
Deciphering Molecular Mechanisms Involved in Salinity Tolerance in Guar (Cyamopsis tetragonoloba (L.) Taub.) Using Transcriptome Analyses


 ,
,
Abstract
:1. Introduction
2. Results
2.1. De Novo Transcript Assembly and Functional Annotation
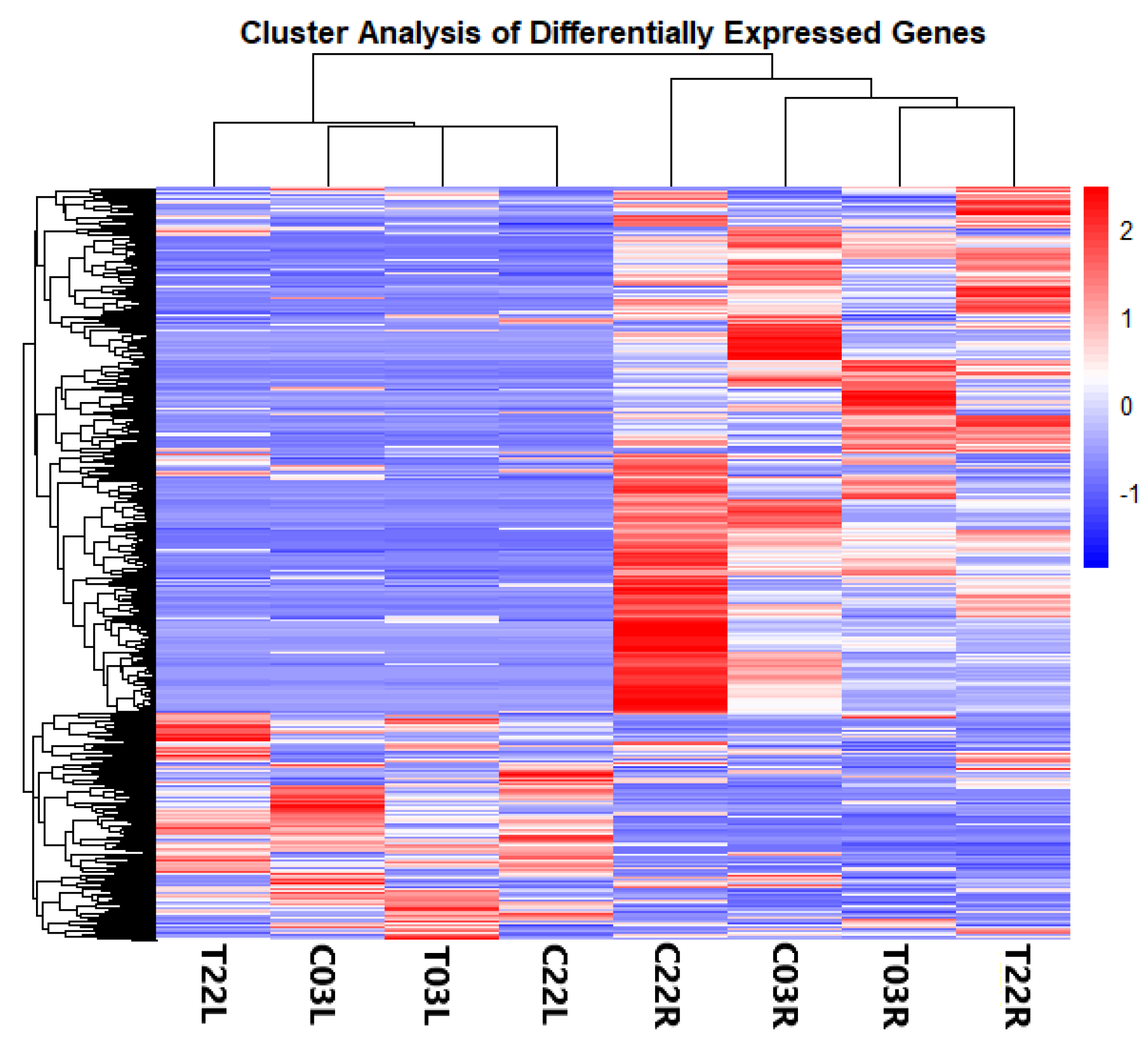
2.2. Differentially Expressed Genes (DEGs) in Response to Salt Stress
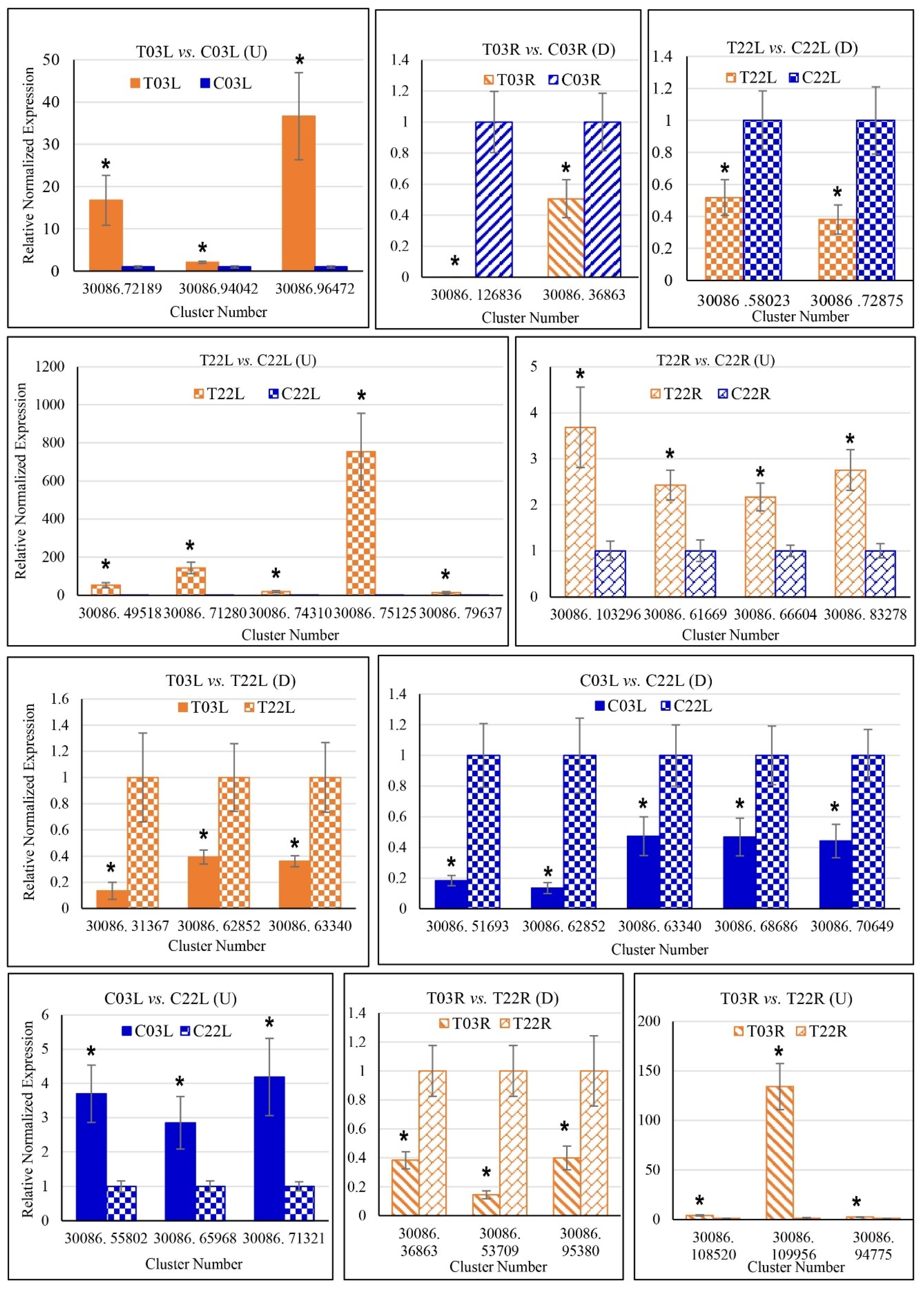
2.3. Verification of DEGs Using Quantitative Reverse Transcription PCR (qRT-PCR)
2.4. Gene Ontology (GO) Enrichment Analysis of DEGs
2.5. KEGG Enrichment Analysis of DEGs
2.6. DEGs Associated with Stress Pathways
2.6.1. Hormonal Signaling
2.6.2. Calcium Signaling
2.6.3. Redox Signaling
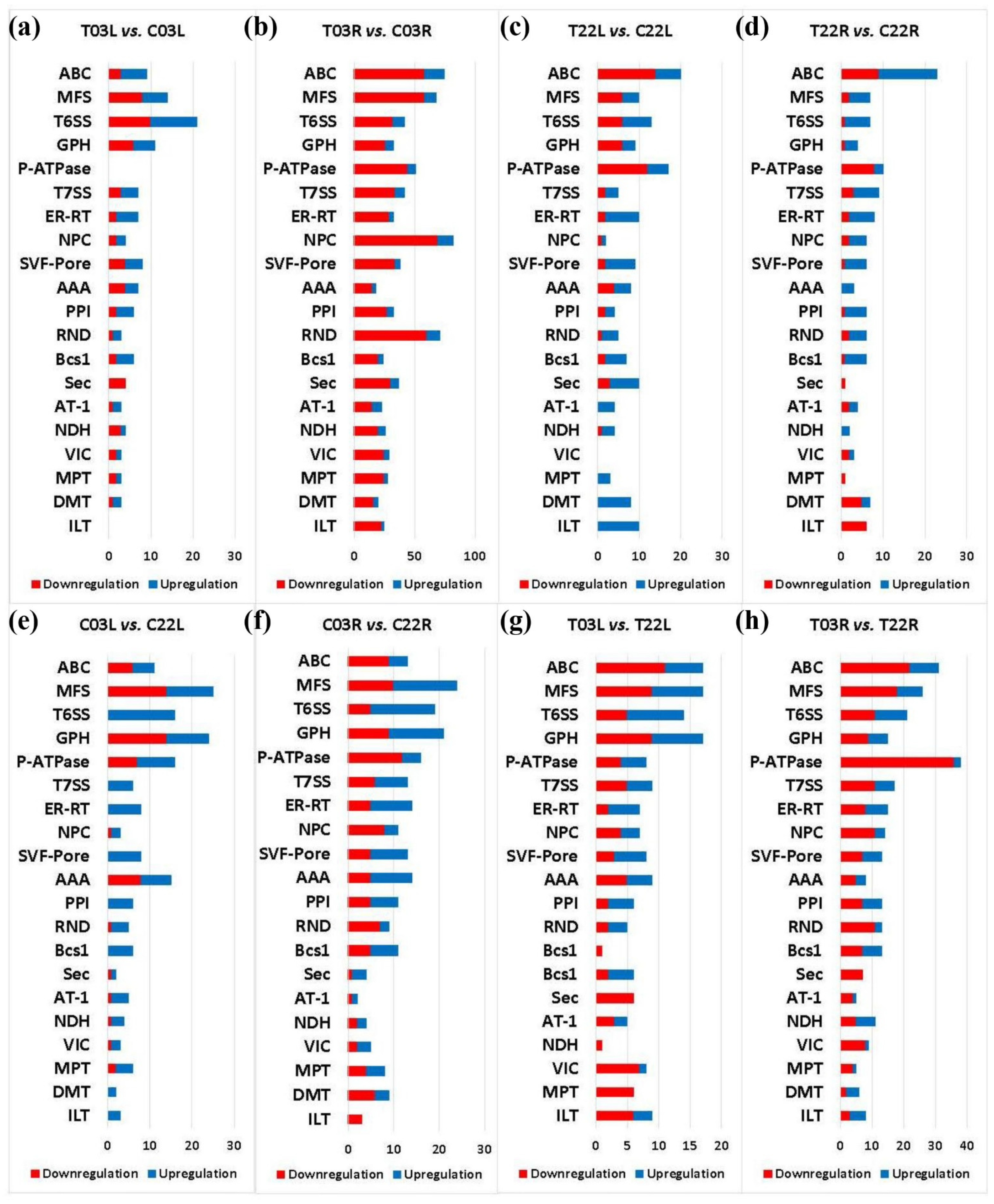
2.7. DEGs Associated with Transporters
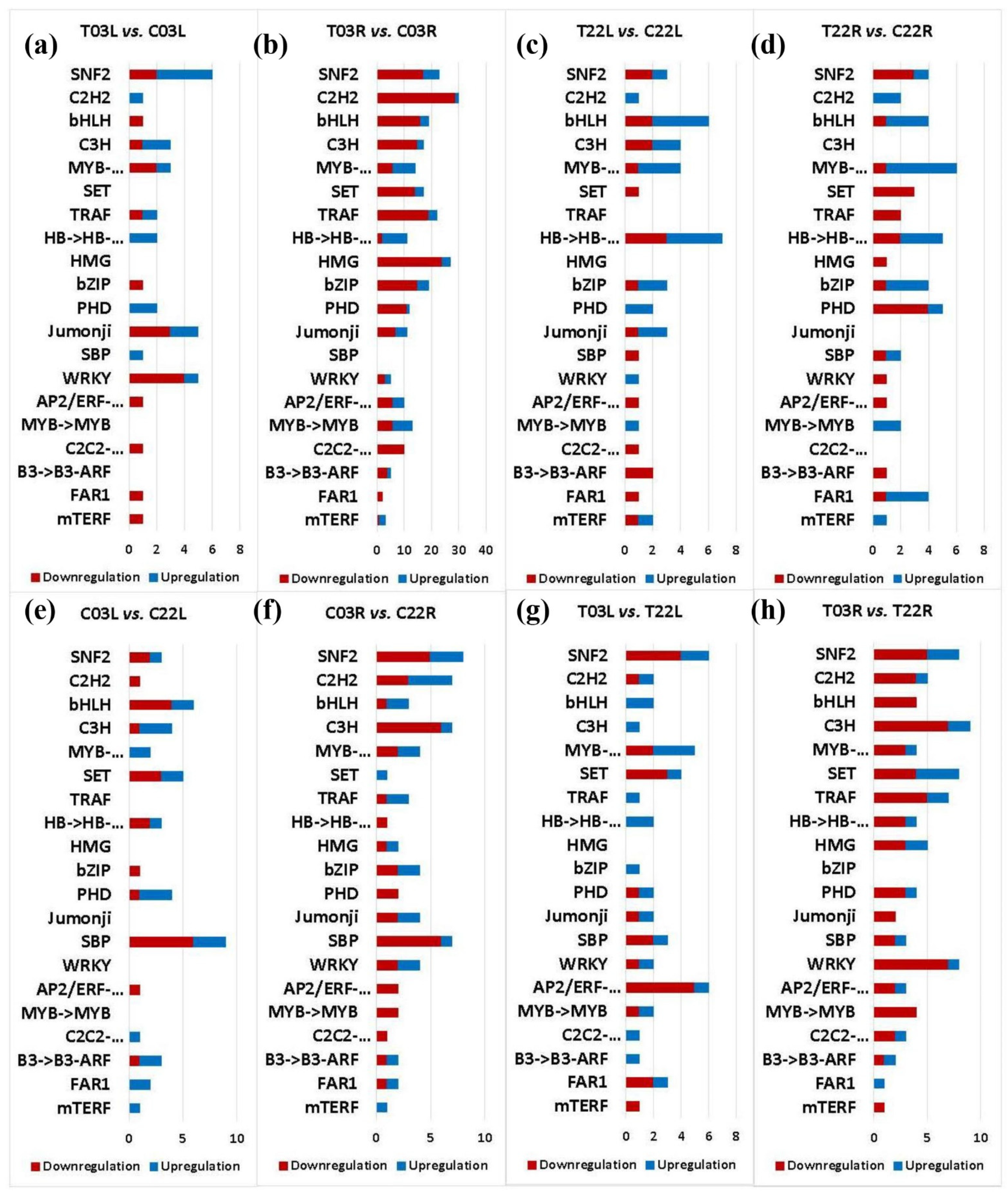
2.8. DEGs Associated with Transcription Factors/Regulators
3. Discussion
4. Materials and Methods
4.1. Plant Material and Salt Treatment
4.2. RNA Extraction and Transcript Sequencing
4.3. De Novo Transcriptome Assembly and Functional Annotation
4.4. Differential Gene Expression Analysis
4.5. Gene Ontology Annotation and Enrichment Analysis of DEGs
4.6. KEGG Pathway Enrichment Analysis of DEGs
4.7. Functional Analysis and Visualization
4.8. Transporter Analysis
4.9. Transcription Factor and Regulator Analyses
4.10. Quantitative Reverse Transcription PCR (qRT-PCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Hassani, A.; Azapagic, A.; Shokri, N. Global predictions of primary soil salinization under changing climate in the 21st century. Nat. Commun. 2021, 12, 6663. