. [Google Scholar] [CrossRef]
- Colwell, S.M.; Murray, C.W.; Handy, N.C.; Amos, R.D. The determination of hyperpolarisabilities using density functional theory. Chem. Phys. Lett. 1993, 210, 261–268. [Google Scholar] [CrossRef]
- Grisafi, A.; Wilkins, D.M.; Csányi, G.; Ceriotti, M. Symmetry-Adapted Machine Learning for Tensorial Properties of Atomistic Systems. Phys. Rev. Lett. 2018, 120, 036002. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, D.M.; Grisafi, A.; Yang, Y.; Lao, K.U.; DiStasio, R.A., Jr.; Ceriotti, M. Accurate molecular polarizabilities with coupled cluster theory and machine learning. Proc. Natl. Acad. Sci. USA 2019, 116, 3401–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, V.H.A.; Lunghi, A. Predicting tensorial molecular properties with equivariant machine learning models. Phys. Rev. B 2022, 105, 165131. [Google Scholar] [CrossRef]
- Amin, M.; Samy, H.; Küpper, J. Robust and Accurate Computational Estimation of the Polarizability Tensors of Macromolecules. J. Phys. Chem. Lett. 2019, 10, 2938–2943. [Google Scholar] [CrossRef] [Green Version]
- Hait, D.; Head-Gordon, M. How accurate are static polarizability predictions from density functional theory? An assessment over 132 species at equilibrium geometry. Phys. Chem. Chem. Phys. 2018, 20, 19800–19810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, T.; Tew, D.; Handy, N. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-basis sets for molecular calculations. 1. Second row atoms, Z = 11 ‒ 18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- McMeekin, T.L.; Groves, M.L.; Wilensky, M. Refractive indices of proteins in relation to amino acid composition and specific volume. Biochem. Biophys. Res. Commun. 1962, 7, 151–156. [Google Scholar] [CrossRef]
- Krishtal, A.; Senet, P.; Alsenoy, C.V. Local softness, softness dipole, and polarizabilities of functional groups: Application to the side chains of the 20 amino acids. J. Chem. Phys. 2009, 131, 044312. [Google Scholar] [CrossRef] [Green Version]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef] [Green Version]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theoret. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Nagle, J.K. 2018 Table of static dipole polarizabilities of the neutral elements in the periodic table. Mol. Phys. 2018, 117, 1200–1225. [Google Scholar] [CrossRef] [Green Version]
- Szabó, P.; Góger, S.; Charry, J.; Karimpour, M.R.; Fedorov, D.V.; Tkatchenko, A. Four-Dimensional Scaling of Dipole Polarizability in Quantum Systems. Phys. Rev. Lett. 2022, 128, 070602. [Google Scholar] [CrossRef] [PubMed]
- Condon, E.U. Handbook of Physics; Condon, E.U., Odishaw, H., Eds.; McGraw-Hill: New York, NY, USA, 1958; pp. 4–22. [Google Scholar]
- Jackson, J.D. Classical Electrodynamics, 2nd ed.; Wiley: New York, NY, USA, 1975; pp. 60–62. [Google Scholar]
- Dmitrieva, I.K.; Plindov, G.I. Dipole Polarizability, Radius and Ionization Potential for Atomic Systems. Phys. Scr. 1983, 27, 402. [Google Scholar] [CrossRef]
- Gough, K.M. Theoretical analysis of molecular polarizabilities and polarizability derivatives in hydrocarbons. J. Chem. Phys. 1989, 91, 2424–2432. [Google Scholar] [CrossRef]
- Laidig, K.E.; Bader, R.F.W. Properties of atoms in molecules: Atomic polarizabilities. J. Chem. Phys. 1990, 93, 7213–7224. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. Polarizability and volume. J. Chem. Phys. 1993, 98, 4305–4306. [Google Scholar]
- Politzer, P.; Jin, P.; Murray, J.S. Atomic polarizability, volume and ionization energy. J. Chem. Phys. 2002, 117, 8197–8202. [Google Scholar] [CrossRef]
