Hyaluronate Nanoparticles as a Delivery System to Carry Neuroglobin to the Brain after Stroke

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Expression and Purification of Recombinant Neuroglobin
2.3. Preparation of Antiserum against NGB
2.4. Synthesis of NPs with NGB
2.5. Characterization of NPs
2.5.1. Particle Size and Polydispersity Index (PDI)
2.5.2. Zeta Potential (ζ-Potential) Measurement
2.5.3. Transmission and Scanning Electron Microscopy (TEM and SEM) Analysis
2.5.4. High Performance Liquid Chromatography (HPLC) Conditions
2.6. Determination of Drug Encapsulation Efficiency (DEE, %)
2.6.1. In Vitro Drug Release Study
2.6.2. In Vitro Drug Release Kinetics
2.7. Stability Studies
2.7.1. Lyophilization of NGB
2.7.2. Lyophilization of NGB–NPs
2.7.3. Stability to 6 °C
2.8. In Vivo Studies
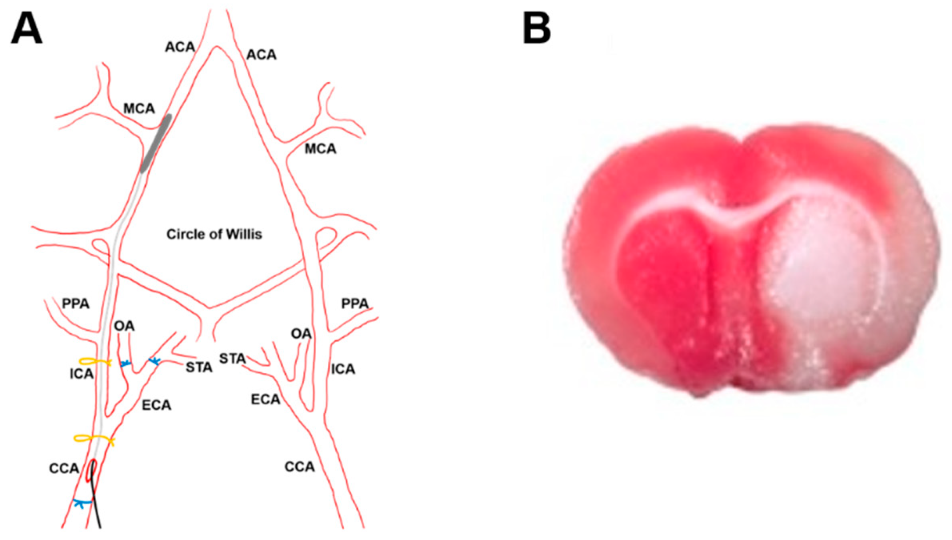
2.8.1. Stroke Model
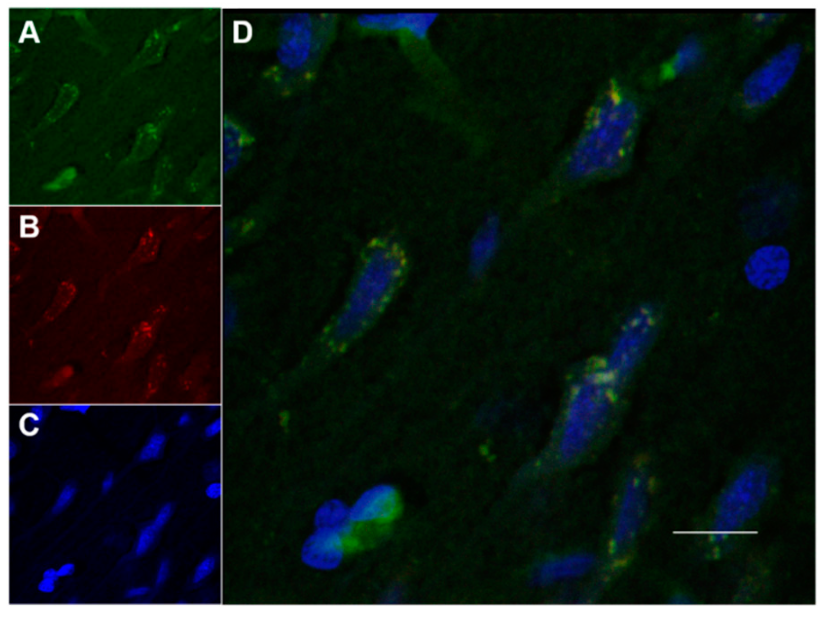
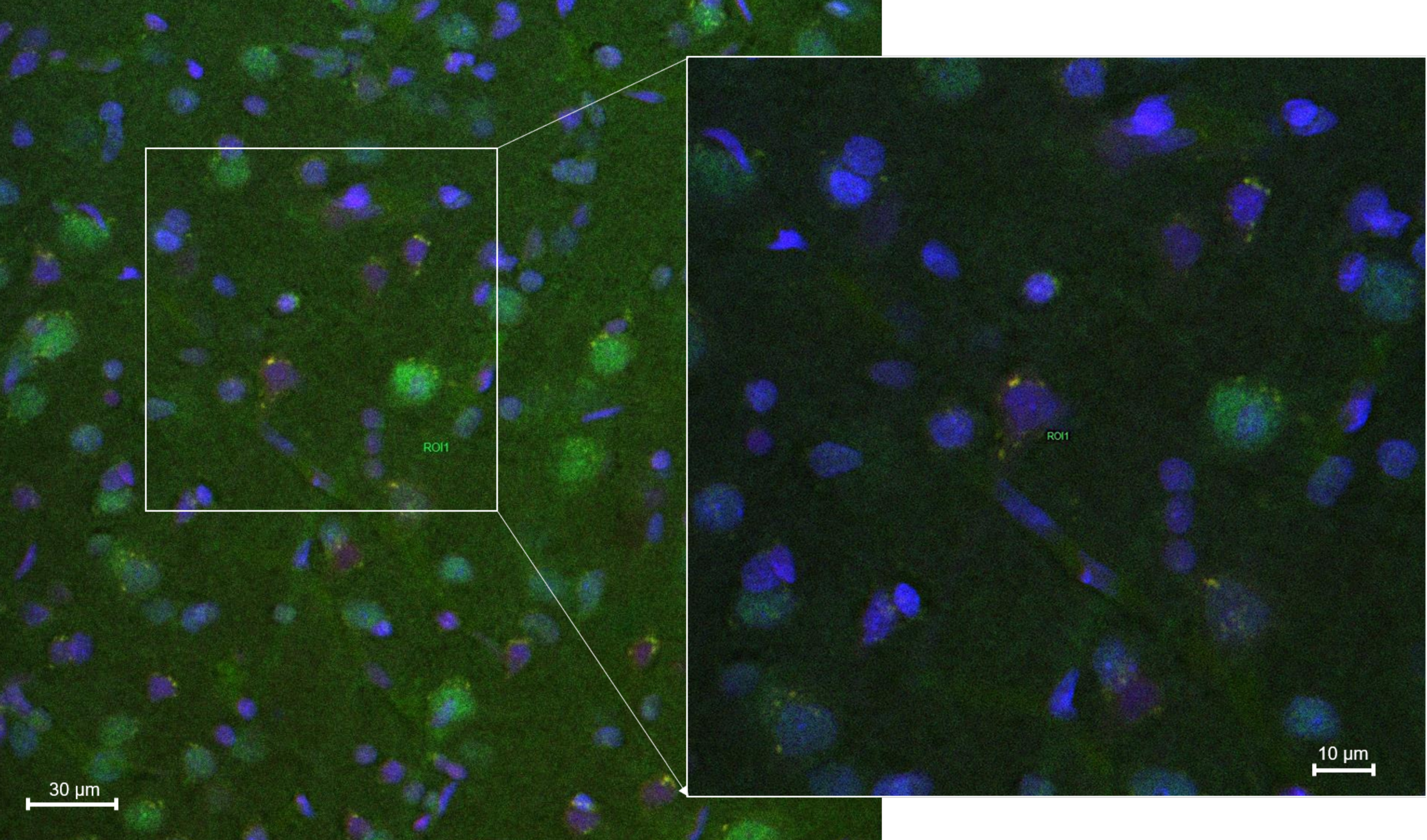
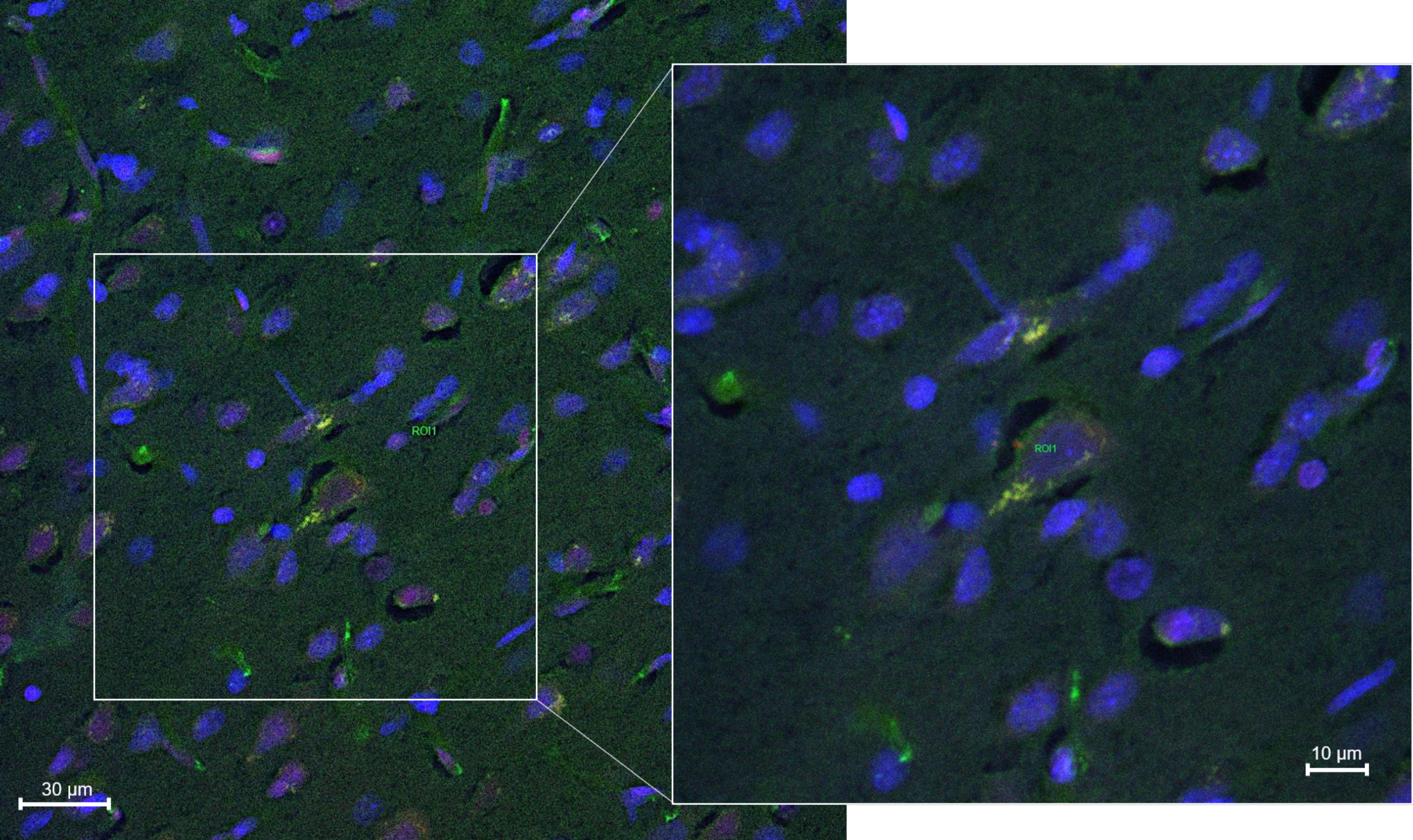
2.8.2. Histology and Microscopy
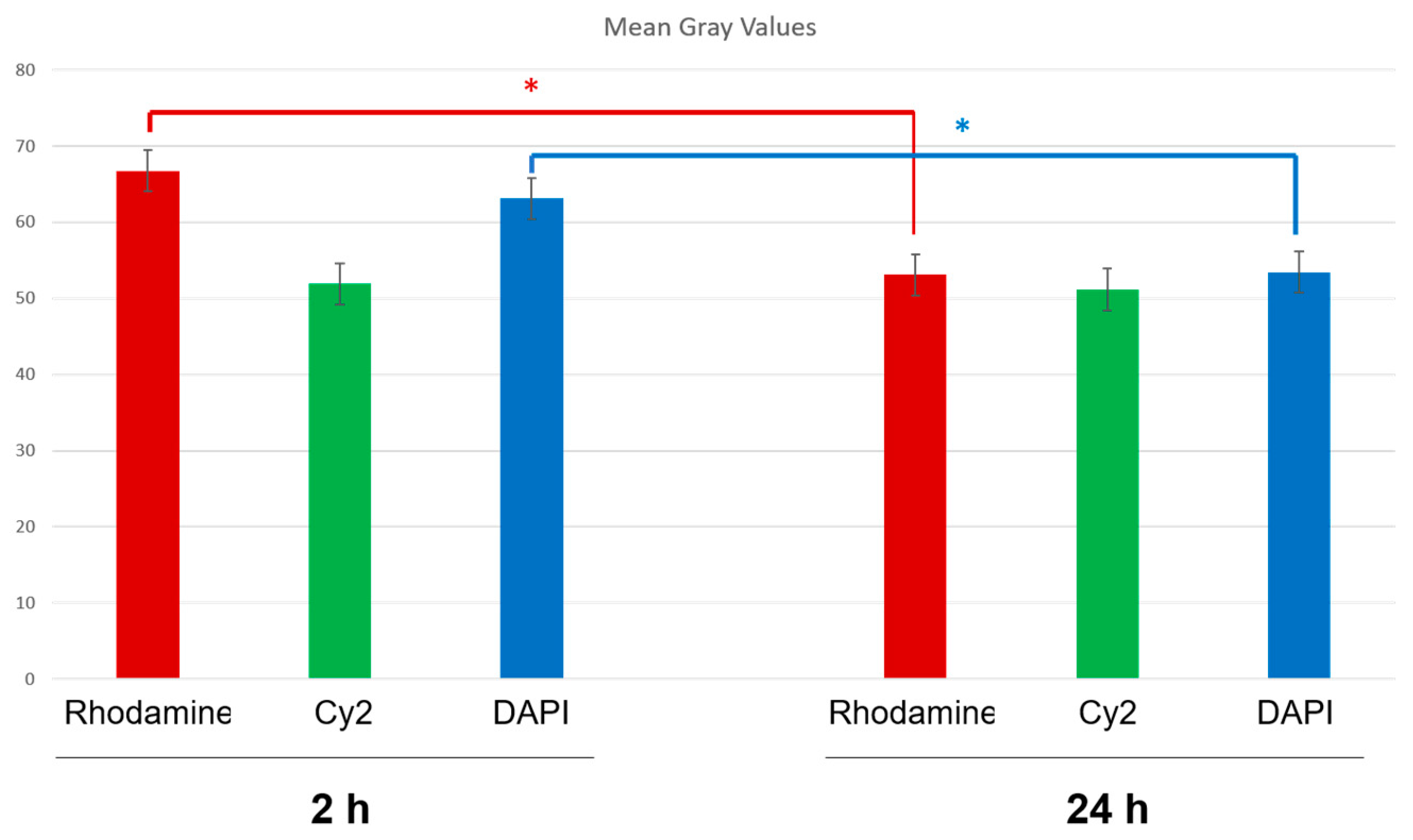
2.8.3. Fluorescence Quantification in the Cerebral Parenchyma
3. Results and Discussion
3.1. Physicochemical Characterization of NGB–NPs
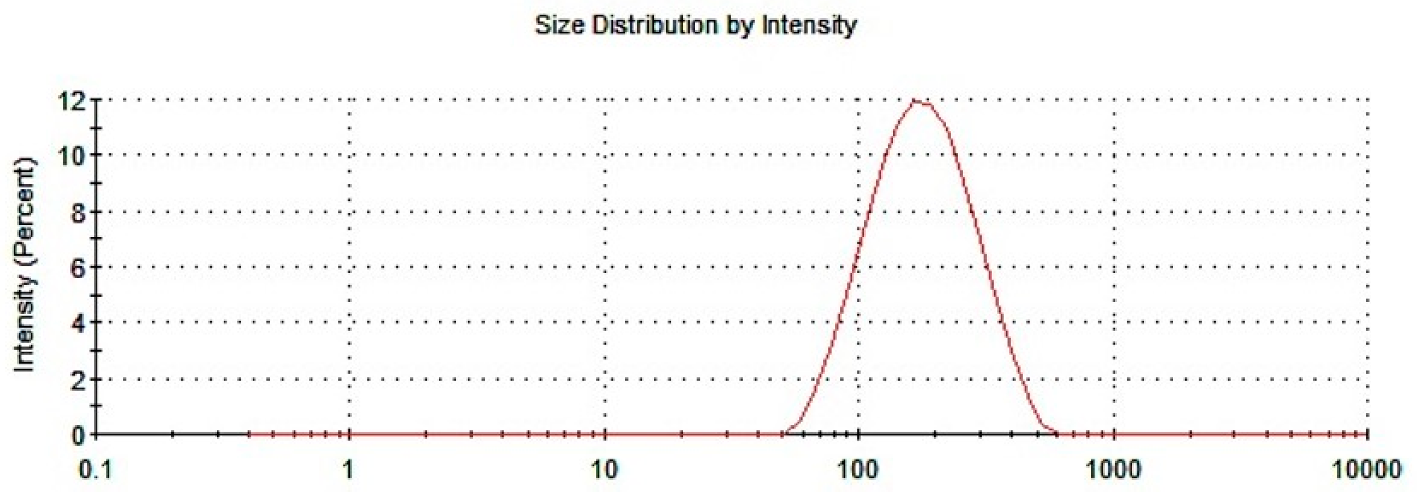
3.1.1. Particle Size and PDI
3.1.2. Zeta Potential
3.1.3. TEM and SEM Analyses
3.2. Drug Encapsulation Efficiency (DEE, %)
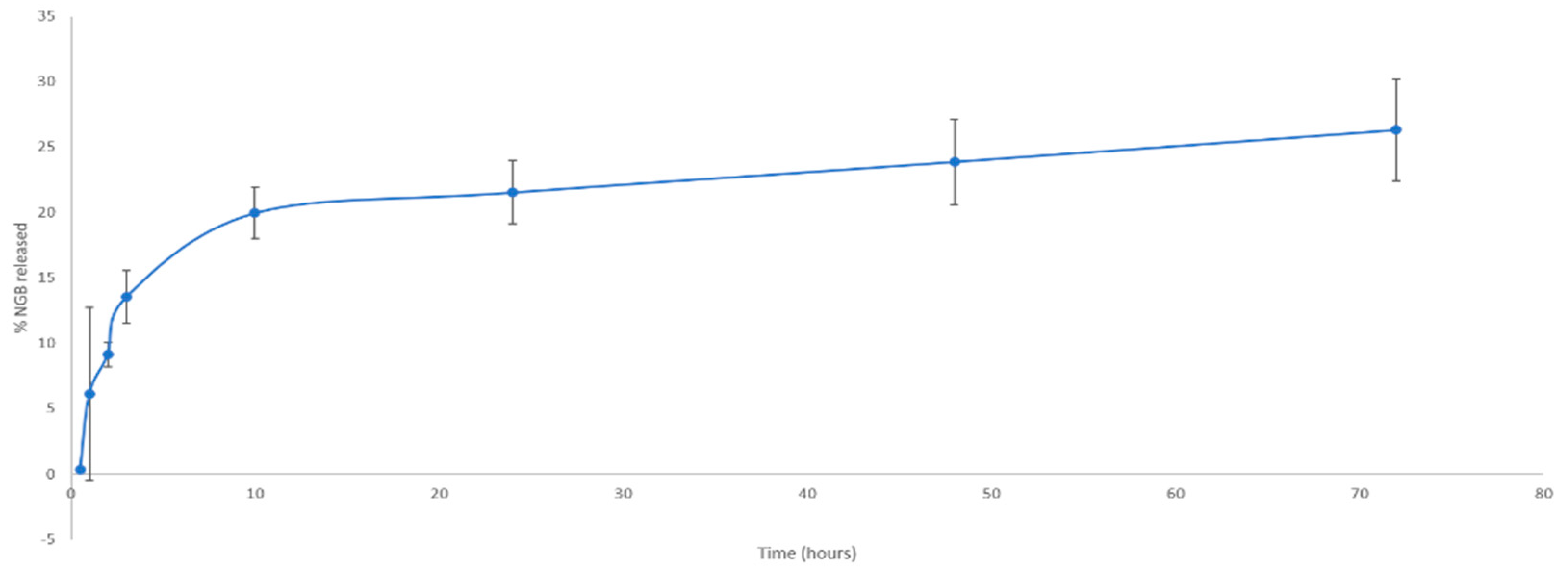
In Vitro NGB–NPs Release
3.3. Studies of Stability of NGB and NPs
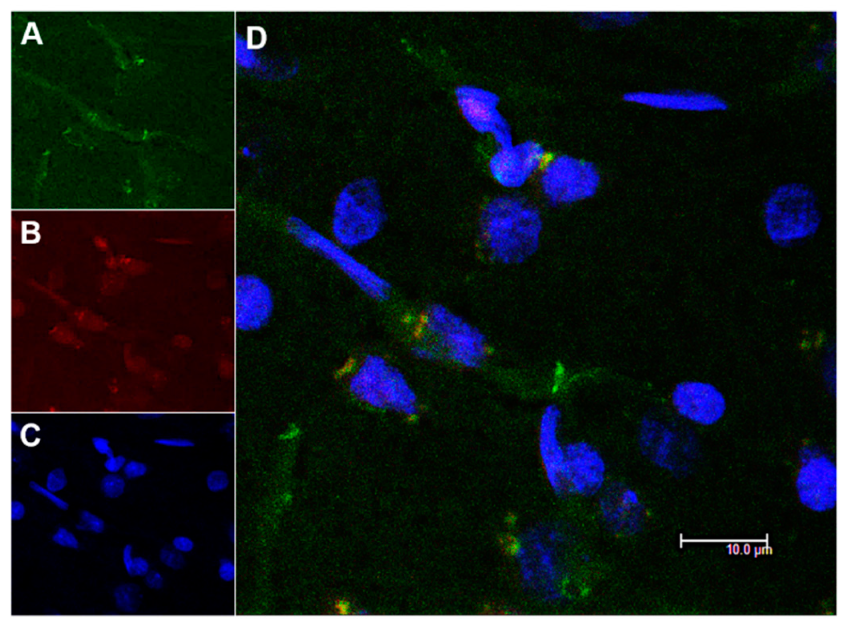
3.4. In Vivo Studies: Penetration of NGB–NPs into the Cerebral Parenchyma
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cai, B.; Lin, Y.; Xue, X.H.; Fang, L.; Wang, N.; Wu, Z.Y. TAT-mediated delivery of neuroglobin protects against focal cerebral ischemia in mice. Exp. Neurol. 2011, 227, 224–231. [Google Scholar] [CrossRef]
