
Fundamental Aspects of Lipid-Based Excipients in Lipid-Based Product Development

 ,
,  , and
, and
Abstract
:1. Introduction
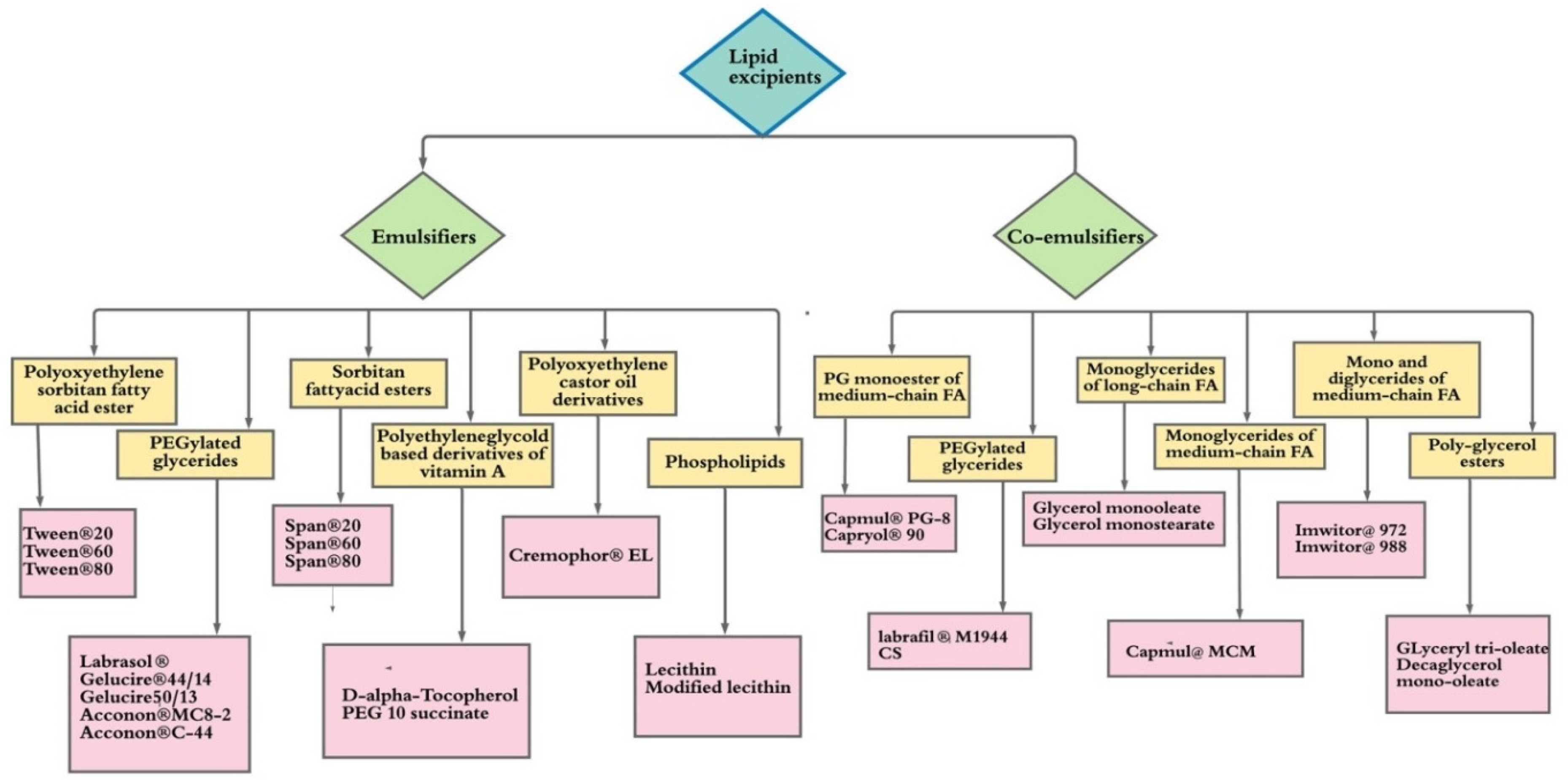
2. Lipid Excipients Used in the Pharmaceutical Product Development
2.1. Glyceryl Dibehenate
2.2. Dynasan
2.3. Glyceryl Distearate
2.4. Glyceryl Caprylate
2.5. Non-Ionic Surfactant (Lauroyl Polyoxyl-32 Glycerides)
2.6. Caprylocaproyl Macrogol-8 Glyceride
2.7. Medium Chain Triglycerides
2.8. Glyceryl Stearate
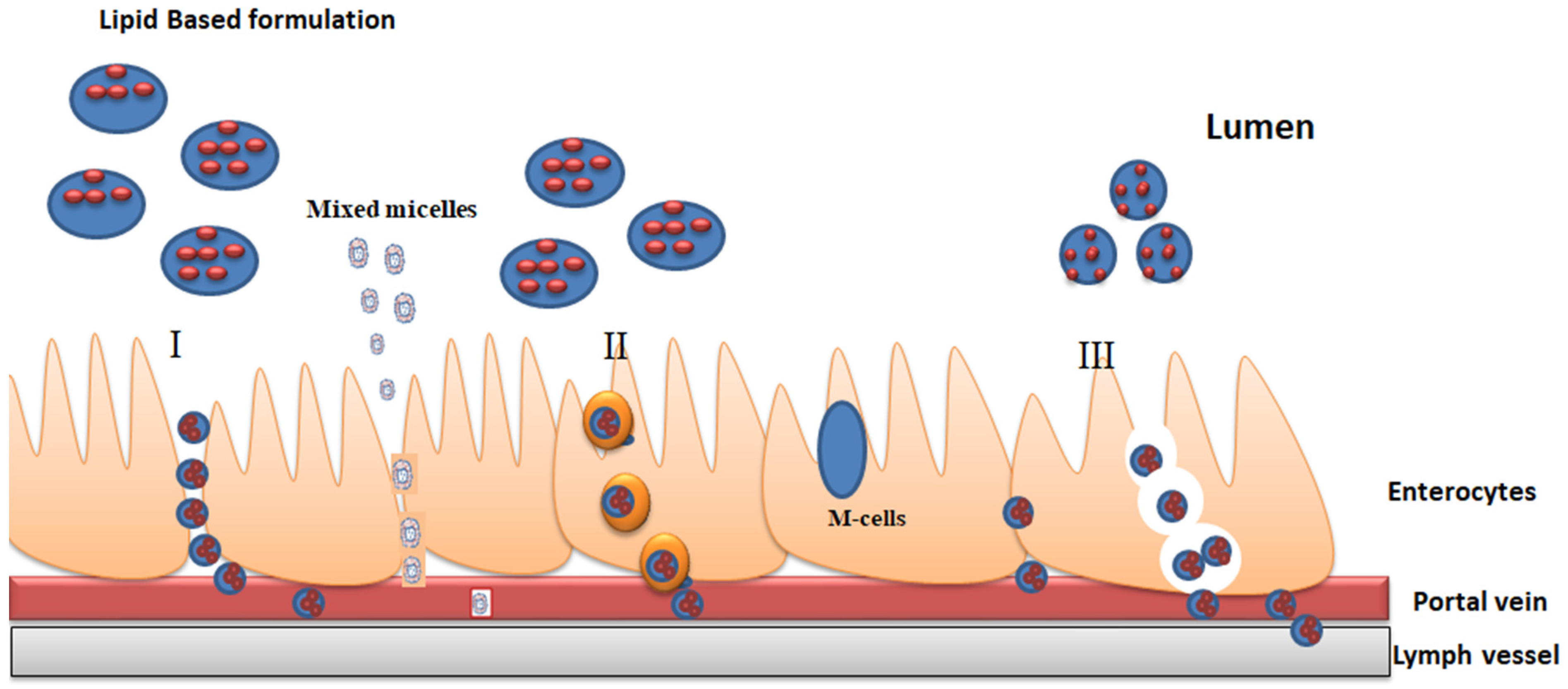
3. Absorption of Lipid Formulations
3.1. Lymphatic Transport Mechanism
3.2. Enterocyte-Based Transport Mechanisms

3.3. Reduced Pre-Systemic Drug Metabolism Mechanism
4. Methodologies for the Formulation of Solid and Semi-Solid Lipid-Based Formulations
4.1. Filling into Capsules (Encapsulation)
4.2. Conversion of Liquid Lipid to Solids
4.3. Carriers Used for the Solidification of Lipid Formulations
4.4. Advanced Formulations Based on Lipids
4.4.1. Self-Emulsifying Drug Delivery Systems
4.4.2. Vesicular Drug Delivery Systems
4.4.3. Solid Lipid Nanoparticles (SLNs)
4.4.4. Nanostructured Lipid Nanocarriers (NLCs)
4.4.5. Mixed Micelles
4.4.6. Lipid Drug Conjugates (LDC)
5. Gaps and Limitations in Development of Lipid-Based Products
6. Characterization of Lipid-Based Product Development
7. Application of Lipid Excipients in Lipid-Based Drug Delivery Systems
7.1. Enhancement of Oral Bioavailability
7.2. p-gp Efflux Inhibition
7.3. Enhanced Permeation
7.4. Photostability
7.5. DNA Delivery
8. Administration Routes of Lipid-Based Formulation
8.1. Oral Delivery
8.2. Topical Delivery
8.3. Pulmonary Delivery
8.4. Parenteral Delivery
8.5. Implantable Drug Delivery System
9. Regulatory Perspectives
10. Computational Prediction
10.1. Formulation Design and Development
10.2. Pharmacokinetics Prediction
10.3. Phase Behaviour during Dispersion
- (i)
- A phase like single reverse micelle comprising included water molecules on the lower concentration of water,
- (ii)
- Single reverse phase of micelle at intermediate and high-water levels and,
- (iii)
- The lamellar glycerides a two-phase system and lose water groups.
11. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, R. Lipid-Based Nanoparticles for Drug-Delivery Systems. Nanocarr. Drug Deliv. 2019, 249–284. [Google Scholar] [CrossRef]
- Mu, H.; Holm, R.; Müllertz, A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int. J. Pharm. 2013, 453, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, J.; Zhao, M.; Xu, H.; Li, L.S.; Wang, S. Loading of tacrolimus containing lipid based drug delivery systems into mesoporous silica for extended release. Asian J. Pharm. Sci. 2016, 11, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Zhang, H.; Wang, X.; He, S.; Zhai, G. Development and evaluation of ibuprofen loaded mixed micelles preparations for topical delivery. J. Drug Deliv. Sci. Technol. 2018, 48, 363–371. [Google Scholar] [CrossRef]
- Small, D. A Classification of Biologic Lipids Based upon Their Interaction in Aqueous Systems. J. Am. Oil Chem. Soc. 1968, 45, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Ohashi, K. Interfacial and emulsifying properties of diacylglycerol. Food Sci. Technol. Res. 2003, 9, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Nanjwade, B.K.; Patel, D.J.; Udhani, R.A.; Manvi, F.V. Functions of Lipids for Enhancement of Oral Bioavailability of Poorly Water-Soluble Drugs. Sci. Pharm. 2011, 79, 705–727. [Google Scholar] [CrossRef] [Green Version]
- Sangsen, Y.; Wiwattanawongsa, K.; Likhitwitayawuid, K.; Sritularak, B.; Graidist, P.; Wiwattanapatapee, R. Influence of surfactants in self-microemulsifying formulations on enhancing oral bioavailability of oxyresveratrol: Studies in Caco-2 cells and in vivo. Int. J. Pharm. 2016, 498, 294–303. [Google Scholar] [CrossRef]
- Ferreira, M.; Chaves, L.L.; Costa, A.; Reis, S. Optimization of nanostructured lipid carriers loaded with methotrexate: A tool for inflammatory and cancer therapy. Int. J. Pharm. 2015, 492, 65–72. [Google Scholar] [CrossRef]
- Espinosa-olivares, M.A.; Delgado-buenrostro, N.L.; Chirino, Y.I.; Trejo-márquez, M.A.; De Estudios, D.; Tecnología, D.P.; De Estudios, F.; Cuautitlán, S.; Nacional, U.; De México, A.; et al. Nanostructured lipid carriers loaded with curcuminoids: Physicochemical characterization, in vitro release, ex vivo skin penetration, stability and antioxidant activity. Eur. J. Pharm. Sci. 2020, 155, 105533. [Google Scholar] [CrossRef]
- Abbas, H.; El-Deeb, N.M.; Zewail, M. PLA-coated Imwitor® 900 K-based herbal colloidal carriers as novel candidates for the intra-articular treatment of arthritis. Pharm. Dev. Technol. 2021, 26, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, A.; Aleksic, I.; Ibric, S.; Parojčić, J.; Cvijic, S. Hot-melt coating with Precirol ATO 5 in a fluidized-bed apparatus: Application of experimental design in the optimization of process parameters. J. Drug Deliv. Sci. Technol. 2018, 46, 274–284. [Google Scholar] [CrossRef]
- Lam, H.T.; Le, N.M.N.; Phan, T.N.Q.; Bernkop-Schnürch, A. Mucolytic self-emulsifying drug delivery systems (SEDDS) containing a hydrophobic ion-pair of proteinase. Eur. J. Pharm. Sci. 2020, 162, 105658. [Google Scholar] [CrossRef] [PubMed]
- Weerapol, Y.; Limmatvapirat, S.; Nunthanid, J.; Sriamornsak, P. Self-Nanoemulsifying Drug Delivery System of Nifedipine: Impact of Hydrophilic—Lipophilic Balance and Molecular Structure of Mixed Surfactants. AAPS PharmSciTech 2014, 15, 456–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, A.; Afouna, M. Enhancement of the in-vitro dissolution and in-vivo oral bioavailability of silymarin from liquid-filled hard gelatin capsules of semisolid dispersion using Gelucire 44/14 as a carrier. Pharmazie 2012, 67, 209–214. [Google Scholar] [CrossRef]