- Blair, S.A.; Thakkar, A.J. Relating polarizability to volume, ionization energy, electronegativity, hardness, moments of momentum, and other molecular properties. J. Chem. Phys. 2014, 141, 074306. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, W.; Fang, T. An Efficient Fragment-Based Approach for Predicting the Ground-State Energies and Structures of Large Molecules. J. Am. Chem. Soc. 2005, 127, 7215–7226. [Google Scholar] [PubMed]
- Li, W.; Li, S.; Jiang, Y. Generalized Energy-Based Fragmentation Approach for Computing the Ground-State Energies and Properties of Large Molecules. J. Phys. Chem. A 2007, 111, 2193–2199. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, W.; Ma, J. Generalized Energy-Based Fragmentation Approach and Its Applications to Macromolecules and Molecular Aggregates. Acc. Chem. Res. 2014, 47, 2712–2720. [Google Scholar]
- Li, W.; Dong, H.; Ma, J.; Li, S. Structures and Spectroscopic Properties of Large Molecules and Condensed-Phase Systems Predicted by Generalized Energy-Based Fragmentation Approach. Acc. Chem. Res. 2021, 54, 169–181. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Jha, K.K.; Munshi, P. Is it Reasonable to Obtain Information on the Polarizability and Hyperpolarizability Only from the Electron Density? Aust. J. Chem. 2018, 71, 295–306. [Google Scholar] [CrossRef]
- Rong, C.Y.; Wang, B.; Zhao, D.B.; Liu, S.B. Information-Theoretic approach in density functional theory and its recent applications to chemical problems. WIREs Comput. Mol. Sci. 2020, 10, e1461. [Google Scholar] [CrossRef]
- Liu, S.B. On the relationship between densities of Shannon entropy and Fisher information for atoms and molecules. J. Chem. Phys. 2007, 126, 191107. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B. Homochirality Originates from Handedness of Helices. J. Phys. Chem. Lett. 2020, 11, 8690–8696. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B. Principle of Chirality Hierarchy in Three-Blade Propeller Systems. J. Phys. Chem. Lett. 2021, 12, 8720–8725. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. (Eds.) GaussView, Version 5; Semichem Inc.: Shawnee, KS, USA, 2009. [Google Scholar]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Adelman, S.; Doll, J. Generalized Langevin equation approach for atom/solid-surface scattering: General formulation for classical scattering off harmonic solids. J. Chem. Phys. 1976, 64, 2375–2388. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Goetz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Goetz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
Figure 1.
Strong correlations between the total energy difference (ΔEtot) and (a) steric hindrance (ΔEs), (b) electrostatic potential (ΔEe), and (c) exchange-correlation effect (ΔExc). The y-axis of (a) spans to (b,c).
Figure 1.
Strong correlations between the total energy difference (ΔEtot) and (a) steric hindrance (ΔEs), (b) electrostatic potential (ΔEe), and (c) exchange-correlation effect (ΔExc). The y-axis of (a) spans to (b,c).

Figure 2.
Two-variable fit of the total energy difference (ΔEtot) with (a) steric hindrance (ΔEs) and electrostatic potential (ΔEe) and (b) electrostatic potential (ΔEs) and exchange-correlation effect (ΔExc).
Figure 2.
Two-variable fit of the total energy difference (ΔEtot) with (a) steric hindrance (ΔEs) and electrostatic potential (ΔEe) and (b) electrostatic potential (ΔEs) and exchange-correlation effect (ΔExc).

Figure 3.
Cross-validation of molecular polarizabilities of 8000 tripeptides carried out at the M06-2X/Def2-TZVP//M06-2X/Def2-SVP and B3LYP/6-31+G(d,p)//B3LYP/6-31G level, respectively.
Figure 3.
Cross-validation of molecular polarizabilities of 8000 tripeptides carried out at the M06-2X/Def2-TZVP//M06-2X/Def2-SVP and B3LYP/6-31+G(d,p)//B3LYP/6-31G level, respectively.

