Repurposing Heparin as Antimalarial: Evaluation of Multiple Modifications Toward In Vivo Application

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Heparin Modification and Characterization
2.2. NMR Experimental Procedure
2.3. P. falciparum Culture and Growth Inhibition Assays
2.4. In Vitro Cytotoxicity Assays
2.5. Hemolysis Assays
2.6. In Vivo Toxicity Assays
2.7. Antimalarial Activity in Mice
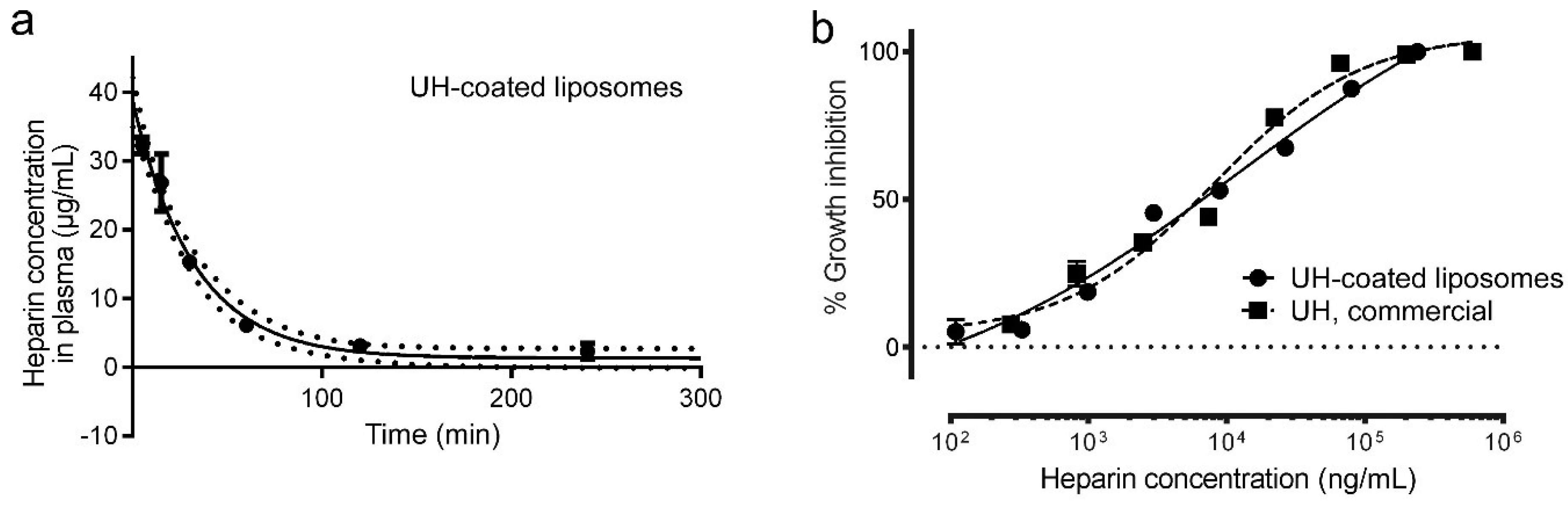
2.8. Preparation of Heparin-Coated Liposomes
2.9. Plasma Half-Life Determination
2.10. Ethics Statement
3. Results
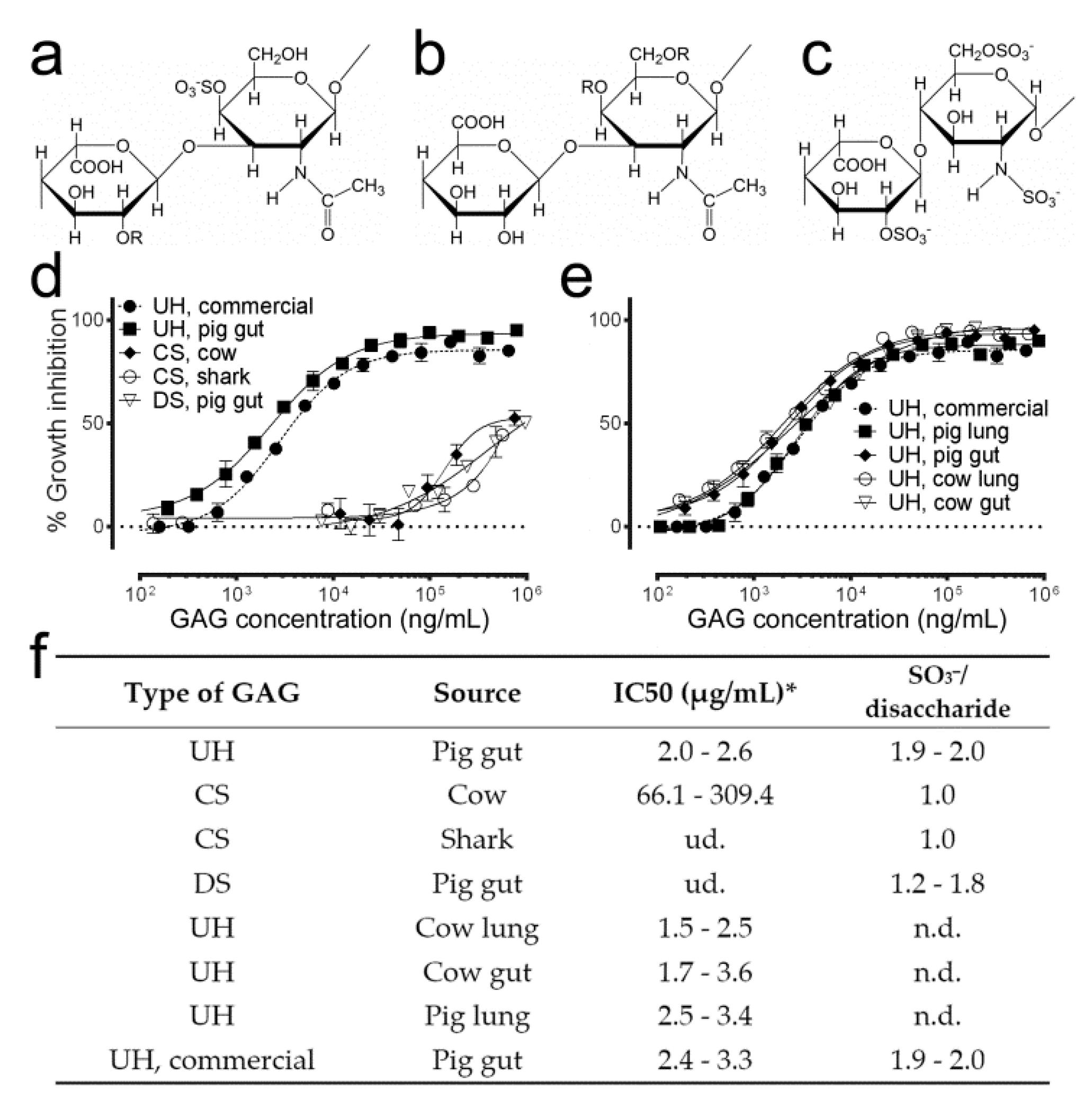
3.1. Antimalarial Activity Determination of Different Natural GAGs
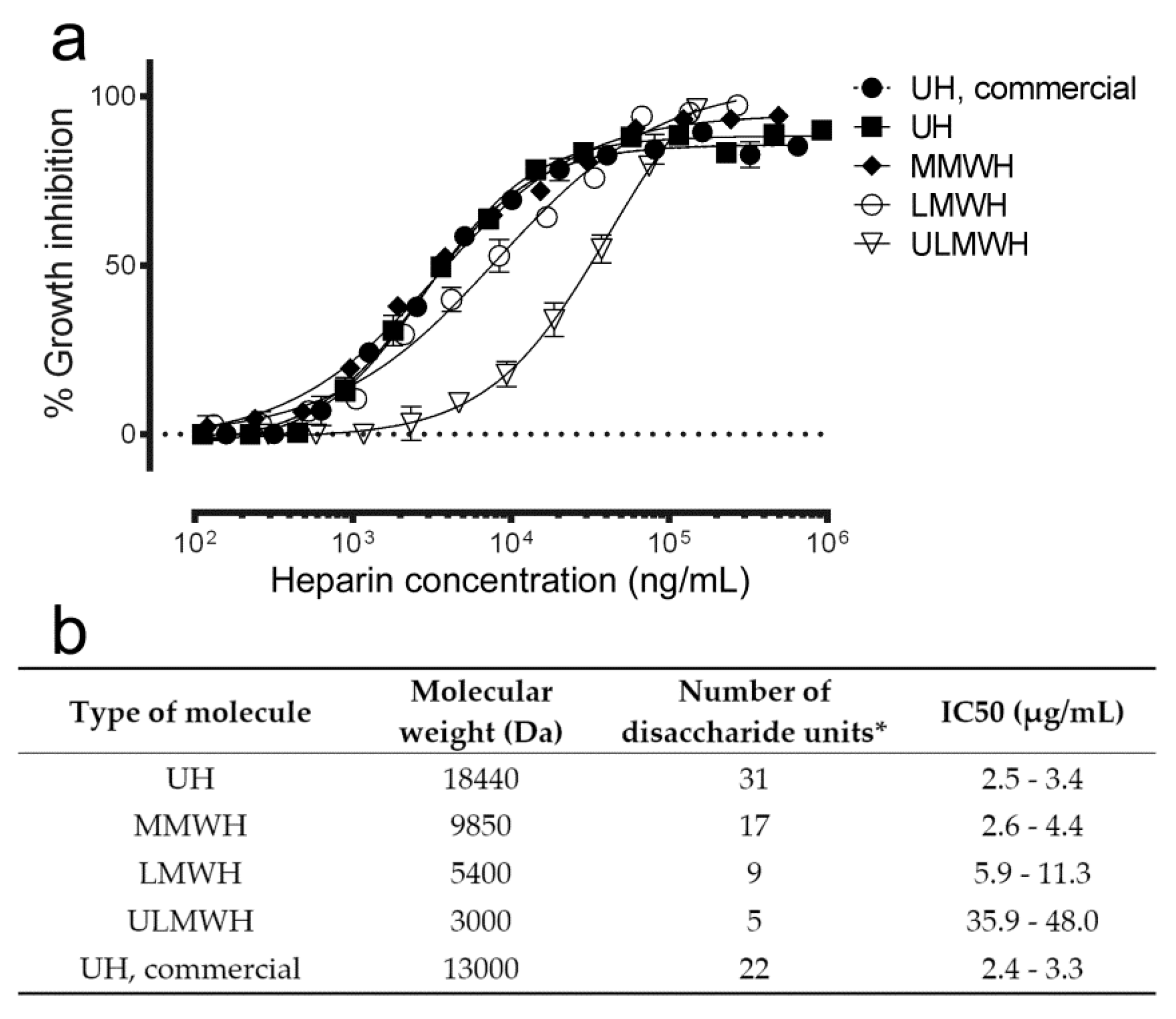
3.2. Effect on Antimalarial Activity of Heparin Molecular Weight
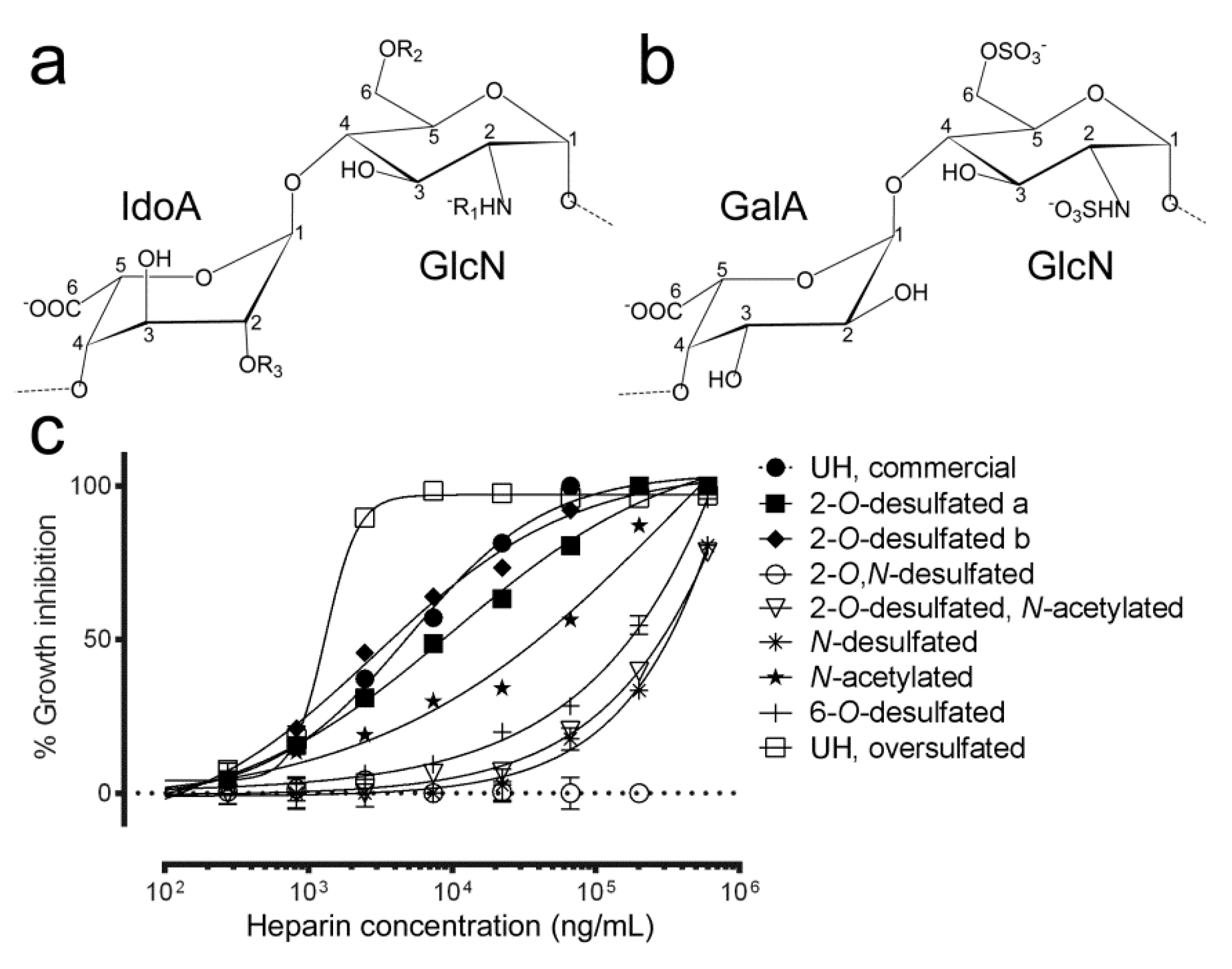
3.3. Effect of Sulfate Group Removal on the Antimalarial Activity of Heparin
3.4. Effect of Glycol-Split on the Antimalarial and Anticoagulant Activities of Heparin
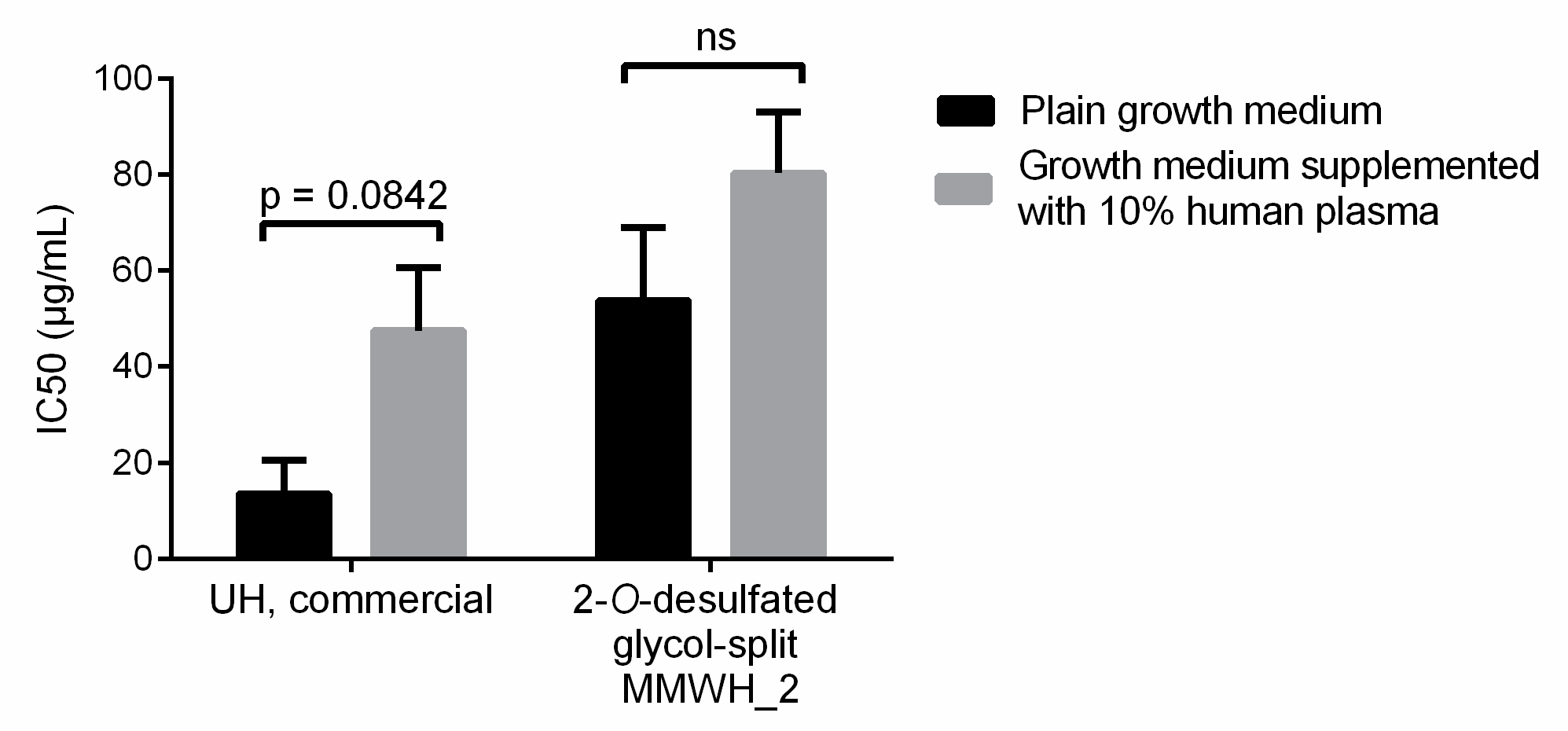
3.5. Selection of Heparin Forms Having Reduced Anticoagulant Activity but Maintaining Significant Plasmodium Growth Inhibition
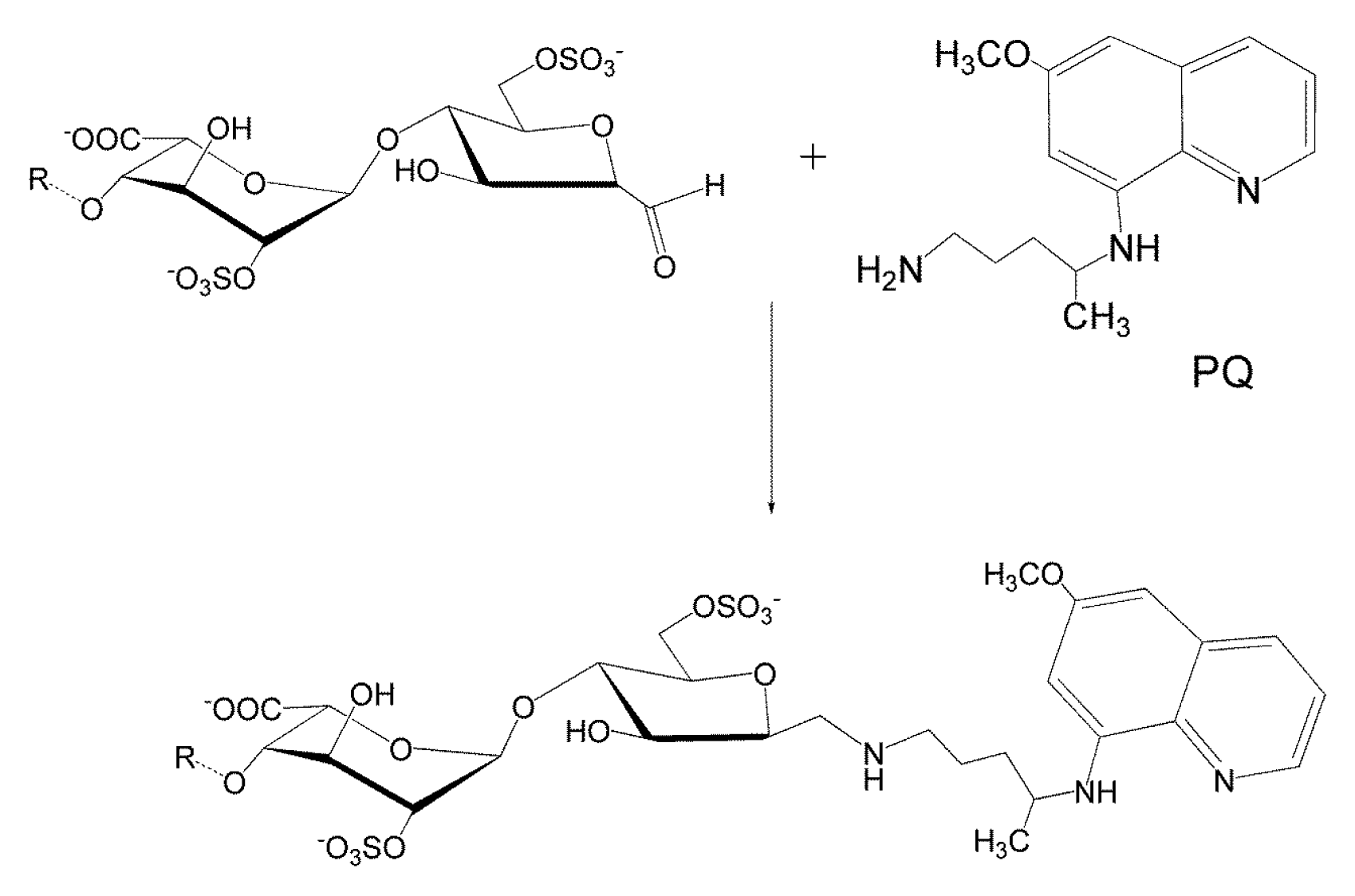
3.6. Conjugation of Heparin with PQ
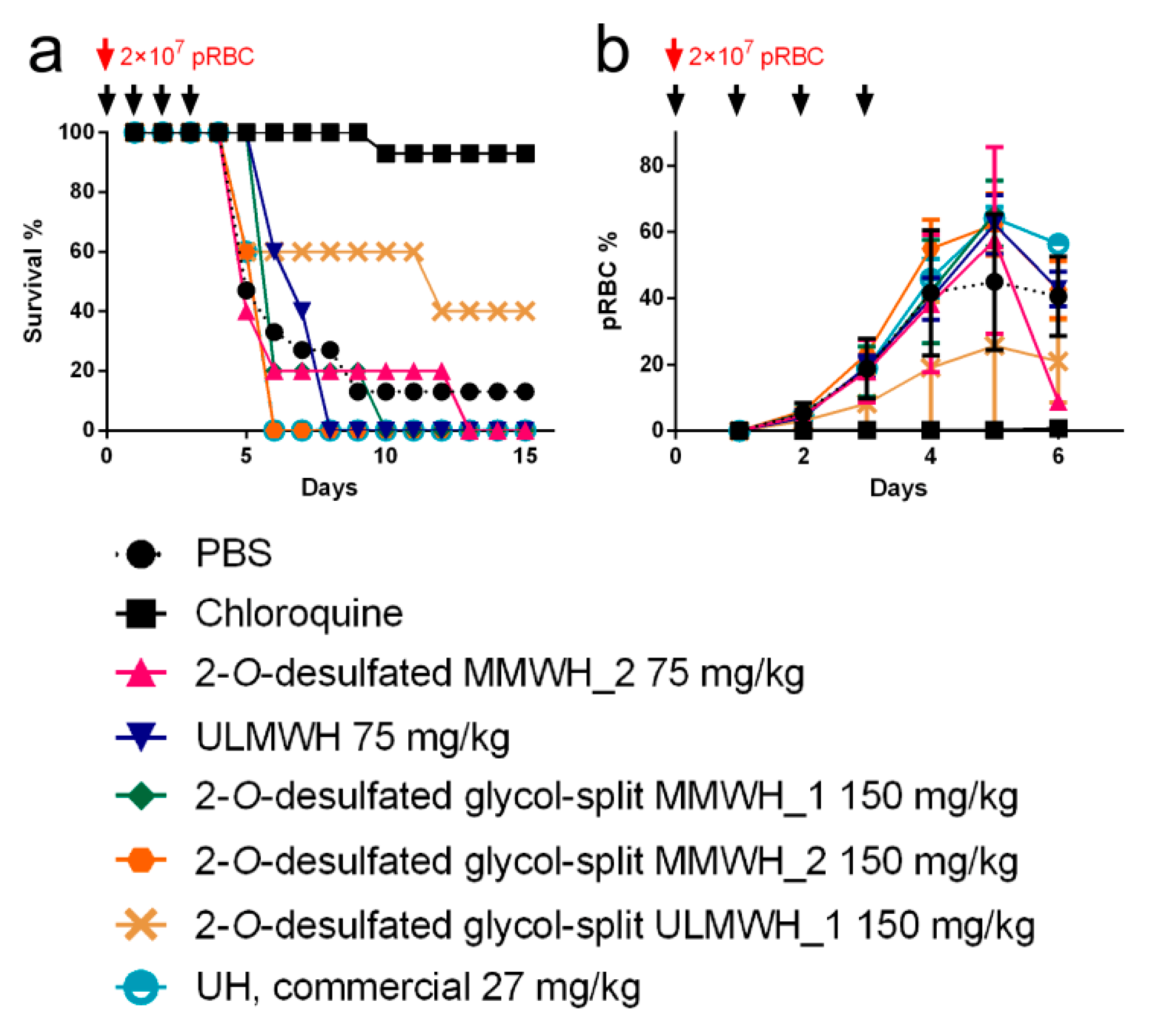
3.7. Antimalarial Activity In Vivo of Heparin-derived Structures
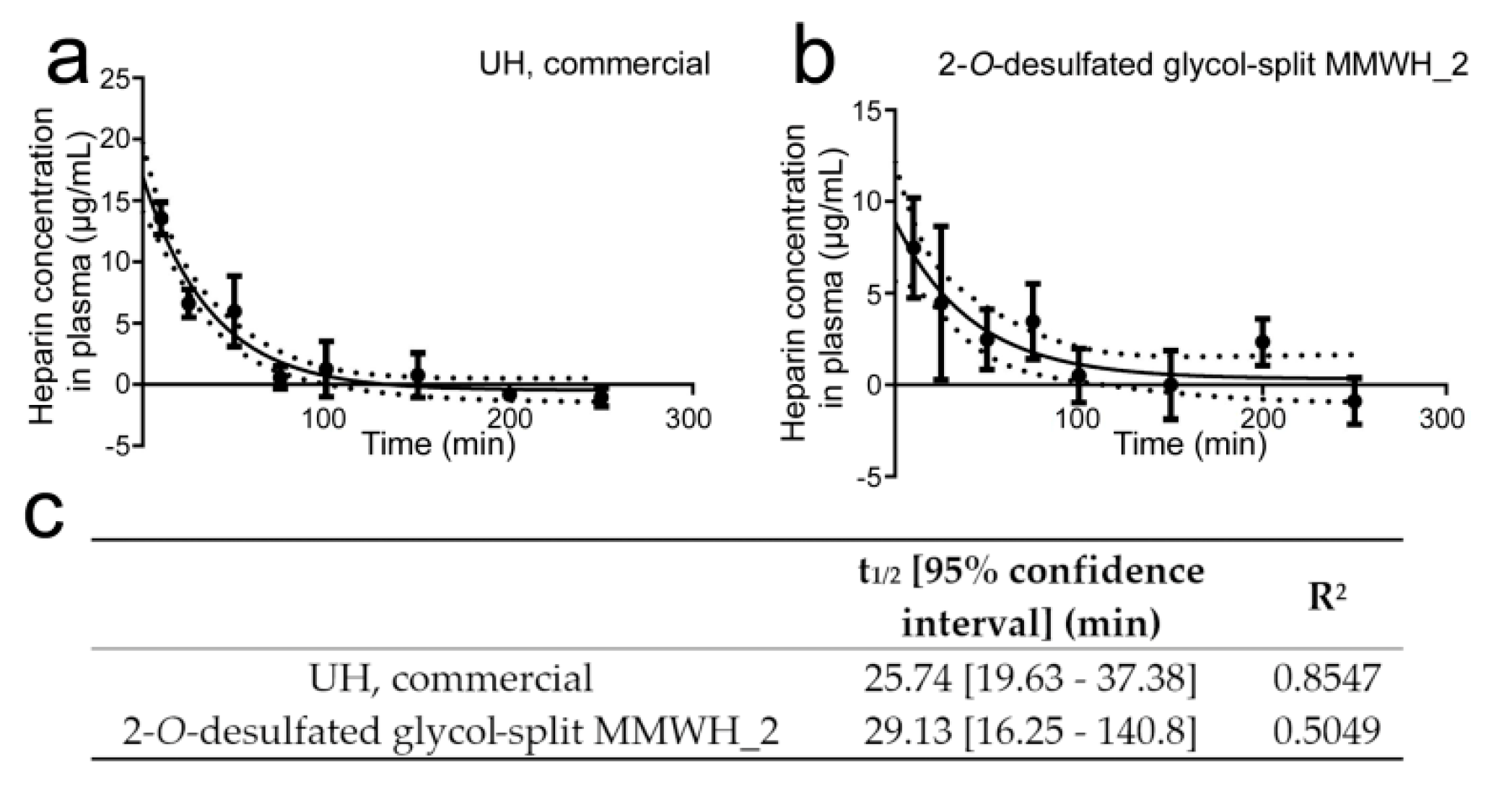
3.8. Determination of the Circulation Time of Intravenously Administered Heparin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Miller, L.H.; Baruch, D.I.; Marsh, K.; Doumbo, O.K. The pathogenic basis of malaria. Nature 2002, 415, 673–679. [Google Scholar] [CrossRef]
- Carlson, J.; Wahlgren, M. Plasmodium falciparum erythrocyte rosetting is mediated by promiscuous lectin-like interactions. J. Exp. Med. 1992, 176, 1311–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, K.S.; Lewis, L.S.; Mali, S.; Parise, M.E. Treatment of malaria in the United States: A systematic review. JAMA 2007, 297, 2264–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeson, J.G.; Andrews, K.T.; Boyle, M.; Duffy, M.F.; Choong, E.K.; Byrne, T.J.; Chesson, J.M.; Lawson, A.M.; Chai, W. Structural basis for binding of Plasmodium falciparum erythrocyte membrane protein 1 to chondroitin sulfate and placental tissue and the influence of protein polymorphisms on binding specificity. J. Biol. Chem. 2007, 282, 22426–22436. [Google Scholar] [CrossRef] [Green Version]