- Kushwaha, J.P.; Baidya, D.; Patil, S. Harmine-loaded galactosylated pluronic F68-gelucire 44/14 mixed micelles for liver targeting. Drug Dev. Ind. Pharm. 2019, 45, 1361–1368. [Google Scholar] [CrossRef]
- Cirri, M.; Mura, P.; Mora, P.C. Liquid spray formulations of xibornol by using self-microemulsifying drug delivery systems. Int. J. Pharm. 2007, 340, 84–91. [Google Scholar] [CrossRef]
- Pando, D.; Caddeo, C.; Manconi, M.; Fadda, A.M.; Pazos, C. Nanodesign of olein vesicles for the topical delivery of the antioxidant resveratrol. J. Pharm. Pharmacol. 2013, 65, 1158–1167. [Google Scholar] [CrossRef]
- Mora-Huertas, C.E.; Garrigues, O.; Fessi, H.; Elaissari, A. Nanocapsules prepared via nanoprecipitation and emulsification—Diffusion methods: Comparative study. Eur. J. Pharm. Biopharm. 2012, 80, 235–239. [Google Scholar] [CrossRef]
- Bertoni, S.; Albertini, B.; Facchini, C.; Prata, C.; Passerini, N. Glutathione-Loaded Solid Lipid Microparticles as Innovative Delivery System for Oral Antioxidant Therapy. Pharmaceutics 2019, 11, 364. [Google Scholar] [CrossRef] [Green Version]
- Abdelbary, G.; Fahmy, R.H. Diazepam-Loaded Solid Lipid Nanoparticles: Design and Characterization. AAPS PharmSciTech 2009, 10, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, G.; Gonçalves, L.; Marto, J.; Carvalho, F.; Sim, S.; Ribeiro, H.; Almeida, A. Increased Therapeutic Efficacy of SLN Containing Etofenamate and Ibuprofen in Topical Treatment of Inflammation. Pharmaceutics 2021, 13, 328. [Google Scholar] [CrossRef] [PubMed]
- Dudhipala, N.; Adel, A. Amelioration of ketoconazole in lipid nanoparticles for enhanced antifungal activity and bioavailability through oral administration for management of fungal infections. Chem. Phys. Lipids 2020, 232, 104953. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, G.; Narender, D.; Kishan, V. Preparation, Characterization and in vivo Evaluation of Rosuvastatin Calcium Loaded Solid Lipid Nanoparticles. Int. J. Pharm. Sci. Nanotechnol. 2015, 8, 2779–2785. [Google Scholar] [CrossRef]
- Shazly, G.A.; AlShehri, S.; Ibrahim, M.A.; Tawfeek, H.M.; Razik, J.A.; Hassan, Y.A.; Shakeel, F. Development of Domperidone Solid Lipid Nanoparticles: In Vitro and In Vivo Characterization. AAPS PharmSciTech 2018, 19, 1712–1719. [Google Scholar] [CrossRef]
- Becker, K.; Salar-Behzadi, S.; Zimmer, A. Solvent-Free Melting Techniques for the Preparation of Lipid-Based Solid Oral Formulations. Pharm. Res. 2015, 32, 1519–1545. [Google Scholar] [CrossRef] [Green Version]
- Forster, S.; Lebo, D. Continuous Melt Granulation for Taste-Masking of Ibuprofen. Pharmaceutics 2021, 13, 863. [Google Scholar] [CrossRef]
- Kim, K.T.; Lee, J.; Park, J.; Cho, H.; Yoon, I.; Kim, D. Capmul MCM/Solutol HS15-Based Microemulsion for Enhanced Oral Bioavailability of Rebamipide. J. Nanosci. Nanotechnol. 2017, 17, 2340–2344. [Google Scholar] [CrossRef]
- Bandivadeka, M.M.; Pancholi, S.S.; Kaul-Ghanekar, R. Self-microemulsifying smaller molecular volume oil (Capmul MCM) using non-ionic surfactants: A delivery system for poorly water-soluble drug. Drug Dev. Ind. Pharm. 2012, 38, 883–892. [Google Scholar] [CrossRef]
- Panigrahi, K.C.; Patra, C.N.; Jena, G.K.; Ghose, D.; Jena, J.; Panda, S.K.; Sahu, M. Gelucire: A versatile polymer for modified release drug delivery system. Future J. Pharm. Sci. 2018, 4, 102–108. [Google Scholar] [CrossRef]
- Mura, P.; Maestrelli, F.; D’Ambrosio, M.; Luceri, C. Evaluation and Comparison of Solid Lipid Nanoparticles (SLNs) and Nanostructured Lipid Carriers (NLCs) as Vectors to Develop Hydrochlorothiazide Effective and Safe Pediatric Oral Liquid Formulations. Pharmaceutics 2021, 13, 437. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, M.; Yang, F.; Liu, C.; Pan, R.; Chang, Q.; Liu, X.; Liao, Y. Involvement of the Inhibition of Intestinal Glucuronidation in Enhancing the Oral Bioavailability of Resveratrol by Labrasol Containing Nanoemulsions. Mol. Pharm. 2015, 12, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Zupancic, O.; Leonaviciute, G.; Lam, H.T.; Partenhauser, A.; Podričnik, S.; Bernkop-schnürch, A. Development and in vitro evaluation of an oral SEDDS for desmopressin. Drug Deliv. 2016, 23, 2074–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulter, I.S.; Griffin, B.T.; Keohane, K. Enhanced colonic delivery of ciclosporin A self-emulsifying drug delivery system encapsulated in coated minispheres. Drug Dev. Ind. Pharm. 2015, 42, 245–253. [Google Scholar] [CrossRef]
- Avasatthi, V.; Pawar, H.; Dora, C.P.; Bansod, P.; Gill, M.S.; Suresh, S. A novel nanogel formulation of methotrexate for topical treatment of psoriasis: Optimization, in vitro and in vivo evaluation. Pharm. Dev. Technol. 2015, 21, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Prombutara, P.; Kulwatthanasal, Y.; Supaka, N.; Sramala, I. Production of nisin-loaded solid lipid nanoparticles for sustained antimicrobial activity. Food Control 2012, 24, 184–190. [Google Scholar] [CrossRef]
- Wang, T.; Luo, Y. Biological fate of ingested lipid-based nanoparticles: Current understanding and future directions. Nanoscale 2019, 11, 11048–11063. [Google Scholar] [CrossRef]
- O’Driscoll, C.M. Lipid-based formulations for intestinal lymphatic delivery. Eur. J. Pharm. Sci. 2002, 15, 405–415. [Google Scholar] [CrossRef]