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Motos, J.R.; Ortuño, M.F.; Bernal-Vicente, A.; Diaz-Vivancos, P.; Sanchez-Blanco, M.J.; Hernandez, J.A. Plant responses to salt stress: Adaptive mechanisms. Agronomy 2017, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Isayenkov, S.V.; Maathuis, F.J.M. Plant salinity stress: Many unanswered questions remain. Front. Plant Sci. 2019, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- van Zelm, E.; Zhang, Y.; Testerink, C. Salt tolerance mechanisms of plants. Annu. Rev. Plant Biol. 2020, 71, 403–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Kim, W.Y.; Yun, D.J. A new insight of salt stress signaling in plant. Mol. Cells 2016, 39, 447–459. [Google Scholar] [CrossRef]
- Acharya, B.R.; Sandhu, D.; Ferreira, J.F.S. Physiological, morphological, biochemical, and genetic responses of alfalfa to salinity. In The Alfalfa Genome; Yu, L.-X., Kole, C., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 145–157. [Google Scholar]
- Jiang, Z.; Zhou, X.; Tao, M.; Yuan, F.; Liu, L.; Wu, F.; Wu, X.; Xiang, Y.; Niu, Y.; Liu, F.; et al. Plant cell-surface GIPC sphingolipids sense salt to trigger Ca2+ influx. Nature 2019, 572, 341–346. [Google Scholar] [CrossRef]
- Zhao, C.; William, D.; Sandhu, D. Isolation and characterization of Salt Overly Sensitive family genes in spinach. Physiol. Plant. 2021, 171, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Quintero, F.J.; Martinez-Atienza, J.; Villalta, I.; Jiang, X.; Kim, W.Y.; Ali, Z.; Fujii, H.; Mendoza, I.; Yun, D.J.; Zhu, J.K.; et al. Activation of the plasma membrane Na/H antiporter Salt-Overly-Sensitive 1 (SOS1) by phosphorylation of an auto-inhibitory C-terminal domain. Proc. Natl. Acad. Sci. USA 2011, 108, 2611–2616. [Google Scholar] [CrossRef] [Green Version]