- Song, X.; Xu, R.; Xie, F.; Zhu, H.; Zhu, J.; Wang, X. Hemin offers neuroprotection through inducing exogenous neuroglobin in focal cerebral hypoxic-ischemia in rats. Int. J. Clin. Exp. Pathol. 2014, 7, 2163–2171. [Google Scholar]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Sims, N.R.; Muyderman, H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta 2010, 1802, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Beslow, L.A.; Smith, S.E.; Vossough, A.; Licht, D.J.; Kasner, S.E.; Favilla, C.G.; Halperin, A.R.; Gordon, D.M.; Jones, C.I.; Cucchiara, A.J.; et al. Hemorrhagic transformation of childhood arterial ischemic stroke. Stroke J. Cereb. Circ. 2011, 42, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Lyden, P. Thrombolytic therapy for acute stroke—Not a moment to lose. N. Engl. J. Med. 2008, 359, 1393–1395. [Google Scholar] [CrossRef] [Green Version]
- Terruso, V.; D’Amelio, M.; Di Benedetto, N.; Lupo, I.; Saia, V.; Famoso, G.; Mazzola, M.A.; Aridon, P.; Sarno, C.; Ragonese, P.; et al. Frequency and determinants for hemorrhagic transformation of cerebral infarction. Neuroepidemiology 2009, 33, 261–265. [Google Scholar] [CrossRef]
- Wiesmann, M.; Zinnhardt, B.; Reinhardt, D.; Eligehausen, S.; Wachsmuth, L.; Hermann, S.; Dederen, P.J.; Hellwich, M.; Kuhlmann, M.T.; Broersen, L.M.; et al. A specific dietary intervention to restore brain structure and function after ischemic stroke. Theranostics 2017, 7, 493–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Liu, N.; Liu, J.; Yang, K.; Wang, X. Neuroglobin, a novel target for endogenous neuroprotection against stroke and neurodegenerative disorders. Int. J. Mol. Sci 2012, 13, 6995–7014. [Google Scholar] [CrossRef] [PubMed]
- Burmester, T.; Weich, B.; Reinhardt, S.; Hankeln, T. A vertebrate globin expressed in the brain. Nature 2000, 407, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Zhu, H.; Tejima, E.; Tsuji, K.; Murata, Y.; Atochin, D.N.; Huang, P.L.; Zhang, C.; Lo, E.H. Effects of neuroglobin overexpression on acute brain injury and long-term outcomes after focal cerebral ischemia. Stroke J. Cereb. Circ. 2008, 39, 1869–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fordel, E.; Geuens, E.; Dewilde, S.; De Coen, W.; Moens, L. Hypoxia/ischemia and the regulation of neuroglobin and cytoglobin expression. IUBMB Life 2004, 56, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Fordel, E.; Thijs, L.; Moens, L.; Dewilde, S. Neuroglobin