- Trevaskis, N.L.; Charman, W.N.; Porter, C.J.H. Lipid-based delivery systems and intestinal lymphatic drug transport: A mechanistic update. Adv. Drug Deliv. Rev. 2008, 60, 702–716. [Google Scholar] [CrossRef]
- Chakraborty, S.; Shukla, D.; Mishra, B.; Singh, S. Lipid—An emerging platform for oral delivery of drugs with poor bioavailability. Eur. J. Pharm. Biopharm. 2009, 73, 1–15. [Google Scholar] [CrossRef]
- van Aken, G.A. Relating Food Emulsion Structure and Composition to the Way It Is Processed in the Gastrointestinal Tract and Physiological Responses: What Are the Opportunities? Food Biophys. 2010, 5, 258–283. [Google Scholar] [CrossRef]
- Hauss, D.J.; Fogal, S.E.; Ficorilli, J.V.; Price, C.A.; Roy, T.; Jayaraj, A.A.; Keirns, J.J. Lipid-Based Delivery Systems for Improving the Bioavailability and Lymphatic Transport of a Poorly Water-Soluble LTB4 Inhibitor. J. Pharm. Sci. 1998, 87, 164–169. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J. Edible lipid nanoparticles: Digestion, absorption, and potential toxicity. Prog. Lipid Res. 2013, 52, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Beloqui, A.; Solinís, M.; Gascón, A.R.; del Pozo-Rodríguez, A.; des Rieux, A.; Préat, V. Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J. Control. Release 2013, 166, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinides, P.P.; Wasan, K.M. Lipid Formulation Strategies for Enhancing Intestinal Transport and Absorption of P-Glycoprotein (P-gp) Substrate Drugs: In vitro/In vivo Case Studies. J. Pharm. Sci. 2007, 96, 235–248. [Google Scholar] [CrossRef]
- Singh, A.; Neupane, Y.R.; Mangla, B.; Kohli, K. Nanostructured Lipid Carriers for Oral Bioavailability Enhancement of Exemestane: Formulation Design, In Vitro, Ex Vivo, and In Vivo Studies. J. Pharm. Sci. 2019, 108, 3382–3395. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, F.; Jiang, S.; Ma, L.; Li, Y. Nano-based Drug Delivery System Enhances the Oral Absorption of Lipophilic Drugs with Extensive Presystemic Metabolism. Curr. Drug Metab. 2012, 13, 1110–1118. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, Y.; Zhong, Q.; Li, X.; Gao, P.; Feng, C.; Chu, Q.; Chen, Y.; Liu, D. In vitro evaluation of the inhibitory potential of pharmaceutical excipients on human carboxylesterase 1A and 2. PLoS ONE 2014, 9, e93819. [Google Scholar] [CrossRef] [Green Version]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Psimadas, D.; Georgoulias, P.; Valotassiou, V.; Loudos, G. Molecular Nanomedicine towards Cancer: 111In-Labeled Nanoparticles. J. Pharm. Sci. 2012, 101, 2271–2280. [Google Scholar] [CrossRef]
- Nazzal, S.; Wang, Y. Characterization of soft gelatin capsules by thermal analysis. Int. J. Pharm. 2001, 230, 35–45. [Google Scholar] [CrossRef]
- Cao, N.; Yang, X.; Fu, Y. Effects of various plasticizers on mechanical and water vapor barrier properties of gelatin films. Food Hydrocoll. 2009, 23, 729–735. [Google Scholar] [CrossRef]
- Jannin, V.; Musakhanian, J.; Marchaud, D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv. Drug Deliv. Rev. 2008, 60, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kusawake, T.; Ishida, M.; Tawa, R.; Shibata, N.; Takada, K. Oral solid gentamicin preparation using emulsifier and adsorbent. J. Control. Release 2005, 105, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W.; Porter, C.J.H. Formulation of lipid-based delivery systems for oral administration: Materials, methods and strategies. Adv. Drug Deliv. Rev. 2008, 60, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Humberstone, A.J.; Charman, W.N. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv. Drug Deliv. Rev. 1997, 25, 103–128. [Google Scholar] [CrossRef]
- Dollo, G.; Le Corre, P.; Guérin, A.; Chevanne, F.; Burgot, J.L.; Leverge, R. Spray-dried redispersible oil-in-water emulsion to improve oral bioavailability of poorly soluble drugs. Eur. J. Pharm. Sci. 2003, 19, 273–280. [Google Scholar] [CrossRef]
- Jang, D.-J.; Jeong, E.J.; Lee, H.-M.; Kim, B.-C.; Lim, S.-J.; Kim, C.-K. Improvement of bioavailability and photostability of amlodipine using redispersible dry emulsion. Eur. J. Pharm. Sci. 2006, 28, 405–411. [Google Scholar] [CrossRef]
- Barbé, C.J.; Bartlett, J.R.; Kong, L.; Finnie, K.S.; Lin, H.Q.; Larkin, M.; Calleja, S.; Bush, A.; Calleja, G. Silica particles: A novel drug-delivery system. Adv. Mater. 2004, 16, 1959–1966. [Google Scholar] [CrossRef]
- Jaganathan, H.; Godin, B. Biocompatibility assessment of Si-based nano- and micro-particles. Adv. Drug Deliv. Rev. 2012, 64, 1800–1819. [Google Scholar] [CrossRef] [Green Version]
- Hentzschel, C.M.; Sakmann, A.; Leopold, C.S. Suitability of various excipients as carrier and coating materials for liquisolid compacts. Drug Dev. Ind. Pharm. 2011, 37, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Rao, S.; Prestidge, C.A. Transforming Lipid-Based Oral Drug Delivery Systems into Solid Dosage Forms: An Overview of Solid Carriers, Physicochemical Properties, and Biopharmaceutical Performance. Pharm. Res. 2013, 30, 2993–3017. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Pedersen, G.; Kristensen, H. Preparation of redispersible dry emulsions by spray drying. Int. J. Pharm. 2001, 212, 187–194. [Google Scholar] [CrossRef]