Figure 4.
Strong correlations between molecular polarizability and (a) molecular volume, (b) SGBP, (c) 2Rr, and (d) G3 for a total of 20 amino acids, 400 dipeptides, and 8000 tripeptides. Computations were carried out at the M06-2X/Def2-TZVP//M06-2X/Def2-SVP level.
Figure 4.
Strong correlations between molecular polarizability and (a) molecular volume, (b) SGBP, (c) 2Rr, and (d) G3 for a total of 20 amino acids, 400 dipeptides, and 8000 tripeptides. Computations were carried out at the M06-2X/Def2-TZVP//M06-2X/Def2-SVP level.

Scheme 1.
Optimized structures of the 30 base pairs. Color code: H in white, C in cyan, N in blue, O in red, and F in pink.
Scheme 1.
Optimized structures of the 30 base pairs. Color code: H in white, C in cyan, N in blue, O in red, and F in pink.

Scheme 2.
Schematic representation of 20 amino acids that form the basis of 400 dipeptides and 8000 tripeptides. Each molecule is named with a three-letter abbreviation and single-letter in the parenthesis. Color code: H in white, C in grey, N in blue, O in red, and S in yellow.
Scheme 2.
Schematic representation of 20 amino acids that form the basis of 400 dipeptides and 8000 tripeptides. Each molecule is named with a three-letter abbreviation and single-letter in the parenthesis. Color code: H in white, C in grey, N in blue, O in red, and S in yellow.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Benchmark results of isotropic polarizabilities (αiso) of several density functionals and basis sets for 20 amino acids. a Units are in Bohr3.
Table 1.
Benchmark results of isotropic polarizabilities (αiso) of several density functionals and basis sets for 20 amino acids. a Units are in Bohr3.
| Methods | 6-311G(d,p) | Def2-SVP | Def2-TZVP | aug-cc-pVTZ | ||||
|---|---|---|---|---|---|---|---|---|
| MUE c | MSE d | MUE | MSE | MUE | MSE | MUE | MSE | |
| M06-2X | −15.3 | 15.3 | −18.4 | 18.4 | −8.9 | 8.9 | −3.7 | 3.8 |
| B3LYP | −10.7 | 10.7 | −13.6 | 13.6 | −4.6 | 4.6 | −0.3 | 1.6 |
| CAM-B3LYP | −13.9 | 13.9 | −1.3 | 2.2 | −7.8 | 7.8 | −4.6 | 4.6 |
| PBE0 | −12.4 | 12.4 | −15.2 | 15.2 | −6.0 | 6.0 | −1.9 | 2.6 |
| ωB97XD | −14.2 | 14.2 | −17.1 | 17.1 | −8.1 | 8.1 | −5.1 | 5.2 |
| MP2 b | −15.6 | 15.6 | ||||||
Table 2.
Total energy difference (ΔE) and its components obtained at the M06-2X/Def2-TZVP level for 30 base pairs, including kinetic (ΔTs), exchange-correlation (ΔExc), electrostatic potential (ΔEe), steric hindrance (ΔEs), and quantum effect (ΔEq). Units are in kcal/mol.
Table 2.
Total energy difference (ΔE) and its components obtained at the M06-2X/Def2-TZVP level for 30 base pairs, including kinetic (ΔTs), exchange-correlation (ΔExc), electrostatic potential (ΔEe), steric hindrance (ΔEs), and quantum effect (ΔEq). Units are in kcal/mol.