and cytoglobin expression in mice. Evidence for a correlation with reactive oxygen species scavenging. FEBS J. 2007, 274, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kastner, R.; Haberkamp, M.; Schmitz, C.; Hankeln, T.; Burmester, T. Neuroglobin mRNA expression after transient global brain ischemia and prolonged hypoxia in cell culture. Brain Res. 2006, 1103, 173–180. [Google Scholar] [CrossRef]
- Shang, A.; Zhou, D.; Wang, L.; Gao, Y.; Fan, M.; Wang, X.; Zhou, R.; Zhang, C. Increased neuroglobin levels in the cerebral cortex and serum after ischemia-reperfusion insults. Brain Res. 2006, 1078, 219–226. [Google Scholar] [CrossRef]
- Ren, C.; Wang, P.; Wang, B.; Li, N.; Li, W.; Zhang, C.; Jin, K.; Ji, X. Limb remote ischemic per-conditioning in combination with post-conditioning reduces brain damage and promotes neuroglobin expression in the rat brain after ischemic stroke. Restor. Neurol. Neurosci. 2015, 33, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Yu, Z.; Gao, X.; Song, Y.S.; Yuan, J.; Xun, Y.; Wang, T.; Yan, F.; Yuan, S.; Zhang, J.; et al. Establishment of Cell-Based Neuroglobin Promoter Reporter Assay for Neuroprotective Compounds Screening. CNS Neurol. Disord.-Drug Targets 2016, 15, 629–639. [Google Scholar] [CrossRef]
- Sun, Y.; Jin, K.; Peel, A.; Mao, X.O.; Xie, L.; Greenberg, D.A. Neuroglobin protects the brain from experimental stroke in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 3497–3500. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, B.; Li, Q.; Zou, L. Neuroglobin up-regulation after ischaemic pre-conditioning in a rat model of middle cerebral artery occlusion. Brain Inj. 2015, 29, 651–657. [Google Scholar] [CrossRef]
- Wen, H.; Liu, L.; Zhan, L.; Liang, D.; Li, L.; Liu, D.; Sun, W.; Xu, E. Neuroglobin mediates neuroprotection of hypoxic postconditioning against transient global cerebral ischemia in rats through preserving the activity of Na+/K+ ATPases. Cell Death Dis. 2018, 9, 635. [Google Scholar] [CrossRef] [Green Version]
