
Genetic Variant ABCC1 rs45511401 Is Associated with Increased Response to Statins in Patients with Familial Hypercholesterolemia

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Patients
2.2. Blood Samples and Laboratory Testing
2.3. Exon-Targeted Gene Sequencing
2.4. Clinical and Pharmacotherapeutic Data
2.5. Pharmacogenetic Analyses
2.6. Molecular Modeling
2.7. Statistical Analyses
3. Results
3.1. Characteristics of the Individuals and Molecular Diagnosis
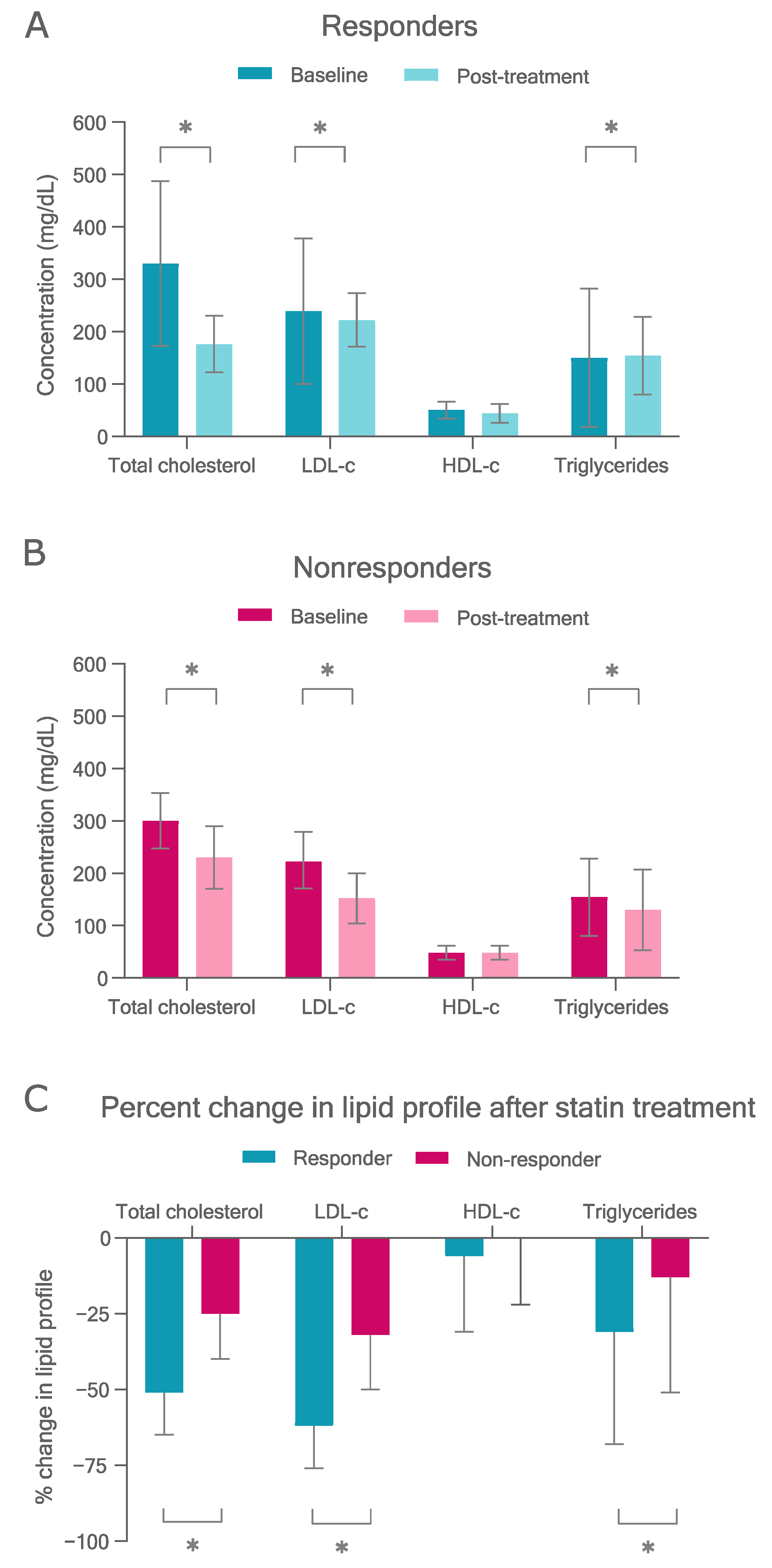
3.2. Statin Response
3.2.1. Therapy Targets
3.2.2. Statin-Related Adverse Events
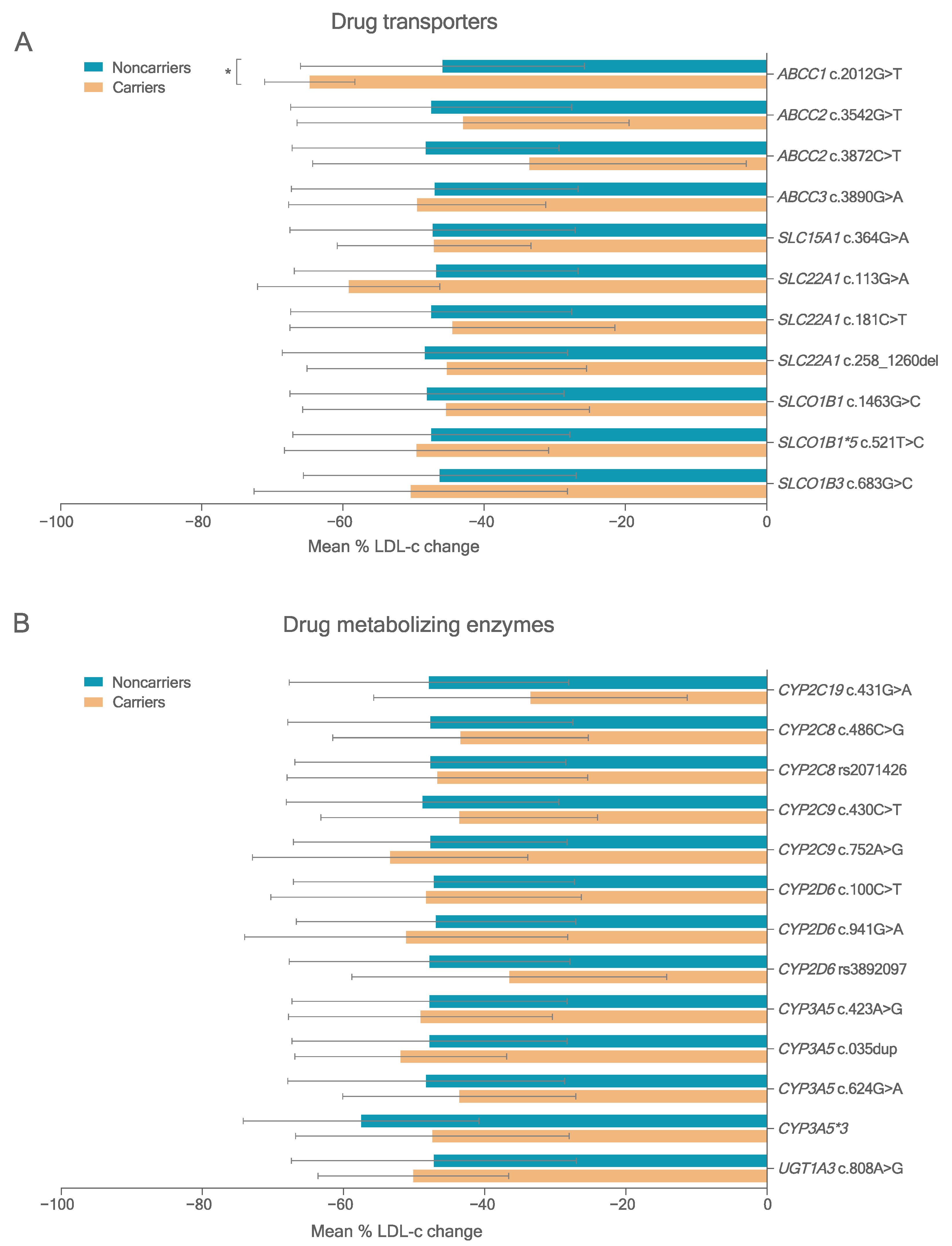
3.3. Variants in PK-Related Genes
3.4. Association Study between Variants in PK Genes and Statin Response
3.4.1. LDL-c Reduction
3.4.2. Molecular Modeling Results
3.4.3. Statin-Related Adverse Events
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef] [PubMed]
- Izar, M.; Giraldez, V.; Bertolami, A.; Santos Filho, R.; Lottenberg, A.; Assad, M.; Saraiva, J.; Chacra, A.; Martinez, T.; Bahia, L.; et al. Update of the Brazilian Guideline for Familial Hypercholesterolemia—2021. Arq. Bras. Cardiol. 2021, 117, 782–844. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, R.D.; Barry, A.R.; Pearson, G.J. Familial Hypercholesterolemia: Review of Diagnosis, Screening, and Treatment. Can. Fam. Phys. 2016, 62, 32–37. [Google Scholar]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity: Mechanistic Insights and Clinical Implications. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Vaz, A.J.; Kondapally Seshasai, S.R.; Cole, D.; Hovingh, G.K.; Kastelein, J.J.P.; Mata, P.; Raal, F.J.; Santos, R.D.; Soran, H.; Watts, G.F.; et al. Familial Hypercholesterolaemia: A Global Call to Arms. Atherosclerosis 2015, 243, 257–259. [Google Scholar] [CrossRef]
- Mach, F.; Ray, K.K.; Wiklund, O.; Corsini, A.; Catapano, A.L.; Bruckert, E.; De Backer, G.; Hegele, R.A.; Hovingh, G.K.; Jacobson, T.A.; et al. Adverse Effects of Statin Therapy: Perception vs. the Evidence—Focus on Glucose Homeostasis, Cognitive, Renal and Hepatic Function, Haemorrhagic Stroke and Cataract. Eur. Heart J. 2018, 39, 2526–2539. [Google Scholar] [CrossRef]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgozoglu, L.; Nordestgaard, B.G.; et al. Statin-Associated Muscle Symptoms: Impact on Statin Therapy--European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur. Heart J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef]
- Karlson, B.W.; Palmer, M.K.; Nicholls, S.J.; Barter, P.J.; Lundman, P. Effects of Age, Gender and Statin Dose on Lipid Levels: Results from the VOYAGER Meta-Analysis Database. Atherosclerosis 2017, 265, 54–59. [Google Scholar] [CrossRef]
- Oni-Orisan, A.; Hoffmann, T.J.; Ranatunga, D.; Medina, M.W.; Jorgenson, E.; Schaefer, C.; Krauss, R.M.; Iribarren, C.; Risch, N. Characterization of Statin Low-Density Lipoprotein Cholesterol Dose-Response Using Electronic Health Records in a Large Population-Based Cohort. Circ. Genom. Precis. Med. 2018, 11, e002043. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, W.D.; Ramsey, L.B.; Johnson, S.G.; Moore, K.G.; Shtutman, M.; Schoonover, J.H.; Kawaguchi-Suzuki, M. Impact of Pharmacogenetics on Efficacy and Safety of Statin Therapy for Dyslipidemia. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2017, 37, 1172–1190. [Google Scholar] [CrossRef] [PubMed]