- Juillerat, A.; Igonet, S.; Vigan-Womas, I.; Guillotte, M.; Gangnard, S.; Faure, G.; Baron, B.; Raynal, B.; Mercereau-Puijalon, O.; Bentley, G.A. Biochemical and biophysical characterisation of DBL1a1-varO, the rosetting domain of PfEMP1 from the VarO line of Plasmodium falciparum. Mol. Biochem. Parasitol. 2010, 170, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Pradel, G.; Garapaty, S.; Frevert, U. Proteoglycans mediate malaria sporozoite targeting to the liver. Mol. Microbiol. 2002, 45, 637–651. [Google Scholar] [CrossRef]
- Coppi, A.; Tewari, R.; Bishop, J.R.; Bennett, B.L.; Lawrence, R.; Esko, J.D.; Billker, O.; Sinnis, P. Heparan sulfate proteoglycans provide a signal to Plasmodium sporozoites to stop migrating and productively invade host cells. Cell Host Microbe 2007, 2, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogerson, S.J.; Reeder, J.C.; Al-Yaman, F.; Brown, G.V. Sulfated glycoconjugates as disrupters of Plasmodium falciparum erythrocyte rosettes. Am. J. Trop. Med. Hyg. 1994, 51, 198–203. [Google Scholar] [CrossRef]
- Angeletti, D.; Sandalova, T.; Wahlgren, M.; Achour, A. Binding of subdomains 1/2 of PfEMP1-DBL1a to heparan sulfate or heparin mediates Plasmodium falciparum rosetting. PLoS ONE 2015, 10, e0118898. [Google Scholar] [CrossRef]
- Vogt, A.M.; Pettersson, F.; Moll, K.; Jonsson, C.; Normark, J.; Ribacke, U.; Egwang, T.G.; Ekre, H.P.; Spillmann, D.; Chen, Q.; et al. Release of sequestered malaria parasites upon injection of a glycosaminoglycan. PLoS Pathog. 2006, 2, e100. [Google Scholar] [CrossRef] [Green Version]
- Sheehy, T.W.; Reba, R.C. Complications of falciparum malaria and their treatment. Ann. Intern. Med. 1967, 66, 807–809. [Google Scholar] [CrossRef]