| Base Pair | ΔTs | ΔEx | ΔEc | ΔExc | ΔEe | ΔEs | ΔEq | ΔE |
|---|---|---|---|---|---|---|---|---|
| C-G-WC | 12.1 | −1.5 | −16.6 | −18.0 | −21.4 | −392.3 | 386.4 | −27.3 |
| G-G-1 | 8.2 | −2.4 | −17.4 | −19.7 | −14.6 | −421.0 | 409.4 | −26.1 |
| C-HX | 11.3 | −0.4 | −13.7 | −14.1 | −18.4 | −300.3 | 297.5 | −21.1 |
| T-G-3 | 7.8 | −1.6 | −13.2 | −14.8 | −14.0 | −325.5 | 318.5 | −21.0 |
| G-G-3 | 6.3 | −0.9 | −12.5 | −13.5 | −11.5 | −310.0 | 302.9 | −18.6 |
| C-C-1 | 13.1 | 0.3 | −13.0 | −12.7 | −20.2 | −268.2 | 268.6 | −19.9 |
| T-G-1 | 4.5 | −0.8 | −12.3 | −13.1 | −7.7 | −298.9 | 290.4 | −16.2 |
| A-G-1 | 9.3 | 1.2 | −13.5 | −12.3 | −13.3 | −273.0 | 270.0 | −16.3 |
| T-G-2 | 4.7 | −0.6 | −11.8 | −12.4 | −8.0 | −286.7 | 279.1 | −15.7 |
| C-A-1 | 10.1 | 0.8 | −11.3 | −10.5 | −14.5 | −235.8 | 235.4 | −14.9 |
| T-A-H | 6.3 | 0.8 | −12.0 | −11.2 | −9.6 | −270.3 | 265.4 | −14.5 |
| T-A-RH | 6.2 | 0.8 | −12.0 | −11.2 | −9.4 | −270.0 | 264.9 | −14.4 |
| C-G-1 | 5.6 | 0.2 | −11.4 | −11.2 | −8.8 | −259.9 | 254.2 | −14.5 |
| C-A-2 | 7.8 | 1.5 | −11.3 | −9.8 | −12.4 | −232.2 | 230.2 | −14.4 |
| FU-A | 6.7 | 0.6 | −12.7 | −12.1 | −9.1 | −277.6 | 272.2 | −14.5 |
| T-A-WC | 6.7 | 0.8 | −12.1 | −11.3 | −9.9 | −270.7 | 266.0 | −14.5 |
| U-A | 6.6 | 0.7 | −12.5 | −11.8 | −8.9 | −272.9 | 267.6 | −14.1 |
| A-G-3 | 8.2 | 2.0 | −12.6 | −10.6 | −12.9 | −249.8 | 247.5 | −15.3 |
| T-A-RWC | 6.1 | 0.8 | −12.0 | −11.2 | −9.4 | −269.9 | 264.9 | −14.4 |
| A-A-1 | 8.7 | 0.9 | −10.2 | −9.3 | −11.3 | −215.8 | 215.1 | −11.9 |
| A-G-4 | 1.9 | 1.5 | −10.1 | −8.6 | −3.7 | −224.8 | 218.1 | −10.4 |
| C-T-2 | 6.5 | 1.3 | −11.9 | −10.6 | −8.4 | −245.6 | 241.6 | −12.4 |
| A-A-2 | 5.7 | 1.4 | −9.3 | −8.0 | −8.7 | −202.1 | 199.8 | −10.9 |
| T-T-1 | 3.6 | 0.1 | −9.7 | −9.5 | −5.8 | −226.1 | 220.3 | −11.7 |
| T-T-2 | 4.6 | 0.0 | −9.8 | −9.8 | −6.7 | −229.4 | 224.1 | −11.9 |
| T-T-3 | 3.8 | 0.2 | −9.5 | −9.3 | −6.1 | −223.2 | 217.7 | −11.6 |
| C-T-1 | 6.1 | 1.5 | −11.7 | −10.2 | −8.0 | −239.4 | 235.3 | −12.0 |
| A-G-2 | 1.9 | 1.5 | −10.1 | −8.6 | −3.7 | −224.4 | 217.8 | −10.4 |
| A-A-3 | 5.3 | 1.4 | −9.3 | −7.9 | −8.3 | −201.8 | 199.2 | −10.9 |
| G-G-4 | −0.9 | 0.8 | −11.0 | −10.1 | 1.1 | −256.2 | 245.2 | −9.9 |
Table 3.