- The SEARCH Collaborative Group. SLCO1B1 Variants and Statin-Induced Myopathy—A Genomewide Study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Li, S.; Li, L.; Li, Y.; Sun, X.; Tian, H. Association Between SLCO1B1 Gene T521C Polymorphism and Statin-Related Myopathy Risk: A Meta-Analysis of Case-Control Studies. Medicine 2015, 94, e1268. [Google Scholar] [CrossRef] [PubMed]
- Voora, D.; Shah, S.H.; Spasojevic, I.; Ali, S.; Reed, C.R.; Salisbury, B.A.; Ginsburg, G.S. The SLCO1B1*5Genetic Variant Is Associated With Statin-Induced Side Effects. J. Am. Coll. Cardiol. 2009, 54, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.C.; Perin, P.M.S.; Purim, S.G.; Silbiger, V.N.; Genvigir, F.D.V.; Willrich, M.A.V.; Arazi, S.S.; Luchessi, A.D.; Hirata, M.H.; Bernik, M.M.S.; et al. Pharmacogenetics of OATP Transporters Reveals That SLCO1B1 c.388A>G Variant Is Determinant of Increased Atorvastatin Response. Int. J. Mol. Sci. 2011, 12, 5815–5827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagli-Hernandez, C.; de Freitas, R.C.C.; da Silva Rodrigues Marçal, E.; Gonçalves, R.M.; Faludi, A.A.; Borges, J.B.; Bastos, G.M.; Los, B.; Mori, A.A.; Bortolin, R.H.; et al. Late Response to Rosuvastatin and Statin-Related Myalgia Due to SLCO1B1, SLCO1B3, ABCB11, and CYP3A5 Variants in a Patient with Familial Hypercholesterolemia: A Case Report. Ann. Transl. Med. 2020, 9, 76. [Google Scholar] [CrossRef]
- Elens, L.; Becker, M.L.; Haufroid, V.; Hofman, A.; Visser, L.E.; Uitterlinden, A.G.; Stricker, B.C.; van Schaik, R.H.N. Novel CYP3A4 Intron 6 Single Nucleotide Polymorphism Is Associated with Simvastatin-Mediated Cholesterol Reduction in The Rotterdam Study. Pharm. Genom. 2011, 21, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Willrich, M.A.V.; Hirata, M.H.; Genvigir, F.D.V.; Arazi, S.S.; Rebecchi, I.M.M.; Rodrigues, A.C.; Bernik, M.M.S.; Dorea, E.L.; Bertolami, M.C.; Faludi, A.A.; et al. CYP3A5*3A Allele Is Associated with Reduced Lowering-Lipid Response to Atorvastatin in Individuals with Hypercholesterolemia. Clin. Chim. Acta 2008, 398, 15–20. [Google Scholar] [CrossRef]
- Dagli-Hernandez, C.; Zhou, Y.; Lauschke, V.M.; Genvigir, F.D.V.; Hirata, T.D.C.; Hirata, M.H.; Hirata, R.D.C. Pharmacogenomics of Statins: Lipid Response and Other Outcomes in Brazilian Cohorts. Pharmacol. Rep. 2022, 74, 47–66. [Google Scholar] [CrossRef]
- Santos, P.C.J.L.; Gagliardi, A.C.M.; Miname, M.H.; Chacra, A.P.; Santos, R.D.; Krieger, J.E.; Pereira, A.C. SLCO1B1 Haplotypes Are Not Associated with Atorvastatin-Induced Myalgia in Brazilian Patients with Familial Hypercholesterolemia. Eur. J. Clin. Pharmacol. 2012, 68, 273–279. [Google Scholar] [CrossRef]