- Leitgeb, A.M.; Blomqvist, K.; Cho-Ngwa, F.; Samje, M.; Nde, P.; Titanji, V.; Wahlgren, M. Low anticoagulant heparin disrupts Plasmodium falciparum rosettes in fresh clinical isolates. Am. J. Trop. Med. Hyg. 2011, 84, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Yang, C.; Patterson, P.S.; Udhayakumar, V.; Lal, A.A. Sulfated polyanions inhibit invasion of erythrocytes by plasmodial merozoites and cytoadherence of endothelial cells to parasitized erythrocytes. Infect. Immun. 1996, 64, 1373–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, M.J.; Richards, J.S.; Gilson, P.R.; Chai, W.; Beeson, J.G. Interactions with heparin-like molecules during erythrocyte invasion by Plasmodium falciparum merozoites. Blood 2010, 115, 4559–4568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeson, J.G.; Drew, D.R.; Boyle, M.J.; Feng, G.; Fowkes, F.J.I.; Richards, J.S. Merozoite surface proteins in red blood cell invasion, immunity and vaccines against malaria. FEMS Microbiol. Rev. 2016, 40, 343–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle-Delgado, J.J.; Urbán, P.; Fernàndez-Busquets, X. Demonstration of specific binding of heparin to Plasmodium falciparum-infected vs. non-infected red blood cells by single-molecule force spectroscopy. Nanoscale 2013, 5, 3673–3680. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.; Moles, E.; Urbán, P.; Prohens, R.; Busquets, M.A.; Sevrin, C.; Grandfils, C.; Fernàndez-Busquets, X. Application of heparin as a dual agent with antimalarial and liposome targeting activities towards Plasmodium-infected red blood cells. Nanomed. NBM 2014, 10, 1719–1728. [Google Scholar] [CrossRef]
- Glushakova, S.; Busse, B.L.; Garten, M.; Beck, J.R.; Fairhurst, R.M.; Goldberg, D.E.; Zimmerberg, J. Exploitation of a newly-identified entry pathway into the malaria parasite-infected erythrocyte to inhibit parasite egress. Sci. Rep. 2017, 7, 12250. [Google Scholar] [CrossRef]
- Boyle, M.J.; Skidmore, M.; Dickerman, B.; Cooper, L.; Devlin, A.; Yates, E.; Horrocks, P.; Freeman, C.; Chai, W.; Beeson, J.G. Identification of heparin modifications and polysaccharide inhibitors of Plasmodium falciparum merozoite invasion that have potential for novel drug development. Antimicrob. Agents Chemother. 2017, 61, e00709–e00717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisano, C.; Aulicino, C.; Vesci, L.; Casu, B.; Naggi, A.; Torri, G.; Ribatti, D.; Belleri, M.; Rusnati, M.; Presta, M. Undersulfated, low-molecular-weight glycol-split heparin as an antiangiogenic VEGF antagonist. Glycobiology 2005, 15, 1C–6C. [Google Scholar] [CrossRef] [Green Version]