- Hansen, T.; Holm, P.; Rohde, M.; Schultz, K. In vivo evaluation of tablets and capsules containing spray-dried o/w-emulsions for oral delivery of poorly soluble drugs. Int. J. Pharm. 2005, 293, 203–211. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.; Zheng, J.; Chen, Z.; Shi, Q.; Liu, H. Development of a solid supersaturatable self-emulsifying drug delivery system of docetaxel with improved dissolution and bioavailability. Biol. Pharm. Bull. 2011, 34, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Mazzeti, A.L.; Oliveira, L.T.; Gonçalves, K.R.; Schaun, G.C.; Mosqueira, V.C.F.; Bahia, M.T. Benznidazole Self-Emulsifying Delivery System: A Novel Alternative Dosage Form for Chagas Disease Treatment. Eur. J. Pharm. Sci. 2020, 145, 105234. [Google Scholar] [CrossRef]
- Garg, T.; Goyal, A.K. Liposomes: Targeted and Controlled Delivery System. Drug Deliv. Lett. 2014, 4, 62–71. [Google Scholar] [CrossRef]
- Nekkanti, V.; Kalepu, S. Recent Advances in Liposomal Drug Delivery: A Review. Pharm. Nanotechnol. 2015, 3, 35–55. [Google Scholar] [CrossRef]
- Joshny, J.; Hari, B.N.V.; Devi, D.R. The purpose of the present investigation is to formulate liposomes of Lornoxicam for topical SC. Eur. J. Pharm. Sci. 2018, 112, 38–51. [Google Scholar] [CrossRef]
- Chime, A.; Onyishi, I.V. Lipid-based drug delivery systems (LDDS): Recent advances and applications of lipids in drug delivery. Afr. J. Pharm. Pharmacol. 2013, 7, 3034–3059. [Google Scholar] [CrossRef] [Green Version]
- Sala, M.; Diab, R.; Elaissari, A.; Fessi, H. Lipid nanocarriers as skin drug delivery systems: Properties, mechanisms of skin interactions and medical applications. Int. J. Pharm. 2017, 535, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ainbinder, D.; Paolino, D.; Fresta, M.; Touitou, E. Drug delivery applications with ethosomes. J. Biomed. Nanotechnol. 2010, 6, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, P.; Tripathi, P.; Gupta, R.; Pandey, S. Niosomes: A review on niosomal research in the last decade. J. Drug Deliv. Sci. Technol. 2020, 56, 101581. [Google Scholar] [CrossRef]
- Mishra, V.; Bansal, K.K.; Verma, A.; Yadav, N.; Thakur, S.; Sudhakar, K.; Rosenholm, J.M. Solid lipid nanoparticles: Emerging colloidal nano drug delivery systems. Pharmaceutics 2018, 10, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doktorovova, S.; Shegokar, R.; Souto, E.B. Role of Excipients in formulation development and biocompatibility of lipid nanoparticles (SLNs/NLCs). Nanostruct. Nov. Ther. 2017, 811–843. [Google Scholar] [CrossRef]
- Dolatabadi, J.E.N.; Omidi, Y. Solid lipid-based nanocarriers as efficient targeted drug and gene delivery systems. Trends Anal. Chem. 2016, 77, 100–108. [Google Scholar] [CrossRef]
- Pin, E.; Ganem-Rondero, A. Lipid Nanocarriers as Skin Drug Delivery Systems; Elsevier Inc.: Amsterdam, The Netherlands, 2019. [Google Scholar] [CrossRef]
- Akhlaghi, S.P.; Loh, W. European Journal of Pharmaceutics and Biopharmaceutics Interactions and release of two palmitoyl peptides from phytantriol cubosomes. Eur. J. Pharm. Biopharm. 2017, 117, 60–67. [Google Scholar] [CrossRef]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems—An overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Negi, P.; Aggarwal, M.; Sharma, G.; Rathore, C.; Sharma, G.; Singh, B.; Katare, O. Niosome-based hydrogel of resveratrol for topical applications: An effective therapy for pain related disorder(s). Biomed. Pharmacother. 2017, 88, 480–487. [Google Scholar] [CrossRef]
- Mastrogiacomo, D.; Lenucci, M.S.; Bonfrate, V.; Di Carolo, M.; Piro, G.; Valli, L.; Rescio, L.; Milano, F.; Comparelli, R.; De Leo, V.; et al. Lipid/detergent mixed micelles as a tool for transferring antioxidant power from hydrophobic natural extracts into bio-deliverable liposome carriers: The case of lycopene rich oleoresins. RSC Adv. 2015, 5, 3081–3093. [Google Scholar] [CrossRef]
- Adhikari, P.; Pal, P.; Das, A.K.; Ray, S.; Bhattacharjee, A.; Mazumder, B. Nano lipid-drug conjugate: An integrated review. Int. J. Pharm. 2017, 529, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Nielsen, K.A.; Nielsen, B.P.; Bøje, N.W.; Rainer, H.; Pyo, S.M. Lipid-drug-conjugate (LDC) solid lipid nanoparticles (SLN) for the delivery of nicotine to the oral cavity—Optimization of nicotine loading efficiency. Eur. J. Pharm. Biopharm. 2018, 128, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Saxena, V.; Panicucci, R.; Joshi, Y.; Garad, S. Developability Assessment in Pharmaceutical Industry: An Integrated Group Approach for Selecting Developable Candidates. J. Pharm. Sci. 2009, 98, 1962–1979. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.S.; Vishwakarma, R.A. Impact of preformulation on drug development. Expert Opin. Drug Deliv. 2013, 10, 1239–1257. [Google Scholar] [CrossRef]
- Holm, R. Bridging the gaps between academic research and industrial product developments of lipid-based formulations. Adv. Drug Deliv. Rev. 2019, 142, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Marioli, M.; Zhang, K. Analytical characterization of liposomes and other lipid nanoparticles for drug delivery. J. Pharm. Biomed. Anal. 2021, 192, 113642. [Google Scholar] [CrossRef]
- Shete, H.; Patravale, V. Long chain lipid based tamoxifen NLC. Part I: Preformulation studies, formulation development and physicochemical characterization. Int. J. Pharm. 2013, 454, 573–583. [Google Scholar] [CrossRef]
- Caputo, F.; Arnould, A.; Bacia, M.; Ling, W.L.; Rustique, E.; Texier, I.; Mello, A.P.; Couffin, A.C. Measuring Particle Size Distribution by Asymmetric Flow Field Flow Fractionation: A Powerful Method for the Preclinical Characterization of Lipid-Based Nanoparticles. Mol. Pharm. 2019, 16, 756–767. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Michniak-Kohn, B. Preparation and characterization of lipid based nanosystems for topical delivery of quercetin. Eur. J. Pharm. Sci. 2013, 48, 442–452. [Google Scholar] [CrossRef]