- Dietz, G.P. Protection by neuroglobin and cell-penetrating peptide-mediated delivery in vivo: A decade of research. Comment on Cai et al: TAT-mediated delivery of neuroglobin protects against focal cerebral ischemia in mice. Exp Neurol. 2011, 227(1): 224–31. Exp. Neurol. 2011, 231, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.X.; Pan, F.; Chen, R.Q.; Hu, D.X.; Qiu, X.Y.; Li, C.Y.; Xie, X.Q.; Tian, B.; Chen, X.Q. Neuroglobin boosts axon regeneration during ischemic reperfusion via p38 binding and activation depending on oxygen signal. Cell Death Dis. 2018, 9, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hundahl, C.; Kelsen, J.; Kjaer, K.; Ronn, L.C.; Weber, R.E.; Geuens, E.; Hay-Schmidt, A.; Nyengaard, J.R. Does neuroglobin protect neurons from ischemic insult? A quantitative investigation of neuroglobin expression following transient MCAo in spontaneously hypertensive rats. Brain Res. 2006, 1085, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Raida, Z.; Hundahl, C.A.; Kelsen, J.; Nyengaard, J.R.; Hay-Schmidt, A. Reduced infarct size in neuroglobin-null mice after experimental stroke in vivo. Exp. Transl. Stroke Med. 2012, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Peralta, S.; Blanco, S.; Hernandez, R.; Castan, H.; Siles, E.; Martinez-Lara, E.; Encarnacion Morales, M.; Angeles Peinado, M.; Adolfina Ruiz, M. Synthesis and characterization of different sodium hyaluronate nanoparticles to transport large neurotherapheutic molecules through blood brain barrier after stroke. Eur. Polym. J. 2019, 112, 433–441. [Google Scholar] [CrossRef]
- Deng, X.; Cao, M.; Zhang, J.; Hu, K.; Yin, Z.; Zhou, Z.; Xiao, X.; Yang, Y.; Sheng, W.; Wu, Y.; et al. Hyaluronic acid-chitosan nanoparticles for co-delivery of MiR-34a and doxorubicin in therapy against triple negative breast cancer. Biomaterials 2014, 35, 4333–4344. [Google Scholar] [CrossRef]
- Saraiva, C.; Praca, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Parveen, S.; Sahoo, S.K. Long circulating chitosan/PEG blended PLGA nanoparticle for tumor drug delivery. Eur. J. Pharmacol. 2011, 670, 372–383. [Google Scholar] [CrossRef]
- Kreuter, J. Nanoparticulate systems for brain delivery of drugs. Adv. Drug Deliv Rev. 2001, 47, 65–81. [Google Scholar] [CrossRef]
