Physiologically Based Pharmacokinetic Modeling of Metoprolol Enantiomers and α-Hydroxymetoprolol to Describe CYP2D6 Drug-Gene Interactions

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Software
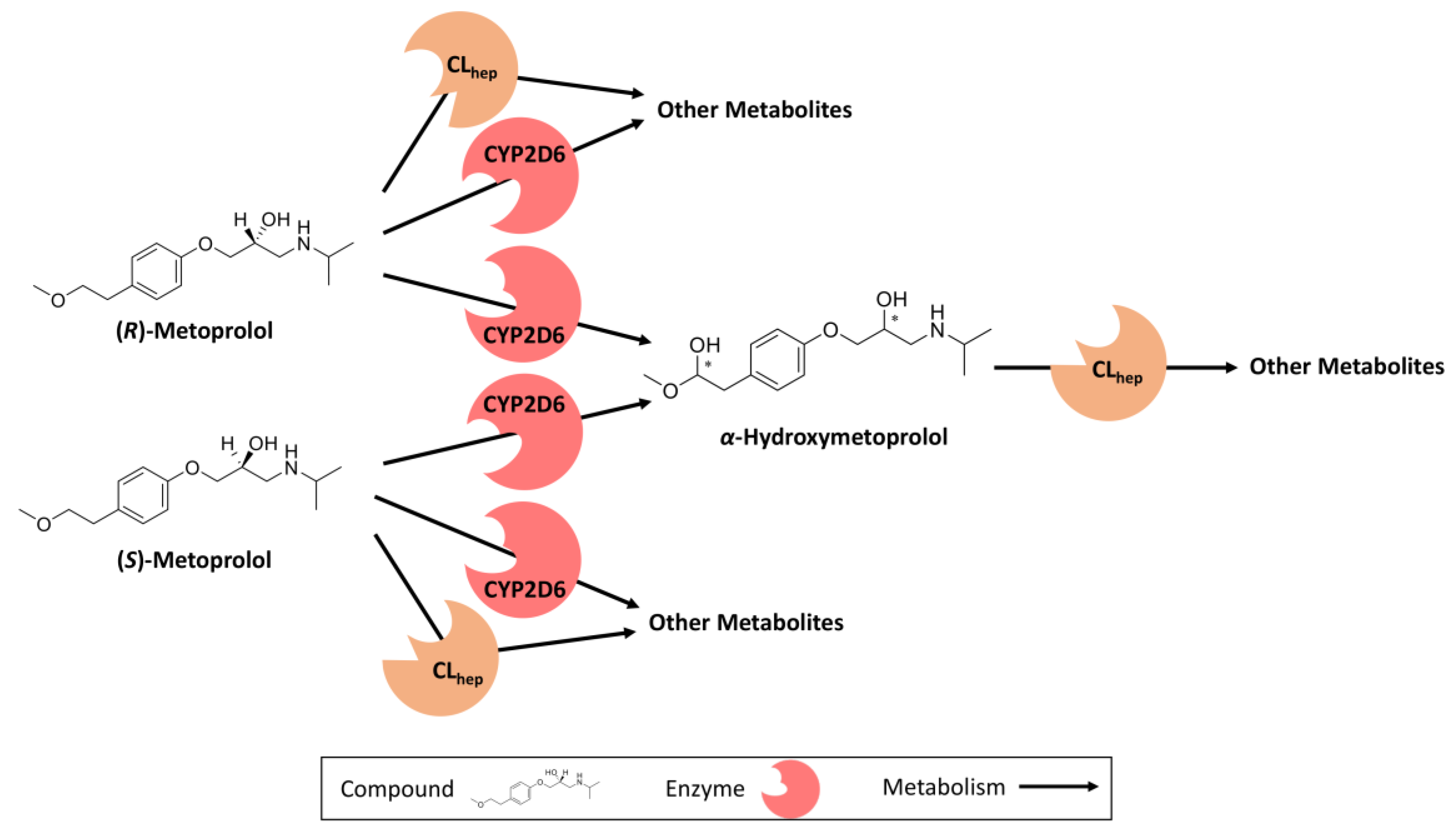
2.2. PBPK Model Building
2.3. DGI Modeling