- Thakkar, H.; Nangesh, J.; Parmar, M.; Patel, D. Formulation and characterization of lipid-based drug delivery system of raloxifene-microemulsion and self-microemulsifying drug delivery system. J. Pharm. Bioallied Sci. 2011, 3, 442–448. [Google Scholar] [CrossRef]
- Jawahar, N.; Hingarh, P.K.; Arun, R.; Selvaraj, J.; Anbarasan, A.; Sathianarayanan, S.; Nagaraju, G. Enhanced oral bioavailability of an antipsychotic drug through nanostructured lipid carriers. Int. J. Biol. Macromol. 2018, 110, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Zheng, B.; Liu, W.; Liu, C. Enhancing nutraceutical bioavailability using excipient emulsions: Influence of lipid droplet size on solubility and bioaccessibility of powdered curcumin. J. Funct. Foods 2015, 15, 72–83. [Google Scholar] [CrossRef]
- Cirri, A.M.; Mennini, N.; Mura, P.; Ghelardini, C.; Cesare, L. Development and in vivo evaluation of an innovative “Hydrochlorothiazide-in Cyclodextrins-in Solid Lipid Nanoparticles” formulation with sustained release and enhanced oral bioavailability for the hypertension treatment in pediatrics. Int. J. Pharm. 2017, 521, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Schultz, H.B.; Wignall, A.D.; Thomas, N.; Prestidge, C.A. Enhancement of Abiraterone Acetate Oral Bioavailability by Supersaturated-Silica Lipid. Int. J. Pharm. 2020, 582, 119264. [Google Scholar] [CrossRef] [PubMed]
- Nornoo, A.O.; Zheng, H.; Lopes, L.B.; Johnson-Restrepo, B.; Kannan, K.; Reed, R. Oral microemulsions of paclitaxel: In situ and pharmacokinetic studies. Eur. J. Pharm. Biopharm. 2009, 71, 310–317. [Google Scholar] [CrossRef]
- Alcantara, K.P.; Zulfakar, M.H.; Castillo, A.L. Development, characterization and pharmacokinetics of mupirocin-loaded nanostructured lipid carriers (NLCs) for intravascular administration. Int. J. Pharm. 2019, 571, 118705. [Google Scholar] [CrossRef]
- Simon, L.; Vincent, M.; Le Saux, S.; Lapinte, V.; Marcotte, N.; Morille, M.; Dorandeu, C.; Devoisselle, J.; Begu, S. Polyoxazolines based mixed micelles as PEG free formulations for an effective quercetin antioxidant topical delivery. Int. J. Pharm. 2019, 570, 118516. [Google Scholar] [CrossRef]
- Caldeira, S.; Ariany, R.; Vinícius, M.; Dutra, L.; Antônio, L.; Eduardo, P.; Tedesco, A.; Bentes, R.; Gontijo, M.; Cristina, M. Photodiagnosis and Photodynamic Therapy Topical photodynamic therapy with chloroaluminum phthalocyanine liposomes is as e ff ective as systemic pentavalent antimony in the treatment of experimental cutaneous leishmaniasis. Photodiagn. Photodyn. Ther. 2019, 28, 210–215. [Google Scholar] [CrossRef]
- Manca, M.L.; Matricardi, P.; Cencetti, C.; Peris, J.E.; Melis, V.; Carbone, C.; Escribano, E.; Zaru, M.; Fadda, A.M.; Manconi, M. Combination of argan oil and phospholipids for the development of an effective liposome-like formulation able to improve skin hydration and allantoin dermal delivery. Int. J. Pharm. 2016, 505, 204–211. [Google Scholar] [CrossRef]
- Lv, X.; Cong, Z.; Liu, Z.; Ma, X.; Xu, M.; Tian, Y.; Zhang, X.; Xu, B.; Zhang, J.; Tang, Z. Improvement of the solubility, photostability, antioxidant activity and UVB photoprotection of trans-resveratrol by essential oil based microemulsions for topical application. J. Drug Deliv. Sci. Technol. 2018, 48, 346–354. [Google Scholar] [CrossRef]
- Ferreira, M.; Barreiros, L.; Segundo, M.; Torres, T.; Selores, M.; Lima, S.; Reis, S. Topical co-delivery of methotrexate and etanercept using lipid nanoparticles: A targeted approach for psoriasis management. Colloids Surf. B Biointerfaces 2017, 159, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.B.; Friedberg, J.S.; Glatstein, E.; Stevenson, J.P.; Sterman, D.H.; Hahn, S.M.; Cengel, K.A. Photodynamic therapy for the treatment of non-small cell lung cancer. J. Thorac. Dis. 2011, 4, 63–75. [Google Scholar] [CrossRef]
- Xiang, Q.; Wang, M.; Chen, F.; Gong, T.; Jian, Y.; Zhang, Z.; Huang, Y. Lung-Targeting Delivery of Dexamethasone Acetate Loaded Solid Lipid Nanoparticles. Arch. Pharm. Res. 2007, 30, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Kumar, Y.; Kuche, K.; Swami, R.; Katiyar, S.S.; Chaudhari, D.; Katare, P.B.; Banerjee, S.; Jain, S. Exploring the potential of novel pH sensitive lipoplexes for tumor targeted gene delivery with reduced toxicity. Int. J. Pharm. 2019, 573, 118889. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Sharma, S.; Khuller, G. Oral solid lipid nanoparticle-based antitubercular chemotherapy. Tuberculosis 2005, 85, 415–420. [Google Scholar] [CrossRef]
- Johnson, B.M.; Charman, W.N.; Porter, C.J.H. An In Vitro Examination of the Impact of Polyethylene Glycol 400, Pluronic P85, and Vitamin E d-a-Tocopheryl Polyethylene Glycol 1000 Succinate on P-Glycoprotein Efflux and Enterocyte-Based Metabolism in Excised Rat Intestine. AAPS PharmSciTech 2002, 4, 193–205. [Google Scholar] [CrossRef]
- AboulFotouh, K.; Allam, A.A.; El-Badry, M.; El-Sayed, A.M. A Self-Nanoemulsifying Drug Delivery System for Enhancing the Oral Bioavailability of Candesartan Cilexetil: Ex Vivo and In Vivo Evaluation. J. Pharm. Sci. 2019, 108, 3599–3608. [Google Scholar] [CrossRef]
- Jain, S.; Patel, N.; Shah, M.K.; Khatri, P.; Vora, N. Recent Advances in Lipid-Based Vesicles and Particulate Carriers for Topical and Transdermal Application. J. Pharm. Sci. 2016, 106, 423–445. [Google Scholar] [CrossRef]
- Pandita, D.; Kumar, S.; Poonia, N.; Lather, V. Solid lipid nanoparticles enhance oral bioavailability of resveratrol, a natural polyphenol. Food Res. Int. 2014, 62, 1165–1174. [Google Scholar] [CrossRef]