- Naggi, A.; Casu, B.; Perez, M.; Torri, G.; Cassinelli, G.; Penco, S.; Pisano, C.; Giannini, G.; Ishai-Michaeli, R.; Vlodavsky, I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J. Biol. Chem. 2005, 280, 12103–12113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shively, J.E.; Conrad, H.E. Formation of anhydrosugars in the chemical depolymerization of heparin. Biochemistry 1976, 15, 3932–3942. [Google Scholar] [CrossRef] [PubMed]
- Cifonelli, J.A. Reaction of heparitin sulfate with nitrous acid. Carbohydr. Res. 1968, 8, 233–242. [Google Scholar] [CrossRef]
- Matsuo, M.; Takano, R.; Kamei-Hayashi, K.; Hara, S. A novel regioselective desulfation of polysaccharide sulfates: Specific 6-O-desulfation with N,O-bis(trimethylsilyl)acetamide. Carbohydr. Res. 1993, 241, 209–215. [Google Scholar] [CrossRef]
- Maruyama, T.; Toida, T.; Imanari, T.; Yu, G.; Linhardt, R.J. Conformational changes and anticoagulant activity of chondroitin sulfate following its O-sulfonation. Carbohydr. Res. 1998, 306, 35–43. [Google Scholar] [CrossRef]
- Casu, B.; Guerrini, M.; Naggi, A.; Perez, M.; Torri, G.; Ribatti, D.; Carminati, P.; Giannini, G.; Penco, S.; Pisano, C.; et al. Short heparin sequences spaced by glycol-split uronate residues are antagonists of fibroblast growth factor 2 and angiogenesis inhibitors. Biochemistry 2002, 41, 10519–10528. [Google Scholar] [CrossRef]
- Bertini, S.; Bisio, A.; Torri, G.; Bensi, D.; Terbojevich, M. Molecular weight determination of heparin and dermatan sulfate by size exclusion chromatography with a triple detector array. Biomacromolecules 2005, 6, 168–173. [Google Scholar] [CrossRef]
- Casu, B.; Gennaro, U. A conductimetric method for the determination of sulphate and carboxyl groups in heparin and other mucopolysaccharides. Carbohydr. Res. 1975, 39, 168–176. [Google Scholar] [CrossRef]