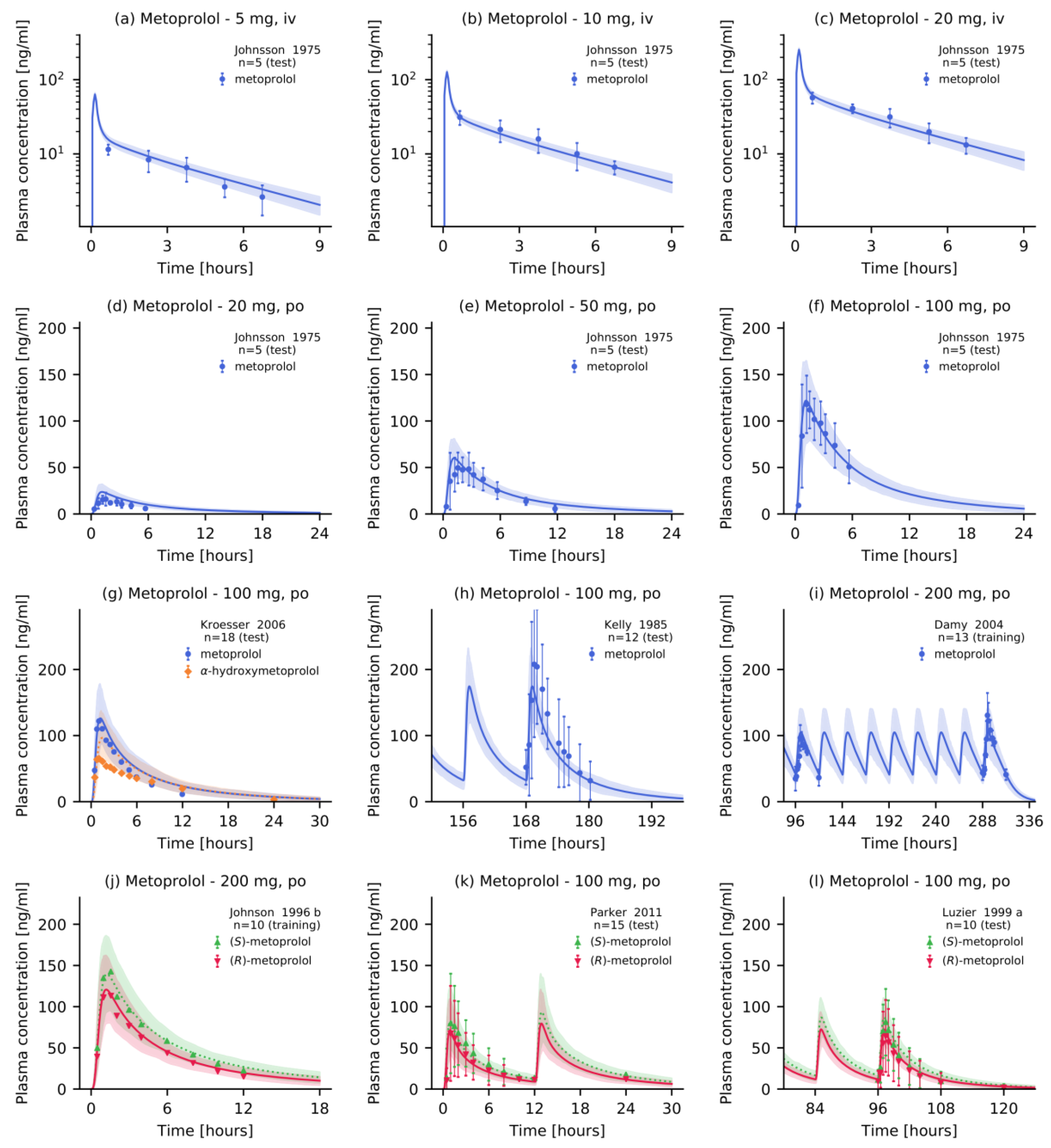
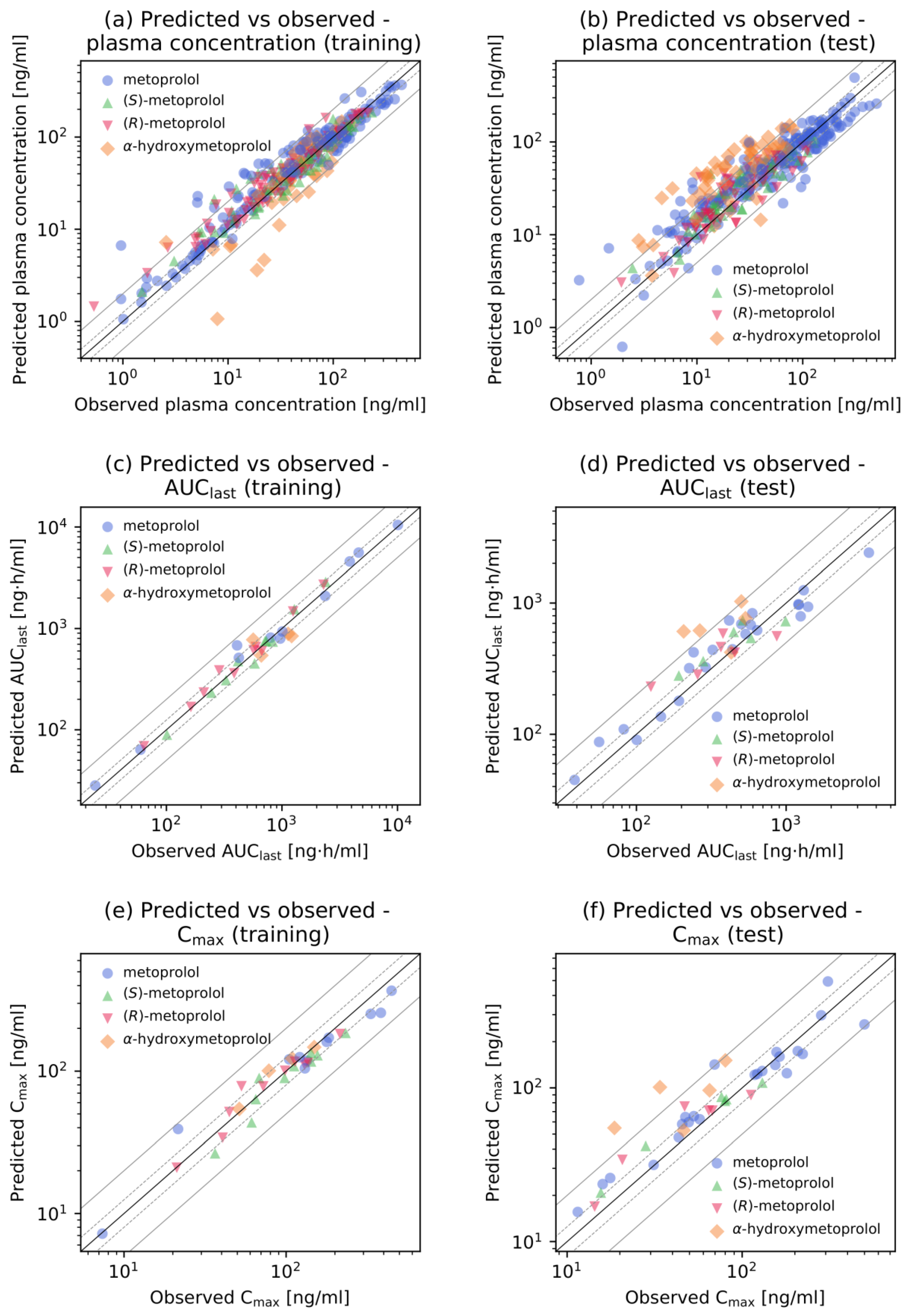
2.4. PBPK Model Evaluation
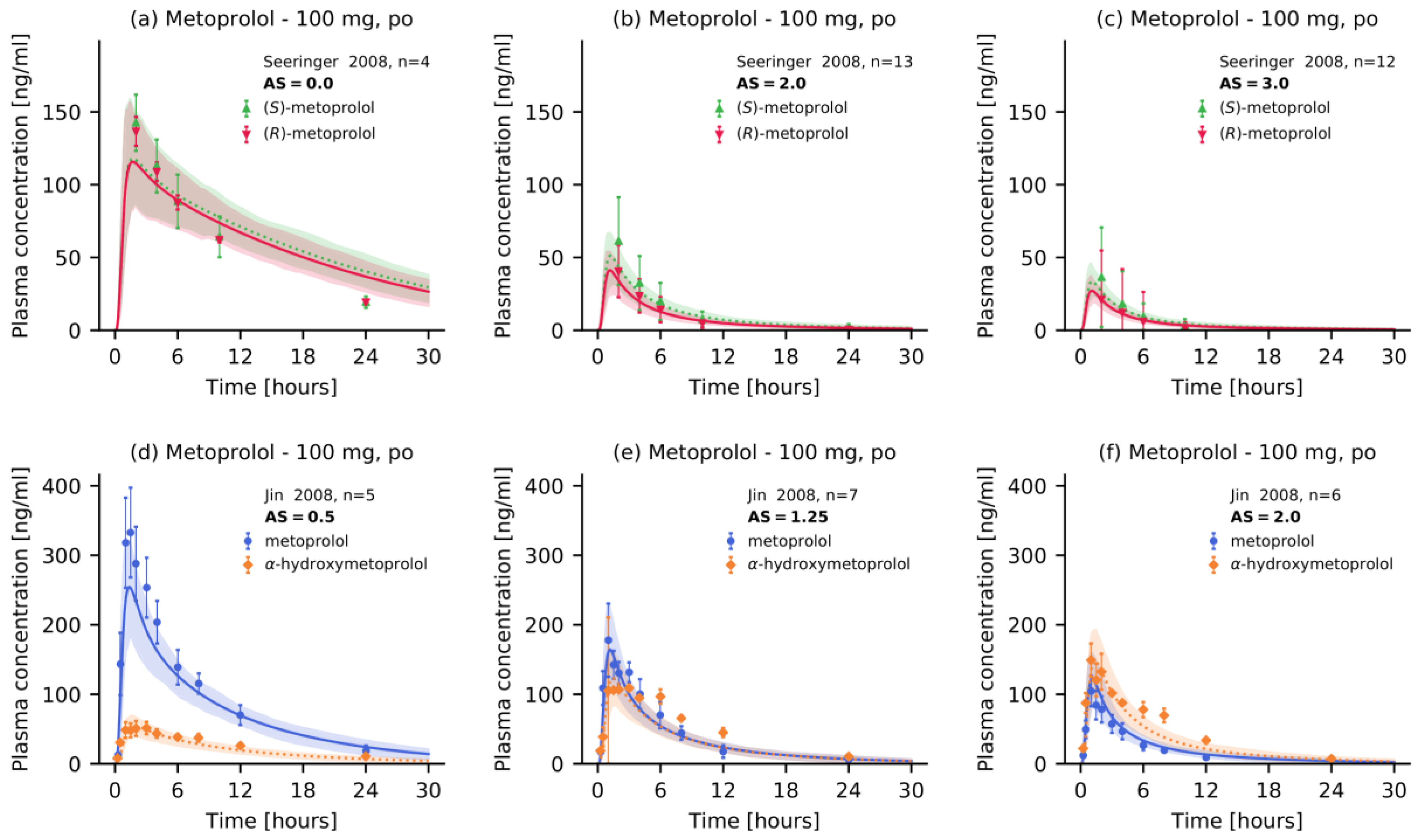
2.5. DGI Modeling Evaluation
3. Results
3.1. Metoprolol PBPK Model Development and Evaluation
3.2. Metoprolol CYP2D6 DGI Model Development and Evaluation