- Borges, J.B.; de Oliveira, V.F.; Ferreira, G.M.; Los, B.; Barbosa, T.K.A.A.; da Silva Rodrigues Marçal, E.; Dagli-Hernandez, C.; de Freitas, R.C.C.; Bortolin, R.H.; Mori, A.A.; et al. Genomics, Epigenomics and Pharmacogenomics of Familial Hypercholesterolemia (FHBGEP): A Study Protocol. Res. Soc. Adm. Pharm. 2021, 17, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Human Genetics Programme. Familial Hypercholesterolaemia (FH): Report of a Second WHO Consultation; WHO: Geneva, Switzerland, 1998. [Google Scholar]
- Friedwald, W.; Levy, R.; Frednickson, D. Estimation of the Concentration of Low Density Lipoprotein Cholesterol in Plasma without Use of the Preparative Ultra Centrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.C.; Hopkins, P.N.; Toth, P.P.; Ballantyne, C.M.; Rader, D.J.; Robinson, J.G.; Daniels, S.R.; Gidding, S.S.; de Ferranti, S.D.; Ito, M.K.; et al. Executive Summary Familial Hypercholesterolemia: Screening, Diagnosis and Management of Pediatric and Adult Patients Clinical Guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia Background and Rationale. J. Clin. Lipidol. 2011, 5, 1–8. [Google Scholar] [CrossRef]
- Chou, R.; Dana, T.; Blazina, I.; Daeges, M.; Jeanne, T.L. Statins for Prevention of Cardiovascular Disease in Adults: Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA—J. Am. Med. Assoc. 2016, 316, 2008–2024. [Google Scholar] [CrossRef]
- Bellosta, S.; Corsini, A. Statin Drug Interactions and Related Adverse Reactions: An Update. Expert Opin. Drug Saf. 2018, 17, 25–37. [Google Scholar] [CrossRef]
- Boretzki, J.; Wolf, E.; Wiese, C.; Noe, S.; Balogh, A.; Meurer, A.; Krznaric, I.; Zink, A.; Lersch, C.; Spinner, C. Highly Specific Reasons for Nonadherence to Antiretroviral Therapy: Results from the German Adherence Study. Patient Prefer. Adherence 2017, 11, 1897–1906. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Mkrtchian, S.; Kumondai, M.; Hiratsuka, M.; Lauschke, V.M. An Optimized Prediction Framework to Assess the Functional Impact of Pharmacogenetic Variants. Pharm. J. 2018, 19, 115–126. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Hoenig, M.R.; Walker, P.J.; Gurnsey, C.; Beadle, K.; Johnson, L. The C3435T Polymorphism in ABCB1 Influences Atorvastatin Efficacy and Muscle Symptoms in a High-Risk Vascular Cohort. J. Clin. Lipidol. 2011, 5, 91–96. [Google Scholar] [CrossRef]
- Su, J.; Xu, H.; Yang, J.; Yu, Q.; Yang, S.; Zhang, J.; Yao, Q.; Zhu, Y.; Luo, Y.; Ji, L.; et al. ABCB1 C3435T Polymorphism and the Lipid-Lowering Response in Hypercholesterolemic Patients on Statins: A Meta-Analysis. Lipids Health Dis. 2015, 14, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebecchi, I.M.M.; Rodrigues, A.C.; Arazi, S.S.; Genvigir, F.D.V.; Willrich, M.A.V.; Hirata, M.H.; Soares, S.A.; Bertolami, M.C.; Faludi, A.A.; Bernik, M.M.S.; et al. ABCB1 and ABCC1 Expression in Peripheral Mononuclear Cells Is Influenced by Gene Polymorphisms and Atorvastatin Treatment. Biochem. Pharmacol. 2009, 77, 66–75. [Google Scholar] [CrossRef]