- Gerotziafas, G.T.; Petropoulou, A.D.; Verdy, E.; Samama, M.M.; Elalamy, I. Effect of the anti-factor Xa and anti-factor IIa activities of low-molecular-weight heparins upon the phases of thrombin generation. J. Thromb. Haemost. 2007, 5, 955–962. [Google Scholar] [CrossRef]
- Monakhova, Y.B.; Diehl, B.W.K.; Do, T.X.; Schulze, M.; Witzleben, S. Novel method for the determination of average molecular weight of natural polymers based on 2D DOSY NMR and chemometrics: Example of heparin. J. Pharm. Biomed. Anal. 2018, 149, 128–132. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef]
- Dluzewski, A.R.; Ling, I.T.; Rangachari, K.; Bates, P.A.; Wilson, R.J. A simple method for isolating viable mature parasites of Plasmodium falciparum from cultures. Trans. R. Soc. Trop. Med. Hyg. 1984, 78, 622–624. [Google Scholar] [CrossRef]
- Fidock, D.A.; Rosenthal, P.J.; Croft, S.L.; Brun, R.; Nwaka, S. Antimalarial drug discovery: Efficacy models for compound screening. Nat. Rev. Drug Discov. 2004, 3, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Frazier, S.B.; Roodhouse, K.A.; Hourcade, D.E.; Zhang, L. The quantification of glycosaminoglycans: A comparison of HPLC, carbazole, and Alcian Blue methods. Open Glycosci. 2008, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Warttinger, U.; Giese, C.; Harenberg, J.; Holmer, E.; Krämer, R. A fluorescent probe assay (Heparin Red) for direct detection of heparins in human plasma. Anal. Bioanal. Chem. 2016, 408, 8241–8251. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Fuster, V. Guide to anticoagulant therapy. Part 1: Heparin. Circulation 1994, 89, 1449–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esko, J.D.; Prestegard, H.; Linhardt, R.J. Proteins that bind sulfated glycosaminoglycans. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017; pp. 493–502. [Google Scholar]
- Naggi, A. Glycol-splitting as a device for modulating inhibition of growth factors and heparanase by heparin and heparin derivatives. In Chemistry and Biology of Heparin and Heparan Sulfate; Garg, H.G., Linhardt, R.J., Hales, C.A., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2005; pp. 461–481. [Google Scholar]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Marques, J.; Vilanova, E.; Mourão, P.A.S.; Fernàndez-Busquets, X. Marine organism sulfated polysaccharides exhibiting significant antimalarial activity and inhibition of red blood cell invasion by Plasmodium. Sci. Rep. 2016, 6, 24368. [Google Scholar] [CrossRef] [Green Version]