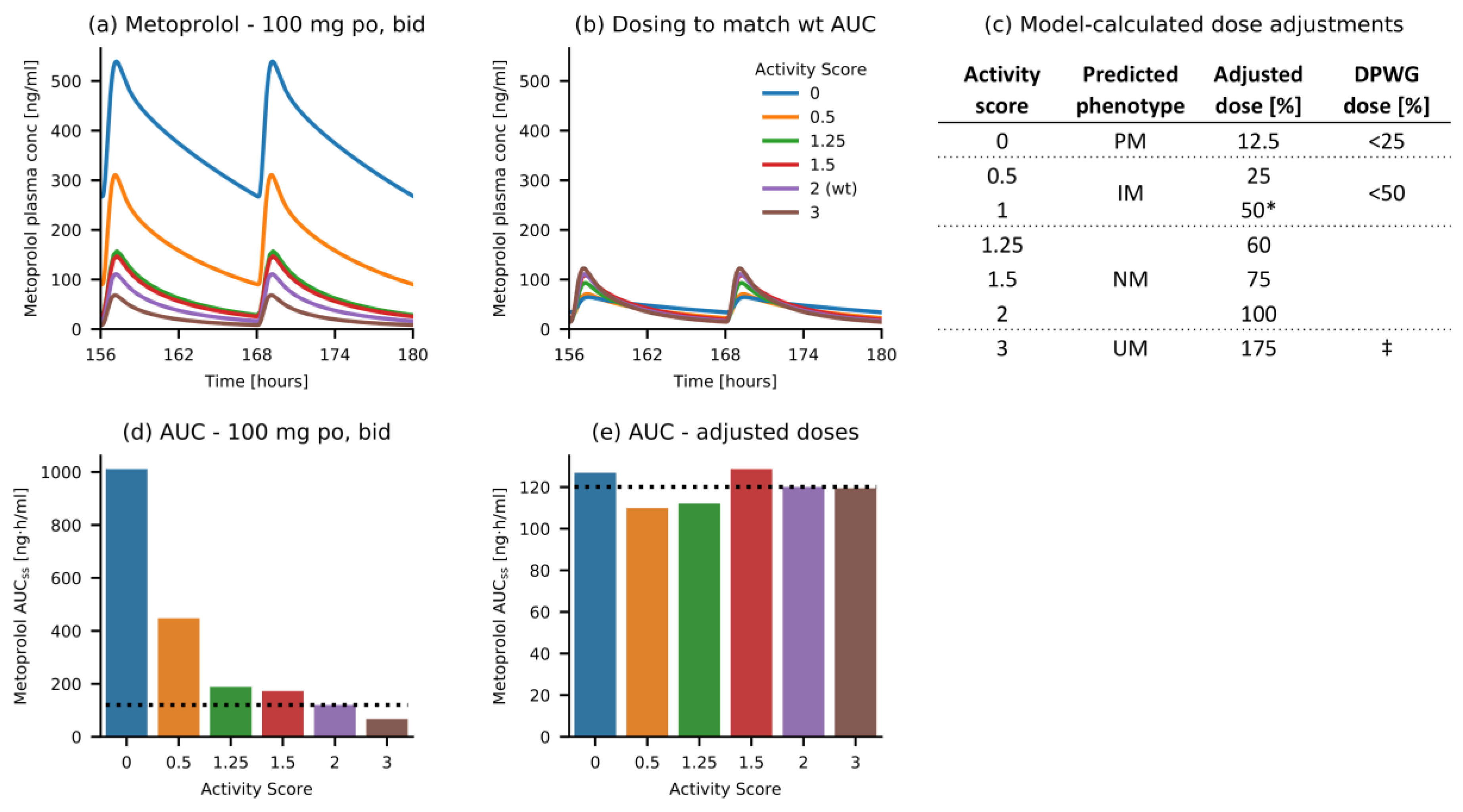
3.3. Metoprolol Dose Adaptation for CYP2D6 DGIs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- ClinCalc LLC. ClinCalc DrugStats Database. Available online: https://clincalc.com/Drugstats/ (accessed on 10 September 2020).
- Novartis Pharmaceuticals Corporation Lopressor® Tablet and Injection—Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/017963s062,018704s021lbl.pdf (accessed on 10 September 2020).
- U.S. Food and Drug Administration Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. FDA. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 10 September 2020).