- Fiegenbaum, M.; da Silveira, F.; Van der Sand, C.; Van der Sand, L.; Ferreira, M.; Pires, R.; Hutz, M. The Role of Common Variants of ABCB1, CYP3A4, and CYP3A5 Genes in Lipid-Lowering Efficacy and Safety of Simvastatin Treatment. Clin. Pharmacol. Ther. 2005, 78, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sew, P.H.; Ambrose, H.; Ryan, S.; Chong, S.S.; Lee, E.J.D.; Lee, C.G.L. Nucleotide Sequence Analyses of the MRP1 Gene in Four Populations Suggest Negative Selection on Its Coding Region. BMC Genom. 2006, 7, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, K.C.; Pereira, B.M.V.; Rodrigues, A.C. An Update on Efflux and Uptake Transporters as Determinants of Statin Response. Expert Opin. Drug Metab. Toxicol. 2018, 14, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Zhou, Y.; Lauschke, V.M. Ethnogeographic and Inter-Individual Variability of Human ABC Transporters. Hum. Genet. 2020, 139, 623–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulsteke, C.; Lambrechts, D.; Dieudonné, A.; Hatse, S.; Brouwers, B.; van Brussel, T.; Neven, P.; Belmans, A.; Schöffski, P.; Paridaens, R.; et al. Genetic Variability in the Multidrug Resistance Associated Protein-1 (Abcc1/Mrp1) Predicts Hematological Toxicity in Breast Cancer Patients Receiving (Neo-)Adjuvant Chemotherapy with 5-Fluorouracil, Epirubicin and Cyclophosphamide (Fec). Ann. Oncol. 2013, 24, 1513–1525. [Google Scholar] [CrossRef] [PubMed]
- Jungsuwadee, P.; Zhao, T.; Stolarczyk, E.I.; Paumi, C.M.; Butterfield, D.A.; St Clair, D.K.; Vore, M. The G671V Variant of MRP1/ABCC1 Links Doxorubicin-Induced Acute Cardiac Toxicity to Disposition of the Glutathione Conjugate of 4-Hydroxy-2-Trans-Nonenal. Pharm. Genom. 2012, 22, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behdad, N.; Kojuri, J.; Azarpira, N.; Masoomi, A.; Namazi, S. Association of ABCB1 (C3435T) and ABCC1 (G2012T) Polymorphisms with Clinical Response to Atorvastatin in Iranian Patients with Primary Hyperlipidemia. Iran. Biomed. J. 2017, 21, 120–125. [Google Scholar] [CrossRef]
- Vohra, M.; Sharma, A.R.; Paul, B.; Bhat, M.K.; Satyamoorthy, K.; Rai, P.S. In Silico Characterization of Functional Single Nucleotide Polymorphisms of Folate Pathway Genes. Ann. Hum. Genet. 2018, 82, 186–199. [Google Scholar] [CrossRef]
- Betts, M.J.; Russell, R.B. Amino Acid Properties and Consequences of Substitutions. In Bioinformatics for Geneticists; Wiley: Hoboken, NJ, USA, 2003. [Google Scholar]
- Rosales, A.; Alvear, M.; Cuevas, A.; Saavedra, N.; Zambrano, T.; Salazar, L.A. Identification of Pharmacogenetic Predictors of Lipid-Lowering Response to Atorvastatin in Chilean Subjects with Hypercholesterolemia. Clin. Chim. Acta 2012, 413, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Postmus, I.; Trompet, S.; Deshmukh, H.A.; Barnes, M.R.; Li, X.; Warren, H.R.; Chasman, D.I.; Zhou, K.; Arsenault, B.J.; Donnelly, L.A.; et al. Pharmacogenetic Meta-Analysis of Genome-Wide Association Studies of LDL Cholesterol Response to Statins. Nat. Commun. 2014, 5, 5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.F.; Hyde, C.L.; Wood, L.S.; Paciga, S.A.; Hinds, D.A.; Cox, D.R.; Hovingh, G.K.; Kastelein, J.J.P. Comprehensive Whole-Genome and Candidate Gene Analysis for Response to Statin Therapy in the Treating to New Targets (TNT) Cohort. Circ. Cardiovasc. Genet. 2009, 2, 173–181. [Google Scholar] [CrossRef]