- Leitgeb, A.M.; Charunwatthana, P.; Rueangveerayut, R.; Uthaisin, C.; Silamut, K.; Chotivanich, K.; Sila, P.; Moll, K.; Lee, S.J.; Lindgren, M.; et al. Inhibition of merozoite invasion and transient de-sequestration by sevuparin in humans with Plasmodium falciparum malaria. PLoS ONE 2017, 12, e0188754. [Google Scholar] [CrossRef]
- Hirsh, J.; Warkentin, T.E.; Shaughnessy, S.G.; Anand, S.S.; Halperin, J.L.; Raschke, R.; Granger, C.; Ohman, E.M.; Dalen, J.E. Heparin and low-molecular-weight heparin: Mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest 2001, 119, 64S–94S. [Google Scholar] [CrossRef] [Green Version]
- Mosqueira, V.C.F.; Loiseau, P.M.; Bories, C.; Legrand, P.; Devissaguet, J.P.; Barratt, G. Efficacy and pharmacokinetics of intravenous nanocapsule formulations of halofantrine in Plasmodium berghei-infected mice. Antimicrob. Agents Chemother. 2004, 48, 1222–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, Y.; Aoyagi, S.; Kusada, Y.; Miyamoto, K. The characteristics of anticoagulation by covalently immobilized heparin. J. Biomed. Mater. Res. 1980, 14, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Najer, A.; Wu, D.; Bieri, A.; Brand, F.; Palivan, C.G.; Beck, H.P.; Meier, W. Nanomimics of host cell membranes block invasion and expose invasive malaria parasites. ACS Nano 2014, 8, 12560–12571. [Google Scholar] [CrossRef] [PubMed]
- Najer, A.; Thamboo, S.; Palivan, C.G.; Beck, H.P.; Meier, W. Giant host red blood cell membrane mimicking polymersomes bind parasite proteins and malaria parasites. Chimia 2016, 70, 288–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, M.; Du, Y.; Ling, L.; Li, X. Artesunate-heparin conjugate based nanocapsules with improved pharmacokinetics to combat malaria. Int. J. Pharm. 2019, 562, 162–171. [Google Scholar] [CrossRef]