- Michaels, S.; Wang, M.Z. The Revised Human Liver Cytochrome P450 “Pie”: Absolute Protein Quantification of CYP4F and CYP3A Enzymes Using Targeted Quantitative Proteomics. Drug Metab. Dispos. 2014, 42, 1241–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Dinh, J.C.; Jeong, H.; Prasad, B.; Leeder, J.S. Ten years’ experience with the CYP2D6 activity score: A perspective on future investigations to improve clinical predictions for precision therapeutics. J. Pers. Med. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaedigk, A.; Simon, S.D.; Pearce, R.E.; Bradford, L.D.; Kennedy, M.J.; Leeder, J.S. The CYP2D6 activity score: Translating genotype information into a qualitative measure of phenotype. Clin. Pharmacol. Ther. 2008, 83, 234–242. [Google Scholar] [CrossRef]
- Bapiro, T.E.; Hasler, J.A.; Ridderström, M.; Masimirembwa, C.M. The molecular and enzyme kinetic basis for the diminished activity of the cytochrome P450 2D6.17 (CYP2D6.17) variant: Potential implications for CYP2D6 phenotyping studies and the clinical use of CYP2D6 substrate drugs in some African populations. Biochem. Pharmacol. 2002, 64, 1387–1398. [Google Scholar] [CrossRef]
- Thomas, C.D.; Mosley, S.A.; Kim, S.; Lingineni, K.; El Rouby, N.; Langaee, T.Y.; Gong, Y.; Wang, D.; Schmidt, S.O.; Binkley, P.F.; et al. Examination of Metoprolol Pharmacokinetics and Pharmacodynamics Across CYP2D6 Genotype-Derived Activity Scores. CPT Pharmacometrics Syst. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Matthaei, J.; Brockmöller, J.; Tzvetkov, M.; Sehrt, D.; Sachse-Seeboth, C.; Hjelmborg, J.; Möller, S.; Halekoh, U.; Hofmann, U.; Schwab, M.; et al. Heritability of metoprolol and torsemide pharmacokinetics. Clin. Pharmacol. Ther. 2015, 98, 611–621. [Google Scholar] [CrossRef]
- Kirchheiner, J.; Heesch, C.; Bauer, S.; Meisel, C.; Seringer, A.; Goldammer, M.; Tzvetkov, M.; Meineke, I.; Roots, I.; Brockmöller, J. Impact of the ultrarapid metabolizer genotype of cytochrome P450 2D6 on metoprolol pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2004, 76, 302–312. [Google Scholar] [CrossRef]
- Plosker, G.L.; Clissold, S.P. Controlled Release Metoprolol Formulations. Drugs 1992, 43, 382–414. [Google Scholar] [CrossRef]
- Regårdh, C.G.; Johnsson, G. Clinical Pharmacokinetics of Metoprolol. Clin. Pharmacokinet. 1980, 5, 557–569. [Google Scholar] [CrossRef]
- Berger, B.; Bachmann, F.; Duthaler, U.; Krähenbühl, S.; Haschke, M. Cytochrome P450 enzymes involved in metoprolol metabolism and use of metoprolol as a CYP2D6 phenotyping probe drug. Front. Pharmacol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, P.M.; Cesarino, E.J.; Mateus, F.H.; Mere, Y.; Santos, S.R.; Lanchote, V.L. Enantioselectivity in the steady-state pharmacokinetics of metoprolol in hypertensive patients. Chirality 1999, 11, 591–597. [Google Scholar] [CrossRef]
- Lennard, M.S.; Tucker, G.T.; Silas, J.H.; Woods, H.F. Debrisoquine polymorphism and the metabolism and action of metoprolol, timolol, propranolol and atenolol. Xenobiotica 1986, 16, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, A.; Başçi, N.B.; Işimer, A.; Sayal, A.; Kayaalp, S.O. Metabolic ratios of four probes of CYP2D6 in Turkish subjects: A cross-over study. Eur. J. Drug Metab. Pharmacokinet. 1996, 21, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Seeringer, A.; Brockmöller, J.; Bauer, S.; Kirchheiner, J. Enantiospecific pharmacokinetics of metoprolol in CYP2D6 ultra-rapid metabolizers and correlation with exercise-induced heart rate. Eur. J. Clin. Pharmacol. 2008, 64, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, J.A. Stereospecificity of beta adrenergic antagonists: R-enantiomers show increased selectivity for beta-2 receptors in ciliary process. J. Pharmacol. Exp. Ther. 1988, 245, 94–101. [Google Scholar]
- Blake, C.M.; Kharasch, E.D.; Schwab, M.; Nagele, P. A Meta-Analysis of CYP2D6 Metabolizer Phenotype and Metoprolol Pharmacokinetics. Clin. Pharmacol. Ther. 2013, 94, 394–399. [Google Scholar] [CrossRef] [Green Version]
- Gaedigk, A.; Sangkuhl, K.; Whirl-Carrillo, M.; Klein, T.; Leeder, J.S. Prediction of CYP2D6 phenotype from genotype across world populations. Genet. Med. 2017, 19, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Del Tredici, A.L.; Malhotra, A.; Dedek, M.; Espin, F.; Roach, D.; Zhu, G.-d.; Voland, J.; Moreno, T.A. Frequency of CYP2D6 alleles including structural variants in the United States. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Gaedigk, A. Complexities of CYP2D6 gene analysis and interpretation. Int. Rev. Psychiatry 2013, 25, 534–553. [Google Scholar] [CrossRef]
- Ke, A.B.; Nallani, S.C.; Zhao, P.; Rostami-Hodjegan, A.; Isoherranen, N.; Unadkat, J.D. A Physiologically Based Pharmacokinetic Model to Predict Disposition of CYP2D6 and CYP1A2 Metabolized Drugs in Pregnant Women. Drug Metab. Dispos. 2013, 41, 801–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chetty, M.; Rose, R.H.; Abduljalil, K.; Patel, N.; Lu, G.; Cain, T.; Jamei, M.; Rostami-Hodjegan, A. Applications of linking PBPK and PD models to predict the impact of genotypic variability, formulation differences, differences in target binding capacity and target site drug concentrations on drug responses and variability. Front. Pharmacol. 2014, 5, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Sharma, V.D.; Lingineni, K.; Farhan, N.; Fang, L.; Zhao, L.; Brown, J.D.; Cristofoletti, R.; Vozmediano, V.; Ait-Oudhia, S.; et al. Evaluating the Clinical Impact of Formulation Variability: A Metoprolol Extended-Release Case Study. J. Clin. Pharmacol. 2019, 59, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Dallmann, A.; Ince, I.; Coboeken, K.; Eissing, T.; Hempel, G. A Physiologically Based Pharmacokinetic Model for Pregnant Women to Predict the Pharmacokinetics of Drugs Metabolized Via Several Enzymatic Pathways. Clin. Pharmacokinet. 2018, 57, 749–768. [Google Scholar] [CrossRef] [PubMed]
- The Royal Dutch Pharmacists Association—Pharmacogenetics Working Group (DPWG) Annotation of DPWG Guideline for Metoprolol and CYP2D6. Available online: https://www.pharmgkb.org/guidelineAnnotation/PA166104995 (accessed on 22 September 2020).