- Floyd, J.S.; Bloch, K.M.; Brody, J.A.; Maroteau, C.; Siddiqui, M.K.; Gregory, R.; Carr, D.F.; Molokhia, M.; Liu, X.; Bis, J.C.; et al. Pharmacogenomics of Statin-Related Myopathy: Meta-Analysis of Rare Variants from Whole-Exome Sequencing. PLoS ONE 2019, 14, e0218115. [Google Scholar] [CrossRef] [Green Version]
- Carr, D.F.; Francis, B.; Jorgensen, A.L.; Zhang, E.; Chinoy, H.; Heckbert, S.R.; Bis, J.C.; Brody, J.A.; Floyd, J.S.; Psaty, B.M.; et al. Genomewide Association Study of Statin-Induced Myopathy in Patients Recruited Using the UK Clinical Practice Research Datalink. Clin. Pharmacol. Ther. 2019, 106, 1353–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Variable a | Total (114) | Responders (58) | Nonresponders (56) | p-Value | |
|---|---|---|---|---|---|
| Age, years | 57.1 (37.9–76.3) | 54.9 (34.7–75.1) | 57.6 (41.9–73.3) | 0.261 | |
| Gender (female), % | 71.9 (82) | 69.0 (40) | 75.0 (42) | 0.611 | |
| Ethnicity, % | White | 53.5 (54) | 58.5 (31) | 48.9 (23) | 0.326 |
| Brown | 31.7 (32) | 24.5 (13) | 38.3 (18) | ||
| Black | 14.9 (15) | 17.0 (9) | 12.8 (6) | ||
| Xanthomas, % | 12.3 (14) | 13.8 (8) | 10.7 (6) | 0.830 | |
| Arcus cornealis, % | 17.9 (20) | 14.0 (8) | 21.8 (12) | 0.407 | |
| FH clinical diagnosis b, % | Defined or probable | 68.4 (78) | 75.9 (44) | 60.7 (34) | 0.124 |
| Possible | 31.6 (36) | 24.1 (14) | 39.3 (22) | ||
| FH molecular | FH variants | 30.7 (35) | 34.5 (20) | 26.8 (15) | 0.491 |
| diagnosis, % | APOB | 0.9 (1) | 0.0 (0) | 1.8 (1) | 0.166 |
| LDLR | 28.3 (32) | 34.5 (20) | 21.4 (12) | ||
| PCSK9 | 1.8 (2) | 0.0 (0) | 3.6 (2) | ||
| LDLRAP1 | 0.0 (0) | 0.0 (0) | 0.0 (0) | ||
| Hypertension, % | 62.5 (70) | 60.3 (35) | 64.8 (35) | 0.770 | |
| Type 2 diabetes, % | 21.6 (24) | 26.3 (15) | 16.7 (9) | 0.316 | |
| Obesity, % | 28.6 (32) | 17.2 (10) | 40.7 (22) | 0.011 | |
| BMI, kg/cm2 | 27.7 (22.5–32.9) | 26.3 (21.4–31.2) | 28.2 (22.5–33.9) | 0.011 | |
| Medical history, % | AMI | 29.2 (33) | 28.1 (16) | 30.4 (17) | 0.952 |
| CAD | 40.0 (42) | 44.0 (22) | 36.4 (20) | 0.550 | |
| CVE | 6.0 (6) | 3.9 (2) | 8.2 (4) | 0.637 | |
| Alcohol consumption, % | 25.0 (22) | 14.6 (7) | 37.5 (15) | 0.007 | |
| Tobacco smoking, % | 14.3 (16) | 17.2 (10) | 11.1 (6) | 0.510 | |
| CAD risk, % | Very high risk | 56.1 (64) | 53.4 (31) | 58.9 (33) | 0.095 |
| High risk | 9.7 (11) | 15.5 (9) | 3.6 (2) | ||
| Intermediate risk | 34.2 (39) | 31.0 (18) | 37.5 (21) | ||
| Lipid-lowering | Atorvastatin | 79.8 (91) | 77.6 (45) | 82.1 (46) | 0.275 |
| treatment, % | Simvastatin | 10.5 (12) | 8.6 (5) | 12.5 (7) | |
| Rosuvastatin | 9.6 (11) | 13.8 (8) | 5.4 (3) | ||
| Statins + Eze | 36.8 (42) | 46.6 (27) | 26.8 (15) | 0.046 | |
| Statin intensity, % | Moderate | 14.0 (16) | 6.9 (4) | 21.4 (12) | 0.050 |
| High | 86.0 (98) | 93.1 (54) | 78.6 (44) | ||