- Manishankar, P.; Wang, N.; Köster, P.; Alatar, A.A.; Kudla, J. Calcium signaling during salt stress and in the regulation of ion homeostasis. J. Exp. Bot. 2018, 69, 4215–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Zhang, H.; Sun, L.; Jiao, Y.; Zhang, G.; Miao, C.; Hao, F. NADPH oxidase AtrbohD and AtrbohF function in ROS-dependent regulation of Na+/K+ homeostasis in Arabidopsis under salt stress. J. Exp. Bot. 2011, 63, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Hasanuzzaman, M.; Bhuyan, M.; Zulfiqar, F.; Raza, A.; Mohsin, S.M.; Mahmud, J.A.; Fujita, M.; Fotopoulos, V. Reactive oxygen species and antioxidant defense in plants under abiotic stress: Revisiting the crucial role of a universal defense regulator. Antioxidants 2020, 9, 681. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Noctor, G. Ascorbate and glutathione: The heart of the redox hub. Plant Physiol. 2011, 155, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Yu, Z.; Duan, X.; Luo, L.; Dai, S.; Ding, Z.; Xia, G. How plant hormones mediate salt stress responses. Trends Plant Sci. 2020, 25, 1117–1130. [Google Scholar] [CrossRef]
- Kaundal, R.; Duhan, N.; Acharya, B.R.; Pudussery, M.V.; Ferreira, J.F.S.; Suarez, D.L.; Sandhu, D. Transcriptional profiling of two contrasting genotypes uncovers molecular mechanisms underlying salt tolerance in alfalfa. Sci. Rep. 2021, 11, 5210. [Google Scholar] [CrossRef]
- Zhao, C.; Sandhu, D.; Ferreira, J.F.S. Transcript analysis of two spinach cultivars reveals the complexity of salt tolerance mechanisms. ACS Agric. Sci. Technol. 2021, 1, 64–75. [Google Scholar] [CrossRef]
- Acharya, B.; Sandhu, D.; Dueñas, C.; Dueñas, M.; Pudussery, M.; Kaundal, A.; Ferreira, J.F.S.; Suarez, D.; Skaggs, T. Morphological, physiological, biochemical, and transcriptome studies reveal the importance of transporters and stress signaling pathways during salinity stress in Prunus. Sci. Rep. 2021. [Google Scholar] [CrossRef]
- Brini, F.; Masmoudi, K. Ion transporters and abiotic stress tolerance in Plants. ISRN Mol. Biol. 2012, 2012, 927436. [Google Scholar] [CrossRef]