- Leite, T.; Hillaireau, H.; Vergnaud, J.; Rivano, M.; Deloménie, C.; Courilleau, D.; Arpicco, S.; Suk, J.S.; Hanes, J.; Fattal, E. Hyaluronic acid-conjugated lipoplexes for targeted delivery of siRNA in a murine metastatic lung cancer model. Int. J. Pharm. 2016, 514, 103–111. [Google Scholar] [CrossRef]
- Islan, G.A.; Martin-Saldaña, S.; Chevalier, M.T.; Alvarez, V.A.; Castro, G.R. ADME. Processes in Pharmaceutical Sciences. In Nanotechnology and Drug Delivery; Springer: Berlin, Germany, 2018. [Google Scholar] [CrossRef]
- Schultz, H.B.; Thomas, N.; Rao, S.; Prestidge, C.A. Supersaturated Silica-Lipid Hybrids (Super-SLH): An Improved Solid-State Lipid-Based Oral Drug Delivery System with Enhanced Drug Loading. Eur. J. Pharm. Biopharm. 2017, 125, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.A.; Serrano, D.R.; Mauger, M.; Dea-ayuela, M.A.; Lalatsa, A. Orally Bioavailable and Effective Buparvaquone Lipid Based Nanomedicines for Visceral Leishmaniasis. Mol. Pharm. 2018, 15, 2570–2583. [Google Scholar] [CrossRef]
- Pandya, N.T.; Jani, P.; Vanza, J.; Tandel, H. Solid lipid nanoparticles as an efficient drug delivery system of olmesartan medoxomil for the treatment of hypertension. Colloids Surf. B Biointerfaces 2018, 165, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Al-Maghrabi, P.M.; Khafagy, E.-S.; Ghorab, M.M.; Gad, S. Influence of formulation variables on miconazole nitrate–loaded lipid based nanocarrier for topical delivery. Colloids Surf. B Biointerfaces 2020, 193, 111046. [Google Scholar] [CrossRef] [PubMed]
- Hamed, R.; Farhan, A.; Abu-Huwaij, R.; Mahmoud, N.N.; Kamal, A. Lidocaine Microemulsion-Laden Organogels as Lipid-Based Systems for Topical Delivery. J. Pharm. Innov. 2019, 15, 521–534. [Google Scholar] [CrossRef]
- Permana, A.D.; Tekko, I.A.; McCrudden, M.T.C.; Anjani, Q.K.; Ramadon, D.; McCarthy, H.O.; Donnelly, R.F. Solid lipid nanoparticle-based dissolving microneedles: A promising intradermal lymph targeting drug delivery system with potential for enhanced treatment of lymphatic filariasis. J. Control. Release 2019, 316, 34–52. [Google Scholar] [CrossRef]
- Mehta, P.; Ghoshal, D.; Pawar, A.P.; Kadam, S.S.; Dhapte-Pawar, V.S. Recent advances in inhalable liposomes for treatment of pulmonary diseases: Concept to clinical stance. J. Drug Deliv. Sci. Technol. 2020, 56, 101509. [Google Scholar] [CrossRef]
- Zhu, X.; Kong, Y.; Liu, Q.; Lu, Y.; Xing, H.; Lu, X.; Yang, Y.; Xu, J.; Li, N.; Zhao, D.; et al. Pulmonary Pharmacology & Therapeutics Inhalable dry powder prepared from folic acid-conjugated docetaxel liposomes alters pharmacodynamic and pharmacokinetic properties relevant to lung cancer chemotherapy. Pulm. Pharmacol. Ther. 2019, 55, 50–61. [Google Scholar] [CrossRef]
- Jaafar-Maalej, C.; Elaissari, A.; Fessi, H. Lipid-based carriers: Manufacturing and applications for pulmonary route. Expert Opin. Drug Deliv. 2012, 9, 1111–1127. [Google Scholar] [CrossRef]
- Umerska, A.; Mugheirbi, N.A.; Kasprzak, A.; Saulnier, P.; Tajber, L. Carbohydrate-based Trojan microparticles as carriers for pulmonary delivery of lipid nanocapsules using dry powder inhalation. Powder Technol. 2020, 364, 507–521. [Google Scholar] [CrossRef]
- Zhang, H.; Leal, J.; Soto, M.; Smyth, H.; Ghosh, D. Aerosolizable lipid nanoparticles for pulmonary delivery of mRNA through design of experiments. Pharmaceutics 2020, 12, 1042. [Google Scholar] [CrossRef] [PubMed]
- Nemati, E.; Mokhtarzadeh, A.; Panahi-Azar, V.; Mohammadi, A.; Hamishehkar, H.; Mesgari-Abbasi, M.; Dolatabadi, J.E.N.; De La Guardia, M. Ethambutol-Loaded Solid Lipid Nanoparticles as Dry Powder Inhalable Formulation for Tuberculosis Therapy. AAPS PharmSciTech 2019, 20, 120. [Google Scholar] [CrossRef] [PubMed]
- Marcano, R.G.D.J.V.; Tominaga, T.T.; Khalil, N.M.; Pedroso, L.S.; Mainardes, R.M. Chitosan functionalized poly (ε-caprolactone) nanoparticles for amphotericin B delivery. Carbohydr. Polym. 2018, 202, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Peng, Q.; San, F.; Luo, J.; Wang, M.; Wu, W. Biomaterials A high-efficiency, low-toxicity, phospholipids-based phase separation gel for long-term delivery of peptides. Biomaterials 2015, 45, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Khan, U.A.; Parveen, U.; Hasan, N.; Ahmed, M.Z.; Saad, S.; Ahmad, F.; Jain, G.K. Parenteral sustained release lipid phase-transition system of ziprasidone: Fabrication and evaluation for schizophrenia therapy. Drug Des. Dev. Ther. 2020, 14, 2237–2247. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Harikumar, K.B. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int. J. Biochem. Cell Biol. 2009, 41, 40–59. [Google Scholar] [CrossRef] [Green Version]
- Saedi, A.; Rostamizadeh, K.; Parsa, M.; Dalali, N.; Ahmadi, N. Preparation and characterization of nanostructured lipid carriers as drug delivery system: Influence of liquid lipid types on loading and cytotoxicity. Chem. Phys. Lipids 2018, 216, 65–72. [Google Scholar] [CrossRef]
- Das, T.; Venkatesh, M.; Kumar, T.P.; Koland, M. SLN based alendronate in situ gel as an implantable drug delivery system—A full factorial design approach. J. Drug Deliv. Sci. Technol. 2019, 55, 101415. [Google Scholar] [CrossRef]
- Chen, M. Lipid excipients and delivery systems for pharmaceutical development: A regulatory perspective. Adv. Drug Deliv. Rev. 2008, 60, 768–777. [Google Scholar] [CrossRef]
- Inactive Ingredient Search for Approved Drug Products. Available online: https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm (accessed on 1 January 2022).