| Drug interactions, % | CYP3A4 inhibitors c | 10 (8.8) | 7 (12.1) | 3 (5.3) | 0.349 |
| CYP3A4 inhibitors + inducers d | 1 (0.01) | 0 (0.0) | 1 (1.9) | - | |
| Reduced adherence, % | Statins | 15.9 (18) | 17.2 (10) | 14.5 (8) | 0.893 |
| Ezetimibe | 10.6 (12) | 13.8 (8) | 7.3 (4) | 0.413 | |
| SRAE, % | SAMS | 16.8 (19) | 29.3 (17) | 3.6 (2) | 0.001 |
| Others e | 21.2 (24) | 34.5 (20) | 7.3 (4) | 0.001 |
| Gene | dbSNP Code | Variant | Amino-Acid Change | Type | In Silico Analysis a | ACMG Classification | Number of Patients (Zygosity) |
|---|---|---|---|---|---|---|---|
| APOB | rs61744153 | c.11477C>T | p.Thr3826Met | Missense | D | LD | 1 (He) |
| LDLR | rs112029328 | c.313+1G>A | - | Splice-site | NA | D | 2 (He) |
| rs121908026 | c.530C>T | p.Ser177Leu | Missense | D | D | 2 (He) | |
| rs875989902 | c.533A>T | p.Asp178Val | Missense | D | LD | 1 (He) | |
| rs121908039 | c.551G>A | p.Cys184Tyr | Missense | D | D | 1 (He) | |
| rs879254797 | c.1118G>A | p.Gly373Asp | Missense | D | LD | 2 (He) | |
| rs28942078 | c.1285G>A | p.Val429Met | Missense | D | D | 1 (He) | |
| rs28942079 | c.1291G>A | p.Ala431Thr | Missense | D | D | 1 (He) | |
| rs879254913 | c.1463T>C | p.Ile488Thr | Missense | D | LD | 2 (He) | |
| rs373646964 | c.1474G>A | p.Asp492Asn | Missense | D | LD | 1 (He) | |
| rs28941776 | c.1646G>A | p.Gly549Asp | Missense | D | D | 2 (He) | |
| rs137929307 | c.1775G>A | p.Gly592Glu | Missense | D | LD | 2 (He) | |
| rs753707206 | c.1801G>C | p.Asp601His | Missense | D | LD | 2 (He) | |
| rs879254687 | c.818-2A>G | - | Splice-site | NA | D | 1 (He) | |
| rs1135402774 | c.1474del | p.Asp492fs | InDel | NA | D | 1 (He) | |
| rs121908031 | c.2043C>A | p.Cys681* | Stop-gain | D | D | 6 (He) | |
| rs752596535 | c.501C>G | p.Cys167* | Stop-gain | D | D | 2 (He) | |
| rs1135402768 | c.487C>T | p.Gln163* | Stop-gain | D | D | 1 (He) | |
| rs875989887 | c.-140C>A | - | 5′UTR | NA | LD | 1 (Ho) | |
| rs387906307 | c.-138del-T | - | 5′UTR | NA | LD | 1 (He) | |
| PCSK9 | rs141502002 | c.1405C>T | p.Arg469Trp | Missense | LN | Conflict b | 2 (He) |
| Gene | Variant | NT Change | AA Change | Type | MAF (%) | MAF (gnomAD a, %) | FPS |
|---|---|---|---|---|---|---|---|
| CYP2C8 | rs1058930 (CYP2C8*4) | c.486C>G | p.Ile162Met | Missense | 4.9 | 2.7 | 0.6 |
| CYP2C9 | rs1799853 (CYP2C9*2) | c.430C>T | p.Arg144Cys | Missense | 8.8 | 6.8 | 1 |
| rs2256871 (CYP2C9*9) | c.752A>G | p.His251Arg | Missense | 2.2 | 0.3 | 0.8 | |
| CYP2C19 | rs17884712 (CYP2C19*9) | c.431G>A | p.Arg144His | Missense | 2.2 | 0.1 | 0.8 |
| CYP2D6 | rs1065852 (CYP2D6*10) | c.100C>T | p.Pro34Ser | Missense | 0.03 b | 12.3 | 1 |
| rs28371703 | c.271C>A | p.Leu91Met | Missense | 1.1 | 5.9 | 0.6 | |
| rs1058172 | c.941G>A | p.Arg314His | Missense | 4.9 | 5.6 | 1 | |
| CYP3A5 | rs6977165 | c.423A>G | p.X141Trp | Stoploss | 5.7 | 8.1 | 1 |
| rs10264272 (CYP3A5*6) | g.19787G>A | p.Lys208 = | Synonymous c | 3.1 | 0.7 | 1.0 | |
| UGT1A3 | rs45449995 | c.808A>G | p.Met270Val | Missense | 2.2 | 1.6 | 0.75 |