Molecular polarizabilities (in Bohr3), molecular volumes (in Bohr3/mol), and ITA quantities (in a.u.) for 30 base pairs.
Table 3.
Molecular polarizabilities (in Bohr3), molecular volumes (in Bohr3/mol), and ITA quantities (in a.u.) for 30 base pairs.
| Base Pair | Polar | Vol | SS | IF | SGBP | E2 | E3 | rR2 | rR3 | G1 | G2 | G3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C-G-WC | 170.2 | 1984.1 | 99.2 | 5854.3 | 924.3 | 866.2 | 48,121.2 | 138.8 | 143.9 | −22.5 | 9.3 | 169.0 |
| G-G-1 | 197.2 | 1954.2 | 109.9 | 6775.8 | 1060.1 | 1002.5 | 55,241.1 | 159.1 | 164.8 | −23.4 | 8.7 | 194.8 |
| C-HX | 159.1 | 1863.3 | 93.6 | 5514.6 | 870.1 | 814.0 | 45,149.3 | 130.6 | 135.6 | −23.6 | 11.2 | 157.8 |
| T-G-3 | 180.1 | 2060.0 | 105.0 | 6215.6 | 978.7 | 927.0 | 53,287.8 | 147.0 | 152.7 | −26.6 | 12.1 | 177.2 |
| G-G-3 | 194.4 | 2184.6 | 110.2 | 6777.3 | 1060.3 | 1002.5 | 55,250.6 | 159.1 | 164.9 | −23.6 | 8.7 | 194.0 |
| C-C-1 | 144.9 | 1731.0 | 88.8 | 4934.0 | 788.7 | 730.0 | 41,006.7 | 118.5 | 123.3 | −21.5 | 9.9 | 142.1 |
| T-G-1 | 177.3 | 2058.5 | 105.1 | 6216.0 | 978.7 | 926.9 | 53,282.0 | 147.0 | 152.7 | −26.7 | 12.5 | 177.1 |
| A-G-1 | 191.7 | 2146.7 | 108.8 | 6327.7 | 1005.6 | 921.3 | 48,236.1 | 151.0 | 156.6 | −24.7 | 10.6 | 186.1 |
| T-G-2 | 177.2 | 2016.1 | 105.1 | 6216.1 | 978.8 | 926.9 | 53,283.8 | 147.0 | 152.7 | −26.7 | 12.3 | 177.0 |
| C-A-1 | 165.7 | 1707.8 | 98.2 | 5406.3 | 869.9 | 785.1 | 41,120.1 | 130.7 | 135.9 | −23.5 | 10.5 | 159.8 |
| T-A-H | 169.8 | 2060.1 | 103.7 | 5766.3 | 924.1 | 845.6 | 46,267.5 | 138.9 | 144.5 | −27.5 | 13.9 | 169.1 |
| T-A-RH | 169.6 | 1997.3 | 103.8 | 5766.3 | 924.1 | 845.7 | 46,272.0 | 138.9 | 144.5 | −27.5 | 13.8 | 169.0 |
| C-G-1 | 147.8 | 1899.5 | 99.6 | 5856.5 | 924.6 | 866.3 | 48,137.5 | 138.8 | 144.0 | −22.8 | 9.7 | 167.6 |
| C-A-2 | 164.4 | 1810.0 | 98.2 | 5406.4 | 869.9 | 785.1 | 41,118.5 | 130.7 | 135.9 | −23.6 | 10.8 | 159.9 |
| FU-A | 158.2 | 1839.0 | 90.1 | 6089.2 | 925.1 | 933.8 | 59,941.1 | 138.6 | 143.4 | −23.2 | 10.5 | 162.2 |