- Apse, M.P.; Blumwald, E. Na+ transport in plants. FEBS Lett. 2007, 581, 2247–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.-L.; Flowers, T.J.; Wang, S.-M. Mechanisms of sodium uptake by roots of higher plants. Plant Soil 2009, 326, 45. [Google Scholar] [CrossRef]
- Golldack, D.; Lüking, I.; Yang, O. Plant tolerance to drought and salinity: Stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant Cell Rep. 2011, 30, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Fernando, V.C.D. Major transcription factor families involved in salinity stress tolerance in plants. In Transcription Factors for Abiotic Stress Tolerance in Plants; Wani, S.H., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 99–109. [Google Scholar]
- Hymowitz, T.; Upadhya, M.D. The Chromosome number of Cyamopsis serrata. Schinz. Curr. Sci. 1963, 32, 427–428. [Google Scholar]
- Alshameri, A.; Al-Qurainy, F.; Gaafar, A.R.; Khan, S.; Nadeem, M.; Alansi, S. Identification of heat-responsive genes in guar [Cyamopsis tetragonoloba (L.) Taub]. Int. J. Genom. 2020, 2020, 3126592. [Google Scholar] [CrossRef]
- Abidi, N.; Liyanage, S.; Auld, D.; Imel, R.K.; Norman, L.; Grover, K.; Angadi, S.; Singla, S.; Trostle, C. Challenges and opportunities for increasing guar production. In Hydraulic Fracturing Impacts and Technologies; Uddameri, V., Morse, A., Tindle, K.J., Eds.; CRC Press: Boca Raton, FL, USA, 2015; pp. 207–226. [Google Scholar]
- Adams, C.B.; Boote, K.J.; Shrestha, R.; MacMillan, J.; Hinson, P.O.; Trostle, C. Growth stages and developmental patterns of guar. Agron. J. 2020, 112, 4990–5001. [Google Scholar] [CrossRef]
- Singla, S.; Grover, K.; Angadi, S.V.; Schutte, B.; VanLeeuwen, D. Guar stand establishment, physiology, and yield responses to planting date in southern New Mexico. Agron. J. 2016, 108, 2289–2300. [Google Scholar] [CrossRef]