- Lippert, J.; Burghaus, R.; Edginton, A.; Frechen, S.; Karlsson, M.; Kovar, A.; Lehr, T.; Milligan, P.; Nock, V.; Ramusovic, S.; et al. Open Systems Pharmacology Community—An Open Access, Open Source, Open Science Approach to Modeling and Simulation in Pharmaceutical Sciences. CPT Pharmacometrics Syst. Pharmacol. 2019, 8, 878–882. [Google Scholar] [CrossRef] [Green Version]
- Wojtyniak, J.G.; Britz, H.; Selzer, D.; Schwab, M.; Lehr, T. Data Digitizing: Accurate and Precise Data Extraction for Quantitative Systems Pharmacology and Physiologically-Based Pharmacokinetic Modeling. CPT Pharmacometrics Syst. Pharmacol. 2020, 9, 322–331. [Google Scholar] [CrossRef]
- Regårdh, C.G.; Borg, K.O.; Johansson, R.; Johnsson, G.; Palmer, L. Pharmacokinetic studies on the selective beta1-receptor antagonist metoprolol in man. J. Pharmacokinet. Biopharm. 1974, 2, 347–364. [Google Scholar] [CrossRef]
- Open Systems Pharmacology Suite Community Open Systems Pharmacology Suite Manual, Version 7.4. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/OpenSystemsPharmacologySuite.pdf (accessed on 12 October 2020).
- Caudle, K.E.; Sangkuhl, K.; Whirl-Carrillo, M.; Swen, J.J.; Haidar, C.E.; Klein, T.E.; Gammal, R.S.; Relling, M.V.; Scott, S.A.; Hertz, D.L.; et al. Standardizing CYP 2D6 Genotype to Phenotype Translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin. Transl. Sci. 2020, 13, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Avdeef, A.; Berger, C.M. pH-metric solubility.: 3. Dissolution titration template method for solubility determination. Eur. J. Pharm. Sci. 2001, 14, 281–291. [Google Scholar] [CrossRef]
- Boldhane, S.; Kuchekar, B. Development and optimization of metoprolol succinate gastroretentive drug delivery system. Acta Pharm. 2010, 60, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Jona, J.; Chow, D.T.; Rong, H.; Semin, D.; Xia, X.; Zanon, R.; Spancake, C.; Maliski, E. High-throughput logP measurement using parallel liquid chromatography/ultraviolet/mass spectrometry and sample-pooling. Rapid Commun. Mass Spectrom. 2002, 16, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Mateus, A.; Matsson, P.; Artursson, P. A High-Throughput Cell-Based Method to Predict the Unbound Drug Fraction in the Brain. J. Med. Chem. 2014, 57, 3005–3010. [Google Scholar] [CrossRef] [PubMed]
- Mautz, D.S.; Nelson, W.L.; Shen, D.D. Regioselective and stereoselective oxidation of metoprolol and bufuralol catalyzed by microsomes containing cDNA-expressed human P4502D6. Drug Metab. Dispos. 1995, 23, 513–517. [Google Scholar]
- Thelen, K.; Coboeken, K.; Willmann, S.; Burghaus, R.; Dressman, J.B.; Lippert, J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, Part 1: Oral solutions. J. Pharm. Sci. 2011, 100, 5324–5345. [Google Scholar] [CrossRef]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modeling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef]
- Johnson, J.A.; Burlew, B.S. Metoprolol metabolism via cytochrome P4502D6 in ethnic populations. Drug Metab. Dispos. 1996, 24, 350–355. [Google Scholar]
- Kelly, J.G.; Salem, S.A.; Kinney, C.D.; Shanks, R.G.; McDevitt, D.G. Effects of ranitidine on the disposition of metoprolol. Br. J. Clin. Pharmacol. 1985, 19, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Damy, T.; Pousset, F.; Caplain, H.; Hulot, J.-S.S.; Lechat, P. Pharmacokinetic and pharmacodynamic interactions between metoprolol and dronedarone in extensive and poor CYP2D6 metabolizers healthy subjects. Fundam. Clin. Pharmacol. 2004, 18, 113–123. [Google Scholar] [CrossRef]
- Austin, R.P.; Barton, P.; Cockroft, S.L.; Wenlock, M.C.; Riley, R.J. The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab. Dispos. 2002, 30, 1497–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnsson, G.; Regärdh, C.-G.; Sölvell, L. Combined pharmacokinetic and pharmacodynamic studies in man of the adrenergic β1-receptor antagonist metoprolol. Acta Pharmacol. Toxicol. (Copenh). 1975, 36, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Krösser, S.; Neugebauer, R.; Dolgos, H.; Fluck, M.; Rost, K.-L.; Kovar, A. Investigation of sarizotan’s impact on the pharmacokinetics of probe drugs for major cytochrome P450 isoenzymes: A combined cocktail trial. Eur. J. Clin. Pharmacol. 2006, 62, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.B.; Soberman, J.E. Effects of paroxetine on the pharmacokinetics and pharmacodynamics of immediate-release and extended-release metoprolol. Pharmacotherapy 2011, 31, 630–641. [Google Scholar] [CrossRef]
- Luzier, A.B.; Killian, A.; Wilton, J.H.; Wilson, M.F.; Forrest, A.; Kazierad, D.J. Gender-related effects on metoprolol pharmacokinetics and pharmacodynamics in healthy volunteers. Clin. Pharmacol. Ther. 1999, 66, 594–601. [Google Scholar] [CrossRef]
- Jin, S.K.; Chung, H.J.; Chung, M.W.; Kim, J.-I.; Kang, J.-H.; Woo, S.W.; Bang, S.; Lee, S.H.; Lee, H.J.; Roh, J. Influence of CYP2D6*10 on the pharmacokinetics of metoprolol in healthy Korean volunteers. J. Clin. Pharm. Ther. 2008, 33, 567–573. [Google Scholar] [CrossRef]
- Guest, E.J.; Aarons, L.; Houston, J.B.; Rostami-Hodjegan, A.; Galetin, A. Critique of the Two-Fold Measure of Prediction Success for Ratios: Application for the Assessment of Drug-Drug Interactions. Drug Metab. Dispos. 2011, 39, 170–173. [Google Scholar] [CrossRef] [Green Version]
- Pharmacogene Variation Consortium (PharmVar) CYP2D6 gene. Available online: https://www.pharmvar.org/gene/CYP2D6 (accessed on 7 October 2020). (Gaedigk et al. 2018, CPT 103:399; Gaedigk et al. 2019, CPT 105:29).