| T-A-WC | 169.7 | 1950.4 | 103.7 | 5766.3 | 924.1 | 845.6 | 46,264.0 | 138.9 | 144.5 | −27.5 | 14.0 | 169.1 |
| U-A | 157.9 | 1892.1 | 93.6 | 5514.7 | 870.1 | 814.0 | 45,157.7 | 130.6 | 135.6 | −23.7 | 11.0 | 157.6 |
| A-G-3 | 190.2 | 2205.6 | 108.9 | 6328.0 | 1005.6 | 921.4 | 48,248.3 | 151.0 | 156.6 | −24.8 | 10.1 | 186.1 |
| T-A-RWC | 169.7 | 2093.3 | 103.8 | 5766.3 | 924.1 | 845.7 | 46,279.4 | 138.9 | 144.5 | −27.5 | 13.4 | 169.0 |
| A-A-1 | 186.9 | 2020.8 | 107.6 | 5878.4 | 951.0 | 840.0 | 41,215.1 | 142.9 | 148.5 | −25.5 | 12.0 | 177.5 |
| A-G-4 | 189.6 | 2119.7 | 109.0 | 6328.3 | 1005.7 | 921.3 | 48,241.8 | 151.0 | 156.6 | −24.7 | 10.4 | 185.3 |
| C-T-2 | 150.7 | 1701.9 | 94.4 | 5294.8 | 843.0 | 790.8 | 46,189.2 | 126.7 | 131.9 | −25.7 | 12.1 | 150.9 |
| A-A-2 | 185.3 | 2157.6 | 107.7 | 5878.6 | 951.0 | 840.0 | 41,219.9 | 142.9 | 148.5 | −25.6 | 11.8 | 177.6 |
| T-T-1 | 158.4 | 1822.9 | 100.1 | 5655.5 | 897.3 | 851.3 | 51,315.6 | 135.0 | 140.6 | −29.8 | 16.3 | 159.7 |
| T-T-2 | 158.2 | 1918.8 | 100.1 | 5655.5 | 897.3 | 851.2 | 51,313.7 | 134.9 | 140.6 | −29.8 | 16.5 | 159.7 |
| T-T-3 | 158.7 | 1961.9 | 100.1 | 5655.5 | 897.3 | 851.3 | 51,318.3 | 135.0 | 140.6 | −29.8 | 16.0 | 159.8 |
| C-T-1 | 150.3 | 1874.4 | 94.4 | 5294.9 | 843.0 | 790.7 | 46,172.2 | 126.7 | 131.9 | −25.7 | 12.9 | 150.8 |
| A-G-2 | 189.6 | 2023.7 | 109.0 | 6328.3 | 1005.7 | 921.2 | 48,235.2 | 151.0 | 156.6 | −24.7 | 10.7 | 185.4 |
| A-A-3 | 185.3 | 2114.5 | 107.7 | 5878.6 | 951.0 | 840.0 | 41,217.5 | 142.9 | 148.5 | −25.6 | 11.9 | 177.4 |
| G-G-4 | 195.4 | 2115.8 | 110.3 | 6777.9 | 1060.4 | 1002.5 | 55,256.4 | 159.0 | 164.7 | −23.7 | 9.2 | 193.4 |
| R2 | 1.000 | 0.632 | 0.834 | 0.722 | 0.824 | 0.544 | 0.048 | 0.826 | 0.828 | 0.007 | 0.141 | 0.904 |
Table 4.
Comparison of molecular polarizabilities (αiso) of 30 base pairs with conventional data as reference. Units of αiso are in Bohr3 and G3 is in a.u.
Table 4.