- Kohli, K.; Chopra, S.; Dhar, D.; Arora, S.; Khar, R.K. Self-emulsifying drug delivery systems: An approach to enhance oral bioavailability. Drug Discov. Today 2010, 15, 958–965. [Google Scholar] [CrossRef]
- Saluja, V.; Sekhon, B.S. The regulation of Pharmaceutical Excipients The regulation of pharmaceutical excipients. J. Excip. Food Chem. 2014, 4, 1049. [Google Scholar]
- Wang, W.; Ye, Z.; Gao, H.; Ouyang, D. Computational pharmaceutics—A new paradigm of drug delivery. J. Control. Release 2021, 338, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Cottura, N.; Howarth, A.; Rajoli, R.K.; Siccardi, M. The Current Landscape of Novel Formulations and the Role of Mathematical Modeling in Their Development. J. Clin. Pharmacol. 2020, 60, S77–S97. [Google Scholar] [CrossRef] [PubMed]
- Abd-Algaleel, S.A.; Abdel-Bar, H.M.; Metwally, A.A.; Hathout, R.M. Evolution of the computational pharmaceutics approaches in the modeling and prediction of drug payload in lipid and polymeric nanocarriers. Pharmaceuticals 2021, 14, 645. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Jia, H.; Dong, J.; Yang, X.; Li, H.; Ouyang, D. Integrated in silico formulation design of self-emulsifying drug delivery systems. Acta Pharm. Sin. B 2021, 11, 3585–3594. [Google Scholar] [CrossRef] [PubMed]
- Bunker, A.; Róg, T. Mechanistic Understanding from Molecular Dynamics Simulation in Pharmaceutical Research 1: Drug Delivery. Front. Mol. Biosci. 2020, 7, 371. [Google Scholar] [CrossRef] [PubMed]
- Aminpour, M.; Montemagno, C.; Tuszynski, J.A. An Overview of Molecular Modeling for Drug Discovery with Specific Illustrative Examples of Applications. Molecules 2019, 24, 1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.P.; Faisal, W.; Ruane-O’Hora, T.; Devine, K.J.; Kostewicz, E.S.; O’Driscoll, C.M.; Griffin, B.T. Lipidic dispersion to reduce food dependent oral bioavailability of fenofibrate: In vitro, in vivo and in silico assessments. Eur. J. Pharm. Biopharm. 2015, 96, 207–216. [Google Scholar] [CrossRef]
- Zheng, W.; Jain, A.; Papoutsakis, D.; Dannenfelser, R.; Panicucci, R.; Garad, S. Selection of oral bioavailability enhancing formulations during drug discovery. Drug Dev. Ind. Pharm. 2012, 38, 235–247. [Google Scholar] [CrossRef]
- Bergström, C.A.; Charman, W.; Porter, C. Computational prediction of formulation strategies for beyond-rule-of-5 compounds. Adv. Drug Deliv. Rev. 2016, 101, 6–21. [Google Scholar] [CrossRef]
- Jones, H.M. Basic Concepts in Physiologically Based Pharmacokinetic Modeling in Drug Discovery and Development. CPT: Pharmacomet. Syst. Pharmacol. 2013, 2, e63. [Google Scholar] [CrossRef] [PubMed]
- Apgar, J.F.; Tang, J.-P.; Singh, P.; Balasubramanian, N.; Burke, J.; Hodges, M.R.; Lasaro, M.A.; Lin, L.; Miliard, B.L.; Moore, K.; et al. Quantitative Systems Pharmacology Model of hUGT1A1-modRNA Encoding for the UGT1A1 Enzyme to Treat Crigler-Najjar Syndrome Type 1. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, D.C.; Yin, F.; Kindt, J.T.; Zhang, H. Molecular Dynamics Simulations of Glycocholate—Oleic Acid Mixed Micelle Assembly. Langmuir 2010, 26, 4687–4692. [Google Scholar] [CrossRef] [PubMed]
- Bogusz, S.; Venable, R.M.; Pastor, R.W. Molecular Dynamics Simulations of Octyl Glucoside Micelles: Structural Properties. J. Phys. Chem. B 2000, 104, 5462–5470. [Google Scholar] [CrossRef]
- Birru, W.A.; Warren, D.B.; Han, S.; Benameur, H.; Porter, C.J.H.; Pouton, C.W.; Chalmers, D.K.; Birru, W.A.; Warren, D.B.; Han, S.; et al. Computational and experimental models of the gastrointestinal environment 2. Phase behavior and drug solubilisation capacity of a Type I lipid-based drug formulation after digestion. Mol. Pharm. 2016, 14, 580–592. [Google Scholar] [CrossRef] [PubMed]
- King, D.T.; Warren, D.B.; Pouton, C.W.; Chalmers, D.K. Using Molecular Dynamics to Study Liquid Phase Behavior: Simulations of the Ternary Sodium Laurate/Sodium Oleate/Water System. Langmuir 2011, 27, 11381–11393. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Warren, D.B.; King, D.; Benameur, H.; Pouton, C.W.; Chalmers, D.K. Glyceride Lipid Formulations: Molecular Dynamics Modeling of Phase Behavior during Dispersion and Molecular Interactions between Drugs and Excipients. Pharm. Res. 2013, 30, 3238–3253. [Google Scholar] [CrossRef]
- Egberts, E.; Marrink, S.; Berendsen, H.J.C. Molecular dynamics simulation of a phospholipid membrane. Eur. Biophys. J. 1994, 22, 423–436. [Google Scholar] [CrossRef]
- Kandt, C.; Ash, W.L.; Tieleman, D.P. Setting up and running molecular dynamics simulations of membrane proteins. Methods 2007, 41, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Poger, D.; Mark, A.E. Turning the growth hormone receptor on: Evidence that hormone binding induces subunit rotation. Proteins Struct. Funct. Bioinform. 2009, 78, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Alskär, L.C.; Bergström, C.A.S. Models for Predicting Drug Absorption from Oral Lipid-Based Formulations. Curr. Mol. Biol. Rep. 2015, 1, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Löbenberg, R. Biorelevant dissolution media as a predictive tool for glyburide a class II drug. Eur. J. Pharm. Sci. 2006, 29, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Aburub, A.; Risley, D.S.; Mishra, D. A critical evaluation of fasted state simulating gastric fluid (FaSSGF) that contains sodium lauryl sulfate and proposal of a modified recipe. Int. J. Pharm. 2008, 347, 16–22. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Chemical Name | Functionalities | Example | Regulatory Status |