- Schulz, J.D.; Gauthier, M.A.; Leroux, J.C. Improving oral drug bioavailability with polycations? Eur. J. Pharm. Biopharm. 2015, 97, 427–437. [Google Scholar] [CrossRef]
- Shukla, S.K.; Mishra, A.K.; Arotiba, O.A.; Mamba, B.B. Chitosan-based nanomaterials: A state-of-the-art review. Int. J. Biol. Macromol. 2013, 59, 46–58. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Molecule of Origin | Structure | Molecular Weight (Da) | aPTT (IU/mg) | IC50 (µg/mL) |
|---|---|---|---|---|---|
| 2-O-desulfated structure a | UH | R1,R2: SO3−; R3: H | 10,721 | 34 | 7.6–13.5 |
| 2-O-desulfated structure b | UH | b | 14,023 | 63 | 2.2–4.1 |
| 2-O-desulfated, N-desulfated | 2-O-desulfated a | R1,R3: H; R2: SO3− | 13,459 | 20 | ud. |
| 2-O-desulfated, N-acetylated | 2-O-desulfated a | R1: Ac; R2: SO3−; R3: H | 10,868 | 24 | >1000 |
| N-desulfated | UH | R2,R3: SO3−; R1: H | 15,771 | 42 | >1000 |
| N-acetylated | N-desulfated | R2,R3: SO3−; R1: Ac | 15,963 | 91 | >300 |
| 6-O-desulfated | UH | R1,R3: SO3−; R2: H | 14,521 | 45 | >1000 |
| UH | - | R1,R2,R3: SO3− | 15,792 | 203 | 2.0–2.6 |
| UH, oversulfated | UH | R1,R2,R3: SO3− | 19,990 | 94 | 1.2–1.4 |
| UH, commercial | - | R1,R2,R3: SO3− | 13,000 | n.d. | 4.1–6.0 |
| Sample | Mw (Da) | SO3−/Disaccharide | IC50 (µg/mL ± SD) | IC90 (µg/mL ± SD) | aPTT (IU/mg) | In Vitro Toxicity 1 (% ± SD) | Hemolysis 1 (% ± SD) |
|---|---|---|---|---|---|---|---|
| MMWH_1 | 10,497 | n.d. | 22.7 ± 2.4 | 178.1 ± 28.4 | 110 | 1.1 ± 3.4 | 0.1 ± 0.2 |
| MMWH_2 | 11,000 | 1.9 | 33.7 ± 21.8 | 374.8 ± 189.3 | 130 | 0.0 ± 6.0 | 0.5 ± 0.2 |
| 2-O-desulfated MMWH_1 | 8771 | 1.6 | 80.7 ± 5.0 | 270.3 ± 42.5 | 38 | 10.0 ± 3.0 | 0.3 ± 0.4 |
| 2-O-desulfated MMWH_2 | 8776 | 1.5 | 91.9 ± 9.0 | 351.9 ± 43.4 | 23 | 0.0 ± 2.3 | 2.0 ± 2.0 |
| 2-O-desulfated MMWH_3 | 10,198 | 1.5 | 59.1 ± 16.5 | 487.9 ± 161.3 | 53 | 17.9 ± 2.1 | 0.5 ± 0.2 |
| 2-O-desulfated MMWH_4 | 11,190 | 1.4 | 68.0 ± 14.5 | 404.8 ± 119.4 | 33 | 7.5 ± 1.8 | 0.0 ± 0.0 |
| 2-O-desulfated glycol-split MMWH_1 | 7388 | 1.8 | 79.6 ± 5.4 | 893.1 ± 321.0 | 6 | 7.3 ± 4.5 | 0.4 ± 0.1 |
| 2-O-desulfated glycol-split MMWH_2 | 7037 | 1.5 | 84.2 ± 13.4 | 303.4 ± 46.2 | 5 | 3.6 ± 5.4 | 0.0 ± 0.0 |
| ULMWH | 4270 | 2.2 | 49.3 ± 6.0 | 236.3 ± 54.9 | 6 | 6.8 ± 9.2 | 0.2 ± 0.1 |