- Kreuter, J.; Alyautdin, R.N. Using nanoparticles to target drugs to the central nervous system. In The Blood-Brain Barrier and Drug Delivery to the CNS; CRC Press: New York, NY, USA, 2000; pp. 205–223. [Google Scholar]
- Kreuter, J.; Alyautdin, R.N.; Kharkevich, D.A.; Ivanov, A.A. Passage of peptides through the blood-brain barrier with colloidal polymer particles (nanoparticles). Brain Res. 1995, 674, 171–174. [Google Scholar] [CrossRef]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood-brain barrier by nanoparticles. J. Control. Release 2012, 161, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Maji, N.; Nayak, A.K.; Sen, K.K.; Basu, S.K. Development of chitosan-based nanoparticles through inter-polymeric complexation for oral drug delivery. Carbohydr. Polym. 2013, 98, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Manna, S.; Nayak, A.K.; Sen, K.K.; Basu, S.K. Carbopol gel containing chitosan-egg albumin nanoparticles for transdermal aceclofenac delivery. Colloids Surf. B Biointerfaces 2014, 114, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Saha, A.; Nayak, A.K.; Sen, K.K.; Basu, S.K. Aceclofenac-loaded chitosan-tamarind seed polysaccharide interpenetrating polymeric network microparticles. Colloids Surf. B Biointerfaces 2013, 105, 303–309. [Google Scholar] [CrossRef]
- Jana, S.; Samanta, A.; Nayak, A.K.; Sen, K.K.; Jana, S. Novel alginate hydrogel core-shell systems for combination delivery of ranitidine HCl and aceclofenac. Int. J. Biol. Macromol. 2015, 74, 85–92. [Google Scholar] [CrossRef]
- Verma, A.; Dubey, J.; Verma, N.; Nayak, A.K. Chitosan-Hydroxypropyl Methylcellulose Matrices as Carriers for Hydrodynamically Balanced Capsules of Moxifloxacin HCl. Curr. Drug Deliv. 2017, 14, 83–90. [Google Scholar] [CrossRef]
- Alyautdin, R.; Khalin, I.; Nafeeza, M.I.; Haron, M.H.; Kuznetsov, D. Nanoscale drug delivery systems and the blood-brain barrier. Int. J. Nanomed. 2014, 9, 795–811. [Google Scholar] [CrossRef] [Green Version]