| ABCC1 | rs45511401 | c.2012G>T | p.Gly671Val | Missense | 3.8 | 1.7 | 0.8 |
| ABCC2 | rs8187692 | c.3542G>T | p.Arg1181Leu | Missense | 2.7 | 0.6 | 0.8 |
| rs17216317 | c.3872C>T | p.Pro1291Leu | Missense | 3.3 | 0.2 | 0.8 | |
| ABCC3 | rs11568591 | c.3890G>A | p.Arg1297His | Missense | 6.5 | 2.9 | 0.8 |
| rs141856639 | c.3971G>A | p.Arg1324His | Missense | 1.1 | 0.01 | 1 | |
| SLC15A1 | rs8187820 | c.364G>A | p.Val122Met | Missense | 1.6 | 0.3 | 0.6 |
| SLC22A1 | rs2282143 | c.1022C>T | p.Pro341Leu | Missense | 1.1 | 4.4 | 0.8 |
| rs35888596 | c.113G>A | p.Gly38Asp | Missense | 2.2 | 0.4 | 1 | |
| rs34059508 | c.1393G>A | p.Gly465Arg | Missense | 1.1 | 0.7 | 0.8 | |
| rs12208357 | c.181C>T | p.Arg61Cys | Missense | 3.8 | 2.3 | 0.6 | |
| SLCO1B1 | rs59502379 | c.1463G>C | p.Gly488Ala | Missense | 1.8 | 0.1 | 0.8 |
| rs4149056 (SLCO1B1*5) | c.521T>C | p.Val174Ala | Missense | 11.0 | 11.2 | 0.8 | |
| SLCO1B3 | rs60140950 | c.767G>C | p.Gly228Ala | Missense | 14.7 | 7.4 | 1 |
| Gene | Variant | NT Change a | Type | MAF (%) | MAF (gnomAD b, %) | Prediction c |
|---|---|---|---|---|---|---|
| Splice-site variants | ||||||
| ABCC1 | rs8187856 | g.16146576C>G | Splice region | 1.1 | 0.3 | B |
| ABCC2 | rs533334893 | g.101552117G>A | Splice donor | 0.5 | 0.0 | D |
| ABCC3 | rs11568607 | g.48745787G>A | Splice region | 2.2 | 0.6 | B |
| ABCG2 | rs34124189 | g.89053790G>A | Splice region | 0.5 | 0.1 | B |
| CYP1A2 | rs1288558234 | g.75041241del | Splice region | 0.5 | 0.1 | B |
| rs913188841 | g.75041242C>G | Splice region | 0.5 | 0.1 | B | |
| CYP2C8 | rs11572078 | g.96827126dup | Splice region | 17.4 | 16.8 | B |
| rs2071426 | g.5932A>G | Splice donor | 23.9 | 15.4 | D | |
| CYP2D6 | rs3892097 (CYP2D6*4) | g.6866G>A | Splice acceptor | 2.2 | 11.1 | D |
| CYP3A5 | rs776746 (CYP3A5*3) | g.12083G>A | Splice acceptor | 49.6 | 20.8 | D |
| SLC15A1 | rs8187827 | g.99354731T>C | Splice region | 0.5 | 1.4 | B |
| SLC22A1 | rs35854239 | c.1275_1276del | Splice acceptor | 45.7 | NR | D |
| SLCO1B1 | rs77271279 | g.21329832G>T | Splice donor | 0.9 | 0.2 | D |
| SLCO1B3 | rs3764009 | g.21013948C>T | Splice region | 16.3 | 79.0 | B |
| rs958332597 | g.21032366C>T | Splice region | 0.5 | 0.0 | B | |
| Frameshift and in-frame variants | ||||||
| ABCC1 | Novel | c.66del | Frameshift variant | 0.5 | NR | D |
| CYP2D6 | rs5030656 | c.88_690del | In-frame deletion | 0.5 | 1.2 | LD |
| c.54del | Frameshift truncation | 1.1 | 0.4 | D | ||
| CYP3A5 | rs200579169 | c.2dup | Frameshift truncation | 0.4 | 0.4 | D |
| rs41303343 | c.1035dup | Frameshift variant | 1.8 | 0.4 | D | |
| rs547253411 | c.372del | Frameshift truncation | 0.4 | 0.03 | D | |
| SLC22A1 | rs72552763 | c.1258_1260del | Disruptive in-frame deletion | 18.5 | 24.3 | LD |
| SLCO1B3 | rs780598056 | c.333del | Frameshift truncation | 0.5 | 0.0 | D |
| rs558592800 | c.19_120insAATT | Frameshift elongation | 0.5 | 0.01 | D | |
| SLCO2B1 | rs60113013 | c._14del | In-frame insertion | 1.6 | 3.1 | LD |
| Variant | Allele | n | β | SE | p-Value |
|---|---|---|---|---|---|