- Sharma, G.; Sharma, S.; Kumar, A.; Al-Muhtaseb, A.H.; Naushad, M.; Ghfar, A.A.; Mola, G.T.; Stadler, F.J. Guar gum and its composites as potential materials for diverse applications: A review. Carbohydr. Polym. 2018, 199, 534–545. [Google Scholar] [CrossRef]
- Sandhu, D.; Pallete, A.; Pudussery, M.V.; Grover, K.K. Contrasting responses of guar genotypes shed light on multiple component traits of salinity tolerance mechanisms. Agronomy 2021, 11, 1068. [Google Scholar] [CrossRef]
- Suthar, J.D.; Rajpar, I.; Ganjegunte, G.K.; Shah, Z.-U.-H. Evaluation of guar (Cyamopsis tetragonoloba L.) genotypes performance under different irrigation water salinity levels: Growth parameters and seed yield. Ind. Crops Prod. 2018, 123, 247–253. [Google Scholar] [CrossRef]
- Suthar, J.D.; Rajpar, I.; Ganjegunte, G.K.; Shah, Z.U.H.; Niu, G.; Grover, K. Germination, growth, and ion uptake of 15 Guar accessions under elevated salinity. Agrosyst. Geosci. Environ. 2019, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Teolis, I.; Liu, W.; Peffley, E.B. Salinity effects on seed germination and plant growth of guar. Crop Sci. 2009, 49, 637–642. [Google Scholar] [CrossRef]
- Khator, K.; Mahawar, L.; Shekhawat, G.S. NaCl induced oxidative stress in legume crops of Indian Thar Desert: An insight in the cytoprotective role of HO1, NO and antioxidants. Physiol. Mol. Biol. Plants 2020, 26, 51–62. [Google Scholar] [CrossRef]
- Rasheed, M.J.Z.; Ahmad, K.; Ashraf, M.; Qurainy, F.A.; Khan, S.; Athar, H.U.R. Screening of diverse local germplasm of guar (Cyamposis tetragonoloba (L.) taub) for salt tolerance: A possible approach to utilize salt-affected soils. Pak. J. Bot. 2015, 47, 1721–1726. [Google Scholar]