- Maduko, C.O.; Akoh, C.C.; Park, Y.W. Enzymatic interesterification of tripalmitin with vegetable oil blends for formulation of caprine milk infant formula analogs. J. Dairy Sci 2007, 90, 594–601. [Google Scholar] [CrossRef] [Green Version]
- Domenech, J.; Martínez, J.; Pla, J.M. (Eds.) Biofarmacia y Farmacocinetica; Síntesis: Madrid, Spain, 1997; Volume II. [Google Scholar]
- Liu, J.; Yu, Z.; Guo, S.; Lee, S.R.; Xing, C.; Zhang, C.; Gao, Y.; Nicholls, D.G.; Lo, E.H.; Wang, X. Effects of neuroglobin overexpression on mitochondrial function and oxidative stress following hypoxia/reoxygenation in cultured neurons. J. Neurosci. Res. 2009, 87, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.; Kronenberg, G.; Balkaya, M.; Farber, K.; Gertz, K.; Kettenmann, H.; Endres, M. Acute neuroprotection by pioglitazone after mild brain ischemia without effect on long-term outcome. Exp. Neurol. 2009, 216, 321–328. [Google Scholar] [CrossRef]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke A J. Cereb. Circ. 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, S.; Azari, H.; McConnell, D.J.; Afzal, A.; Mocco, J. Intraluminal middle cerebral artery occlusion (MCAO) model for ischemic stroke with laser doppler flowmetry guidance in mice. J. Vis. Exp. JoVE 2011, 85, e2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decuzzi, P.; Pasqualini, R.; Arap, W.; Ferrari, M. Intravascular delivery of particulate systems: Does geometry really matter? Pharm. Res. 2009, 26, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hans, M.L.; Lowman, A.M. Biodegradable nanoparticles for drug delivery and targeting. Curr. Opin. Solid State Mat. Sci. 2002, 6, 319–327. [Google Scholar] [CrossRef]
- Muller, R.H.; Jacobs, C.; Kayser, O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv. Drug Deliv Rev. 2001, 47, 3–19. [Google Scholar] [CrossRef]
- Ritsema, J.A.S.; Herschberg, E.M.A.; Borgos, S.E.; Lovmo, C.; Schmid, R.; Te Welscher, Y.M.; Storm, G.; van Nostrum, C.F. Relationship between polarities of antibiotic and polymer matrix on nanoparticle formulations based on aliphatic polyesters. Int. J. Pharm 2018, 548, 730–739. [Google Scholar] [CrossRef]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Qian, J.; Zheng, S.; Changyi, Y.; Zhang, J.; Ju, S.; Zhu, J.; Li, C. Overcoming the blood-brain barrier for delivering drugs into the brain by using adenosine receptor nanoagonist. ACS Nano 2014, 8, 3678–3689. [Google Scholar] [CrossRef]