- Gonzalez, D.; Rao, G.G.; Bailey, S.C.; Brouwer, K.L.R.; Cao, Y.; Crona, D.J.; Kashuba, A.D.M.; Lee, C.R.; Morbitzer, K.; Patterson, J.H.; et al. Precision Dosing: Public Health Need, Proposed Framework, and Anticipated Impact. Clin. Transl. Sci. 2017, 10, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; He, M.M.; Liu, H.; Wrighton, S.A.; Wang, L.; Guo, B.; Li, C. Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab. Dispos. 2007, 35, 1292–1300. [Google Scholar] [CrossRef]
- Goryachkina, K.; Burbello, A.; Boldueva, S.; Babak, S.; Bergman, U.; Bertilsson, L. CYP2D6 is a major determinant of metoprolol disposition and effects in hospitalized Russian patients treated for acute myocardial infarction. Eur. J. Clin. Pharmacol. 2008, 64, 1163–1173. [Google Scholar] [CrossRef]
- Rau, T.; Heide, R.; Bergmann, K.; Wuttke, H.; Werner, U.; Feifel, N.; Eschenhagen, T. Effect of the CYP2D6 genotype on metoprolol metabolism persists during long-term treatment. Pharmacogenetics 2002, 12, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Hamadeh, I.S.; Langaee, T.Y.; Dwivedi, R.; Garcia, S.; Burkley, B.M.; Skaar, T.C.; Chapman, A.B.; Gums, J.G.; Turner, S.T.; Gong, Y.; et al. Impact of CYP2D6 polymorphisms on clinical efficacy and tolerability of metoprolol tartrate. Clin. Pharmacol. Ther. 2014, 96, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijl, M.J.; Visser, L.E.; Van Schaik, R.H.N.; Kors, J.A.; Witteman, J.C.M.; Hofman, A.; Vulto, A.G.; Van Gelder, T.; Stricker, B.H.C. Genetic variation in the CYP2D6 gene is associated with a lower heart rate and blood pressure in β-blocker users. Clin. Pharmacol. Ther. 2009, 85, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Huang, Z.; Yang, G.; Lv, H.; Sang, H.; Yao, Y. Effects of Polymorphism of the β 1 Adrenoreceptor and CYP2D6 on the Therapeutic Effects of Metoprolol. J. Int. Med. Res. 2008, 36, 1354–1362. [Google Scholar] [CrossRef]
- Fux, R.; Mörike, K.; Pröhmer, A.M.T.T.; Delabar, U.; Schwab, M.; Schaeffeler, E.; Lorenz, G.; Gleiter, C.H.; Eichelbaum, M.; Kivistö, K.T. Impact of CYP2D6 genotype on adverse effects during treatment with metoprolol: A prospective clinical study. Clin. Pharmacol. Ther. 2005, 78, 378–387. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activity Score | Projected Phenotype | Examples of Relevant CYP2D6 Genotypes |
|---|---|---|
| 0 | PM | *3/*3, *3/*4, *4/*4, *5/*6 |
| 0.25 | IM | *4/*10, *5/*10 |
| 0.5 | *4/*41, *5/*17, *10/*10 | |
| 0.75 | *17/*10, *41/*10 | |
| 1 | *1/*4, *2/*5, *17/*17, *17/*41 | |
| 1.25 | NM | *1/*10, *2/*10, *35/*10 |
| 1.5 | *1/*41, *2/*17, *35/*41 | |
| 2 | *1/*1, *1/*2, *2/*35 | |
| 2.25 | *1x2/*17, *35x2/*41 | |
| >2.25 | UM | *1/*1x3, *1/*35x2, *2x2/*9 |
| Parameter | Unit | (R)-Metoprolol | (S)-Metoprolol | Description | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Value | Source | Literature | Reference | Value | Source | Literature | Reference | |||
| MW | g/mol | 267.36 | Lit. | 267.36 | [34] | 267.36 | Lit. | 267.36 | [34] | Molecular weight |
| pKa (base) | - | 9.7 | Lit. | 9.70 | [34] | 9.7 | Lit. | 9.70 | [34] | Acid dissociation constant |
| Solubility tart. (pH 7.4) | g/mL | 1.00 | Lit. | 1.00 | [35] | 1.00 | Lit. | 1.00 | [35] | Solubility |
| Solubility succ. (pH 5.5) | g/mL | 0.16 | Lit. | 0.16 | [36] | 0.16 | Lit. | 0.16 | [36] | Solubility |
| logP | - | 1.77 | Lit. | 1.77 | [37] | 1.77 | Lit. | 1.77 | [37] | Lipophilicity |