|---|---|---|---|
| Caprylocaproyl macrogol-8 glycerides | Solubilizer | SMEDDS [8] | FDA IID |
| Hard Fat | Taste masking, Hot melt coating | HME, Melt granulation, NLCs [9] | USFDA |
| Glycerol dibehenate | Sustained-release, Lubricant, API protection | SR tablet, SLN and NLCs [10] | FDA IID |
| Glyceryl Monostearate | Lubricant, Emulsifier | Colloidal carriers [11] | USFDA |
| Glycerol distearate | Sustained release, Lubricant, Taste-masking | SR Tablet, Lipid nanoparticles, Hot melt coating [12] | FDA IID |
| Glyceryl Monocaprylate | Permeability enhancer, Emulsifier, Solubilizer | SEDDS [13] | USP/NF |
| Propylene Glycol Monocaprylate | Emulsifier, Solubilizer | SEDDS, Nanoparticles [13] | USFDA |
| Glycerol Monocaprylocaprate | Solubilizer, penetration enhancer | SNEDDS [14] | USFDA |
| Succinic Triglycerides | Super fatting Agent | Creams [15] | USFDA |
| Lauroyl macrogol-32 glycerides | Bioavailability enhancer | Hard gelatin capsule [15] (SNEDDS, SMEDDS), Mixed micelles [16] | FDA IID |
| Oleoyl macrogrol-32 glyceride | Bioavailability enhancer, Solubilizer | Oral solutions, SMEDDS [17] | FDA IID |
| Polyglyceryl-3 dioleate | Surfactant | Niosomes [18] | FDA IID |
| Propylene glycol dicaprylocaprate | Oily vehicle | Emulsions and Nanocapsules [19] | FDA IID |
| Trimyristin | Controlled release, Lubricant, taste masking | SLNs [20], HME, SEDDS, SLMs [20] | USFDA |
| Tristearin/Glyceryl Tristearate | Controlled release, Lubricant, lipid carrier | HME, Hot melt coating as a taste masking agent, SMEDDS and SLMs [20,21] | USFDA |
| Glyceryl Caprylate | Permeability enhancer, Emulsifier, Solubilizer | SMEDDS [22,23] | IIG/USFDA |
| Polyoxyl stearate | Bioavailability enhancer | (SNEDDS, SMEDDS) and Solid dispersion [23,24,25,26] | FDA IID |
| Stearoylmacrogol-32 glyceride | Bioavailability enhancer | Solid dispersion [26] | FDA IID |
| S. No. | Drug | Formulation | Excipients | Application | References |
|---|---|---|---|---|---|
| 1 | Olanzapine | NLCs | Glyceryl monostearate, Tripalmitate, polyoxypropylenepolyoxyethylene block copolymer (pluronic F-68) | Enhancement in oral bioavailability bypassing pre systemic metabolism | Jawahar N. et al. [92] |
| 2 | Curcumin | Emulsion | Corn oil, polyethylene sorbitan monooleate | Improved bioavailability | Liqiang et al. [93] |
| 3 | Hydrochlorothiazide | SLN | Glyceryl distearate, polyoxypropylenepolyoxyethylene block copolymer (pluronic F-68) and polyethylene sorbitan monooleate | Increased diuretic effect and bioavailability | Cirri et al. [94] |
| 4 | Abiraterone acetate | Silica-lipid hybrids | Propylene glycol monocaprylate, glyceryl caprylate, PEG-35 castor oil | Exceeded the bioavailability by 1.43 fold | Schultz et al. [95] |
| 5 | Paclitaxel | Microemulsion | Distilled diacetylated monoglyceride, lecithin, glyceryl caprylate | p-gp efflux inhibition, increase in in situ permeability | Nornoo et al. [96] |
| 6 | Mupirocin | NLC | Cetyl palmitate, caprylic/caprylic acid, polyethylene sorbitan monooleate, phosphatidylcholine | preventing enzymatic degradation | Alcantar et al. [97] |
| 7 | Quercetin | Mixed micelles | Polyoxazolines, phosphatidylcholine | Improved antioxidant effect | Simon et al. [98] |
| 8 | Ibuprofen | Mixed micelles | Sodium deoxycholate, polyethylene sorbitan monooleate | Enhanced topical delivery | Sun et al. [4] |
| 9 | Chloraluminumphthalocyanine | Liposomes | egg phosphatidylcholine | Reduced parasitic load in liver and spleen | Lopes SC et al. [99] |
| 10 | Allantoin | Liposomes | Argan oil, phospholipids | Enhanced penetration | Manca et al. [100] |
| 11 | trans-resveratrol | Microemulsions | PEG-35 castor oil,1,2-propanediol, essential oils | Enhanced skin permeation | Cong et al. [101] |
| 12 | Methotrexate and etanercept | SLNs | Cetyl palmitate | improved drug therapeutic effect | Ferreira et al. [102] |
| 13 | Ferrous chlorophyllin | Ethosomes | Soya Lecithin, ceteareth-25 | Increased skin penetration | Simone et al. [103] |
| 14 | Dexamethasone acetate | SLNs | Soybean lecithin, glycerol tristearate, pluronic F-68 | Higher uptake by the lung | Xiang et al. [104] |
| 15 | Lipofectamine® | Lipoplexes | 1,2-dioleoyloxy-3(trimethylammonium)propane, dioleoylphosphatidylethanolamin, DNase I | 1.3-fold higher tumor transfection | Kumar et al. [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakmode, D.; Bhavana, V.; Thakor, P.; Madan, J.; Singh, P.K.; Singh, S.B.; Rosenholm, J.M.; Bansal, K.K.; Mehra, N.K. Fundamental Aspects of Lipid-Based Excipients in Lipid-Based Product Development. Pharmaceutics 2022, 14, 831. https://doi.org/10.3390/pharmaceutics14040831
Nakmode D, Bhavana V, Thakor P, Madan J, Singh PK, Singh SB, Rosenholm JM, Bansal KK, Mehra NK. Fundamental Aspects of Lipid-Based Excipients in Lipid-Based Product Development. Pharmaceutics. 2022; 14(4):831. https://doi.org/10.3390/pharmaceutics14040831
Chicago/Turabian StyleNakmode, Deepa, Valamla Bhavana, Pradip Thakor, Jitender Madan, Pankaj Kumar Singh, Shashi Bala Singh, Jessica M. Rosenholm, Kuldeep K. Bansal, and Neelesh Kumar Mehra. 2022. "Fundamental Aspects of Lipid-Based Excipients in Lipid-Based Product Development" Pharmaceutics 14, no. 4: 831. https://doi.org/10.3390/pharmaceutics14040831