- Jallouli, Y.; Paillard, A.; Chang, J.; Sevin, E.; Betbeder, D. Influence of surface charge and inner composition of porous nanoparticles to cross blood-brain barrier in vitro. Int. J. Pharm. 2007, 344, 103–109. [Google Scholar] [CrossRef]
- Sanchez, M.T.; Ruiz, M.A.; Lasserrot, A.; Hormigo, M.; Morales, M.E. An improved ionic gelation method to encapsulate Lactobacillus spp. bacteria: Protection, survival and stability study. Food Hydrocoll. 2017, 69, 67–75. [Google Scholar] [CrossRef]
- Song, H.; Yu, W.; Gao, M.; Liu, X.; Ma, X. Microencapsulated probiotics using emulsification technique coupled with internal or external gelation process. Carbohydr. Polym. 2013, 96, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Anal, A.K.; Singh, H. Recent advances in microencapsulation of probiotics for industrial applications and targeted delivery. Trends Food Sci. Technol. 2007, 18, 240–251. [Google Scholar] [CrossRef]
- Rathore, S.; Desai, P.M.; Liew, C.V.; Chan, L.W.; Lieng, P.W.S. Microencapsulation of microbial cells. J. Food Eng. 2013, 116, 369–381. [Google Scholar] [CrossRef]
- Chen, F.; Lu, J.; Chen, F.; Lin, Z.; Lin, Y.; Yu, L.; Su, X.; Yao, P.; Cai, B.; Kang, D. Recombinant neuroglobin ameliorates early brain injury after subarachnoid hemorrhage via inhibiting the activation of mitochondria apoptotic pathway. Neurochem. Int. 2018, 112, 219–226. [Google Scholar] [CrossRef]
- Nayak, A.K. Controlled release drug delivery systems. Sci. J. UBU 2011, 2, 1–8. [Google Scholar]
- Jin, K.; Mao, X.O.; Xie, L.; Khan, A.A.; Greenberg, D.A. Neuroglobin protects against nitric oxide toxicity. Neurosci. Lett. 2008, 430, 135–137. [Google Scholar] [CrossRef] [Green Version]
- Fordel, E.; Thijs, L.; Martinet, W.; Lenjou, M.; Laufs, T.; Van Bockstaele, D.; Moens, L.; Dewilde, S. Neuroglobin and cytoglobin overexpression protects human SH-SY5Y neuroblastoma cells against oxidative stress-induced cell death. Neurosci. Lett. 2006, 410, 146–151. [Google Scholar] [CrossRef]