- Ghaffarian, M.R.; Yadavi, A.; Movahhedi Dehnavi, M.; Dabbagh Mohammadi Nassab, A.; Salehi, M. Improvement of physiological indices and biological yield by intercropping of Kochia (Kochia scoparia), Sesbania (Sesbania aculeata) and Guar (Cyamopsis tetragonoliba) under the salinity stress of irrigation water. Physiol. Mol. Biol. Plants 2020, 26, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Francois, L.E.; Donovan, T.J.; Maas, E.V. Salinity effects on emergence, vegetative growth, and seed yield of guar. Agron. J. 1990, 82, 587–592. [Google Scholar] [CrossRef]
- Ashraf, M.Y.; Akhtar, K.; Sarwar, G.; Ashraf, M. Role of the rooting system in salt tolerance potential of different guar accessions. Agron. Sustain. Dev. 2005, 25, 243–249. [Google Scholar] [CrossRef]
- Tanwar, U.K.; Pruthi, V.; Randhawa, G.S. RNA-Seq of guar (Cyamopsis tetragonoloba, L. Taub.) leaves: De novo transcriptome assembly, functional annotation and development of genomic resources. Front. Plant Sci. 2017, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Rawal, H.C.; Kumar, S.; Mithra, S.V.A.; Solanke, A.U.; Nigam, D.; Saxena, S.; Tyagi, A.; Sureshkumar, V.; Yadav, N.R.; Kalia, P.; et al. High quality unigenes and microsatellite markers from tissue specific transcriptome and development of a database in clusterbean (Cyamopsis tetragonoloba, L. Taub). Genes 2017, 8, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, A.; Nigam, D.; Mithra, S.V.A.; Solanke, A.U.; Singh, N.K.; Sharma, T.R.; Gaikwad, K. Genome-wide discovery of tissue-specific miRNAs in clusterbean (Cyamopsis tetragonoloba) indicates their association with galactomannan biosynthesis. Plant Biotechnol. J. 2018, 16, 1241–1257. [Google Scholar] [CrossRef] [Green Version]
- Thakur, O.; Randhawa, G.S. Identification and characterization of SSR, SNP and InDel molecular markers from RNA-Seq data of guar (Cyamopsis tetragonoloba, L. Taub.) roots. BMC Genom. 2018, 19, 951. [Google Scholar] [CrossRef] [Green Version]