| fu | % | 88 | Lit. | 88 | [38] | 88 | Lit. | 88 | [38] | Fraction unbound |
| CYP2D6 Km ⭢ αHM | µmol/L | 10.08 | Lit. | 10.08 ‡ | [39] | 10.75 | Lit. | 10.75 ‡ | [39] | Michaelis-Menten constant |
| CYP2D6 kcat ⭢ αHM | 1/min | 6.02 | Optim. † | 7.50 | [39] | 6.55 | Optim. † | 8.27 | [39] | Catalytic rate constant |
| CYP2D6 Km ⭢ ODM | µmol/L | 8.82 | Lit. | 8.82 ‡ | [39] | 12.43 | Lit. | 12.43 ‡ | [39] | Michaelis-Menten constant |
| CYP2D6 kcat ⭢ ODM | 1/min | 9.87 | Optim. † | 12.30 | [39] | 8.21 | Optim. † | 10.37 | [39] | Catalytic rate constant |
| CLhep., unsp. | 1/min | 0.08 | Optim. | - | - | 0.09 | Optim. | - | - | Unspecific hepatic clearance |
| GFR fraction | - | 1.00 | Asm. | - | - | 1.00 | Asm. | - | - | Filtered drug in the urine |
| EHC continuous fraction | - | 1.00 | Asm. | - | - | 1.00 | Asm. | - | - | Bile fraction cont. released |
| Intestinal permeability | cm/min | 4.14 × 10−5 | Optim. | 1.12 × 10−5 | Calc. [40] | 4.14 × 10−5 | Optim. | 1.12 × 10−5 | Calc. [40] | Transcellular intestinal perm. |
| Cellular permeability | cm/min | 4.64 × 10−3 | Calc. | PK-Sim | [32] | 4.64 × 10−3 | Calc. | PK-Sim | [32] | Perm. into the cellular space |
| Partition coefficients | - | Diverse | Calc. | R&R | [41,42] | Diverse | Calc. | R&R | [41,42] | Cell to plasma partitioning |
| NR Weibull time parameter | min | 12.31 | Optim. | - | [43,44] | 12.31 | Optim. | - | [43,44] | Dissolution time (50%) |
| NR Weibull shape parameter | - | 0.72 | Optim. | - | [43,44] | 0.72 | Optim. | - | [43,44] | Dissolution profile shape |
| CR Weibull time parameter | min | 331.92 | Optim. | - | [45] | 331.92 | Optim. | - | [45] | Dissolution time (50%) |
| CR Weibull shape parameter | - | 1.53 | Optim. | - | [45] | 1.53 | Optim. | - | [45] | Dissolution profile shape |
| Activity Score | (R)-Metoprolol | (S)-Metoprolol | kcat, rel | ||
|---|---|---|---|---|---|
| kcat ⭢ αHM | kcat ⭢ ODM | kcat ⭢ αHM | kcat ⭢ ODM | ||
| 0 | 0.00 1/min | 0.00 1/min | 0.00 1/min | 0.00 1/min | 0% |
| 0.5 | 1.65 1/min | 2.70 1/min | 1.82 1/min | 2.27 1/min | 19% |
| 1.25 | 5.73 1/min | 9.40 1/min | 6.30 1/min | 7.89 1/min | 64% |
| 1.5 | 6.38 1/min | 10.48 1/min | 7.03 1/min | 8.81 1/min | 72% |
| 2 | 10.17 1/min | 16.69 1/min | 11.19 1/min | 14.02 1/min | 100% |
| 3 | 19.03 1/min | 31.22 1/min | 20.93 1/min | 26.23 1/min | 213% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rüdesheim, S.; Wojtyniak, J.-G.; Selzer, D.; Hanke, N.; Mahfoud, F.; Schwab, M.; Lehr, T. Physiologically Based Pharmacokinetic Modeling of Metoprolol Enantiomers and α-Hydroxymetoprolol to Describe CYP2D6 Drug-Gene Interactions. Pharmaceutics 2020, 12, 1200. https://doi.org/10.3390/pharmaceutics12121200
Rüdesheim S, Wojtyniak J-G, Selzer D, Hanke N, Mahfoud F, Schwab M, Lehr T. Physiologically Based Pharmacokinetic Modeling of Metoprolol Enantiomers and α-Hydroxymetoprolol to Describe CYP2D6 Drug-Gene Interactions. Pharmaceutics. 2020; 12(12):1200. https://doi.org/10.3390/pharmaceutics12121200
Chicago/Turabian StyleRüdesheim, Simeon, Jan-Georg Wojtyniak, Dominik Selzer, Nina Hanke, Felix Mahfoud, Matthias Schwab, and Thorsten Lehr. 2020. "Physiologically Based Pharmacokinetic Modeling of Metoprolol Enantiomers and α-Hydroxymetoprolol to Describe CYP2D6 Drug-Gene Interactions" Pharmaceutics 12, no. 12: 1200. https://doi.org/10.3390/pharmaceutics12121200