- Li, R.C.; Pouranfar, F.; Lee, S.K.; Morris, M.W.; Wang, Y.; Gozal, D. Neuroglobin protects PC12 cells against beta-amyloid-induced cell injury. Neurobiol. Aging 2008, 29, 1815–1822. [Google Scholar] [CrossRef]
- Xue, L.; Chen, H.; Lu, K.; Huang, J.; Duan, H.; Zhao, Y. Clinical significance of changes in serum neuroglobin and HIF-1alpha concentrations during the early-phase of acute ischemic stroke. J. Neurol. Sci. 2017, 375, 52–57. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Size Average (nm) | Polydispersion | ζ-Potential (mV) | |
|---|---|---|---|
| NGB–NPs | 145.77 ± 1.14 | 0.2628 ± 0.0046 | 27.55 ± 1.017 |
| Order 1 | Order 2 | Root 2 | Root 3 |
|---|---|---|---|
| 50.41 ± 0.59 | 49.63 ± 0.55 | 49.97 ± 0.48 | 47.51 ± 0.31 |
| Day 0 | Day 15 | Day 30 | |
|---|---|---|---|
| Percentage of NGB | 95.05% | 94.68% | 96.07% |
| Time (Days) | % Lost |
|---|---|
| 0 | 0.00% |
| 8 | 0.36% |
| 15 | 3.70% |
| 30 | 5.45% |
| 60 | 16.32% |
| 90 | 23.97% |
| Size Average (nm) | Polydispersion | ζ-Potential (mV) | |
|---|---|---|---|
| NGB–NPs | 143.98 ± 2.01 | 0.2331 ± 0.085 | 29.11 ± 1.971 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco, S.; Peralta, S.; Morales, M.E.; Martínez-Lara, E.; Pedrajas, J.R.; Castán, H.; Peinado, M.Á.; Ruiz, M.A. Hyaluronate Nanoparticles as a Delivery System to Carry Neuroglobin to the Brain after Stroke. Pharmaceutics 2020, 12, 40. https://doi.org/10.3390/pharmaceutics12010040
Blanco S, Peralta S, Morales ME, Martínez-Lara E, Pedrajas JR, Castán H, Peinado MÁ, Ruiz MA. Hyaluronate Nanoparticles as a Delivery System to Carry Neuroglobin to the Brain after Stroke. Pharmaceutics. 2020; 12(1):40. https://doi.org/10.3390/pharmaceutics12010040
Chicago/Turabian StyleBlanco, Santos, Sebastián Peralta, María Encarnación Morales, Esther Martínez-Lara, José Rafael Pedrajas, Herminia Castán, María Ángeles Peinado, and María Adolfina Ruiz. 2020. "Hyaluronate Nanoparticles as a Delivery System to Carry Neuroglobin to the Brain after Stroke" Pharmaceutics 12, no. 1: 40. https://doi.org/10.3390/pharmaceutics12010040