| CYP2C8 rs2071426 g.5932A>G | G allele | 92 | 2.8 | 3.8 | 0.456 |
| CYP3A5*3 rs776746 g.12083G>A | A allele | 114 | 12.9 | 7.7 | 0.096 |
| ABCC1 rs45511401 c.2012G>T | T allele | 92 | −14.4 | 6.8 | 0.038 |
| SLC22A1 rs72552763 c.1260_1262del | Deletion | 92 | −1.48 | 4.1 | 0.718 |
| SLCO1B1 rs4149056 c.521T>C | C allele | 114 | −4.7 | 4.4 | 0.288 |
| SLCO1B3 rs60140950 c.767G>C | C allele | 92 | −8.2 | 4.5 | 0.070 |
| Variable | No SRAE, % (90) | SRAE, % (24) | OR (95% CI) | p-Value | |
|---|---|---|---|---|---|
| CYP2C8*4 rs1058930 | A allele | 45.5 (35) | 35.7 (5) | 0.70 (0.19–2.37) | 0.574 |
| CYP2C9*2 rs1799853 | T allele | 16.9 (15) | 12.5 (3) | 0.54 (0.1–2.2) | 0.428 |
| CYP2C9*9 rs2256871 | G allele | 2.2 (2) | 12.5 (3) | 3.03 (0.35–29.74) | 0.309 |
| CYP3A5*6 rs10264272 | A allele | 3.4 (3) | 4.2 (1) | 1.34 (0.06–13.48) | 0.817 |
| CYP3A5 rs6977165 | G allele | 11.2 (10) | 12.5 (3) | 1.11 (0.22–4.44) | 0.886 |
| CYP3A5*3 rs776746 | A allele | 93.3 (83) | 95.8 (23) | 2.7 (0.33–60.01) | 0.418 |
| ABCC1 rs45511401 | T allele | 6.5 (5) | 14.3 (2) | 1.65 (0.2–9.46) | 0.594 |
| ABCC2 rs17216317 | T allele | 5.2 (4) | 14.3 (2) | 6.12 (0.72–41.6) | 0.067 |
| ABCC2 rs8187692 | T allele | 5.2 (4) | 7.1 (1) | 1.28 (0.06–11.08) | 0.841 |
| ABCC3 rs11568591 | A allele | 13 (10) | 14.3 (2) | 0.72 (0.07–4.06) | 0.734 |
| SLC22A1 rs35888596 | A allele | 3.9 (3) | 7.1 (1) | 3.44 (0.16–32.63) | 0.317 |
| SLC22A1 rs35854239 | Deletion | 37.7 (29) | 14.3 (2) | 0.27 (0.04–1.19) | 0.122 |
| SLCO1B1*5 rs4149056 | C allele | 21.3 (19) | 25.0 (6) | 1.23 (0.36–3.85) | 0.727 |
| SLCO1B1 rs59502379 | C allele | 3.4 (3) | 4.2 (1) | 2.4 (0.11–22.59) | 0.479 |
| SLCO1B3 rs60140950 | C allele | 26 (20) | 14.3 (2) | 0.36 (0.05–1.68) | 0.252 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagli-Hernandez, C.; Borges, J.B.; Marçal, E.d.S.R.; de Freitas, R.C.C.; Mori, A.A.; Gonçalves, R.M.; Faludi, A.A.; de Oliveira, V.F.; Ferreira, G.M.; Bastos, G.M.; et al. Genetic Variant ABCC1 rs45511401 Is Associated with Increased Response to Statins in Patients with Familial Hypercholesterolemia. Pharmaceutics 2022, 14, 944. https://doi.org/10.3390/pharmaceutics14050944
Dagli-Hernandez C, Borges JB, Marçal EdSR, de Freitas RCC, Mori AA, Gonçalves RM, Faludi AA, de Oliveira VF, Ferreira GM, Bastos GM, et al. Genetic Variant ABCC1 rs45511401 Is Associated with Increased Response to Statins in Patients with Familial Hypercholesterolemia. Pharmaceutics. 2022; 14(5):944. https://doi.org/10.3390/pharmaceutics14050944
Chicago/Turabian StyleDagli-Hernandez, Carolina, Jéssica Bassani Borges, Elisangela da Silva Rodrigues Marçal, Renata Caroline Costa de Freitas, Augusto Akira Mori, Rodrigo Marques Gonçalves, Andre Arpad Faludi, Victor Fernandes de Oliveira, Glaucio Monteiro Ferreira, Gisele Medeiros Bastos, and et al. 2022. "Genetic Variant ABCC1 rs45511401 Is Associated with Increased Response to Statins in Patients with Familial Hypercholesterolemia" Pharmaceutics 14, no. 5: 944. https://doi.org/10.3390/pharmaceutics14050944