Comparison of molecular polarizabilities (αiso) of 30 base pairs with conventional data as reference. Units of αiso are in Bohr3 and G3 is in a.u.
| Base Pair | αiso | Other Work a | This Work | ||
|---|---|---|---|---|---|
| Becke | Hirshfeld | avg. | G3 | ||
| C-G-WC | 170.2 | 128.7 | 188.1 | 158.4 | 169.0 |
| G-G-1 | 197.2 | 144.9 | 213.1 | 179.0 | 194.8 |
| C-HX | 159.1 | 122.0 | 177.4 | 149.7 | 157.8 |
| T-G-3 | 180.1 | 135.3 | 198.3 | 166.8 | 177.2 |
| G-G-3 | 194.4 | 145.2 | 212.6 | 178.9 | 194.0 |
| C-C-1 | 144.9 | 112.9 | 163.1 | 138.0 | 142.1 |
| T-G-1 | 177.3 | 135.5 | 198.2 | 166.9 | 177.1 |
| A-G-1 | 191.7 | 142.3 | 207.9 | 175.1 | 186.1 |
| T-G-2 | 177.2 | 135.5 | 198.3 | 166.9 | 177.0 |
| C-A-1 | 165.7 | 126.2 | 182.9 | 154.5 | 159.8 |
| T-A-H | 169.8 | 132.3 | 193.4 | 162.8 | 169.1 |
| T-A-RH | 169.6 | 132.3 | 193.4 | 162.9 | 169.0 |
| C-G-1 | 169.9 | 129.2 | 187.5 | 158.4 | 167.7 |
| C-A-2 | 164.4 | 126.3 | 183.2 | 154.7 | 159.9 |
| FU-A | 158.2 | 122.1 | 177.8 | 150.0 | 162.2 |
| T-A-WC | 169.7 | 132.3 | 193.4 | 162.8 | 169.1 |
| U-A | 157.9 | 122.0 | 177.4 | 149.7 | 157.6 |
| A-G-3 | 190.2 | 142.4 | 208.0 | 175.2 | 186.1 |
| T-A-RWC | 169.7 | 132.3 | 193.4 | 162.9 | 169.0 |
| A-A-1 | 186.9 | 139.4 | 202.6 | 171.0 | 177.5 |
| A-G-4 | 189.6 | 142.5 | 207.5 | 175.0 | 185.3 |
| C-T-2 | 150.7 | 119.3 | 173.4 | 146.4 | 150.9 |
| A-A-2 | 185.3 | 139.5 | 202.8 | 171.2 | 177.6 |
| T-T-1 | 158.4 | 125.8 | 183.6 | 154.7 | 159.7 |
| T-T-2 | 158.2 | 125.8 | 183.6 | 154.7 | 159.7 |
| T-T-3 | 158.7 | 125.8 | 183.7 | 154.7 | 159.8 |
| C-T-1 | 150.3 | 119.4 | 173.4 | 146.4 | 150.8 |
| A-G-2 | 189.6 | 142.5 | 207.5 | 175.0 | 185.4 |
| A-A-3 | 185.3 | 139.5 | 202.8 | 171.2 | 177.4 |
| G-G-4 | 195.4 | 145.5 | 212.1 | 178.8 | 193.4 |
| MUE(%) b | −23.4 | 11.6 | −5.9 | −1.2 | |
| MSE(%) c | 23.4 | 11.6 | 5.9 | 1.5 | |
a Tkatchenko–Scheffler approach on top of Becke or Hirshfeld partitions. b MUE: mean unsigned error. c MSE: mean signed error.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhao, D.; Liu, S.; Chen, D. A Density Functional Theory and Information-Theoretic Approach Study of Interaction Energy and Polarizability for Base Pairs and Peptides. Pharmaceuticals 2022, 15, 938. https://doi.org/10.3390/ph15080938
AMA Style
Zhao D, Liu S, Chen D. A Density Functional Theory and Information-Theoretic Approach Study of Interaction Energy and Polarizability for Base Pairs and Peptides. Pharmaceuticals. 2022; 15(8):938. https://doi.org/10.3390/ph15080938
Chicago/Turabian StyleZhao, Dongbo, Shubin Liu, and Dahua Chen. 2022. "A Density Functional Theory and Information-Theoretic Approach Study of Interaction Energy and Polarizability for Base Pairs and Peptides" Pharmaceuticals 15, no. 8: 938. https://doi.org/10.3390/ph15080938
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.