| 2-O-desulfated ULMWH_1 | 4150 | 1.6 | 140.9 ± 15.9 | 322.5 ± 27.5 | ud. | 20.0 ± 16.7 | 0.0 ± 0.0 |
| 2-O-desulfated ULMWH_2 | 4450 | 1.4 | 129.2 ± 13.1 | 262.5 ± 16.1 | ud. | 37.0 ± 9.9 | 0.2 ± 0.0 |
| 2-O-desulfated glycol-split ULMWH_1 | 4024 | 1.7 | 104.4 ± 6.0 | 192.4 ± 16.2 | ud. | 2.5 ± 2.3 | 0.0 ± 0.2 |
| 2-O-desulfated glycol-split ULMWH_2 | 3800 | 1.6 | 130.3 ± 15.5 | 200.5 ± 111.6 | ud. | 11.5 ± 8.0 | 0.0 ± 0.1 |
| UH, commercial | 13,000 | 1.9–2.0 | 9.4 ± 4.4 | 135.5 ± 12.8 | 197 | 0.0 ± 6.0 | 0.0 ± 0.0 |
| Sample | Mw (Da) | IC50 (µM) ± SD |
|---|---|---|
| MMWH-PQ | 13,945 | 1.16 ± 0.40 |
| MMWH_1 | 11,000 | 3.1 ± 2.0 1 |
| PQ | 259 | 5.20 ± 1.33 |
| Sample | IC50 (µg/mL ± SD) | aPTT (IU/mg) | In Vitro Toxicity 1 (% ± SD) | Hemolysis 1 (% ± SD) | In Vitro Toxicity 2 |
|---|---|---|---|---|---|
| 2-O-desulfated MMWH_2 | 91.95 ± 8.97 | 23 | 0.00 ± 2.34 | 1.99 ± 2.05 | >320 mg/kg |
| ULMWH | 49.31 ± 5.97 | 6 | 6.83 ± 9.18 | 0.21 ± 0.13 | >320 mg/kg |
| 2-O-desulfated glycol-split MMWH_1 | 79.60 ± 5.38 | 6 | 7.26 ± 4.55 | 0.36 ± 0.09 | >750 mg/kg |
| 2-O-desulfated glycol-split MMWH_2 | 84.20 ± 13.45 | 5 | 3.61 ± 5.42 | 0.00 ± 0.03 | >750 mg/kg |
| 2-O-desulfated glycol-split ULMWH_1 | 104.40 ± 6.03 | 0 | 2.49 ± 2.35 | 0.00 ± 0.22 | >750 mg/kg |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lantero, E.; Aláez-Versón, C.R.; Romero, P.; Sierra, T.; Fernàndez-Busquets, X. Repurposing Heparin as Antimalarial: Evaluation of Multiple Modifications Toward In Vivo Application. Pharmaceutics 2020, 12, 825. https://doi.org/10.3390/pharmaceutics12090825
Lantero E, Aláez-Versón CR, Romero P, Sierra T, Fernàndez-Busquets X. Repurposing Heparin as Antimalarial: Evaluation of Multiple Modifications Toward In Vivo Application. Pharmaceutics. 2020; 12(9):825. https://doi.org/10.3390/pharmaceutics12090825
Chicago/Turabian StyleLantero, Elena, Carlos Raúl Aláez-Versón, Pilar Romero, Teresa Sierra, and Xavier Fernàndez-Busquets. 2020. "Repurposing Heparin as Antimalarial: Evaluation of Multiple Modifications Toward In Vivo Application" Pharmaceutics 12, no. 9: 825. https://doi.org/10.3390/pharmaceutics12090825