- Al-Qurainy, F.; Alshameri, A.; Gaafar, A.-R.; Khan, S.; Nadeem, M.; Alameri, A.A.; Tarroum, M.; Ashraf, M. Comprehensive stress-based de novo transcriptome assembly and annotation of Guar (Cyamopsis tetragonoloba (L.) Taub.): An important industrial and forage crop. Int. J. Genom. 2019, 2019, 7295859. [Google Scholar] [CrossRef] [Green Version]
- Alshameri, A.; Al-Qurainy, F.; Gaafar, A.R.; Khan, S.; Nadeem, M.; Alansi, S.; Shaikhaldein, H.O.; Salih, A.M. Identification of differentially expressed drought-responsive genes in guar [Cyamopsis tetragonoloba (L.) Taub]. Int. J. Genom. 2020, 2020, 4147615. [Google Scholar] [CrossRef]
- Hu, H.; Wang, H.; Zhang, Y.; Kan, B.; Ding, Y.; Huang, J. Characterization of genes in guar gum biosynthesis based on quantitative RNA-sequencing in guar bean (Cyamopsis tetragonoloba). Sci. Rep. 2019, 9, 10991. [Google Scholar] [CrossRef] [Green Version]
- Rajaprakasam, S.; Rahman, H.; Karunagaran, S.; Bapu, K.; Kulandivelu, G.; Kambale, R.; Ramanathan, V.; Muthurajan, R. Comparative transcriptome and metabolome profiling in the maturing seeds of contrasting cluster bean (Cyamopsis tetragonoloba L. Taub) cultivars identified key molecular variations leading to increased gum accumulation. Gene 2021, 791, 145727. [Google Scholar] [CrossRef] [PubMed]
- Dubrovina, A.S.; Kiselev, K.V. The role of calcium-dependent protein kinase genes VaCPK1 and VaCPK26 in the response of Vitis amurensis (in vitro) and Arabidopsis thaliana (in vivo) to abiotic Stresses. Russ. J. Genet. 2019, 55, 319–329. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, X.; Yang, Y.; Li, X.; Cheng, Y.; Yang, Y. Expression of Stipa purpurea SpCIPK26 in Arabidopsis thaliana enhances salt and drought tolerance and regulates abscisic acid signaling. Int. J. Mol. Sci. 2016, 17, 966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Cai, X.T.; Du, J.; Zhao, T.L.; Wang, P.F.; Zhao, P.X.; Liu, R.; Xie, Q.; Cao, X.F.; Xiang, C.B. PARAQUAT TOLERANCE3 is an E3 ligase that switches off activated oxidative response by targeting histone-modifying protein methyltransferase4b. PLoS Genet. 2016, 12, e1006332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.C.; Augé, R.M.; Dong, C.; Cheng, Z.M. Increased salt tolerance with overexpression of cation/proton antiporter 1 genes: A meta-analysis. Plant Biotechnol. J. 2017, 15, 162–173. [Google Scholar] [CrossRef]
- Kim, W.Y.; Ali, Z.; Park, H.J.; Park, S.J.; Cha, J.Y.; Perez-Hormaeche, J.; Quintero, F.J.; Shin, G.; Kim, M.R.; Qiang, Z.; et al. Release of SOS2 kinase from sequestration with GIGANTEA determines salt tolerance in Arabidopsis. Nat. Commun. 2013, 4, 1352. [Google Scholar] [CrossRef] [Green Version]
- Ke, Q.; Kim, H.S.; Wang, Z.; Ji, C.Y.; Jeong, J.C.; Lee, H.-S.; Choi, Y.-I.; Xu, B.; Deng, X.; Yun, D.-J.; et al. Down-regulation of GIGANTEA-like genes increases plant growth and salt stress tolerance in poplar. Plant Biotechnol. J. 2017, 15, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Chhillar, H.; Chopra, P.; Khanna, R.R.; Khan, M.I.R. Potassium: A track to develop salinity tolerant plants. Plant Physiol. Biochem. 2021, 167, 1011–1023. [Google Scholar] [CrossRef]
- Ahn, S.J.; Shin, R.; Schachtman, D.P. Expression of KT/KUP genes in Arabidopsis and the role of root hairs in K+ uptake. Plant Physiol. 2004, 134, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Wei, Y.; Sun, K.; Wu, J.; Wang, Y.; Wu, K. The maize AAA-type protein SKD1 confers enhanced salt and drought stress tolerance in transgenic tobacco by interacting with Lyst-interacting protein 5. PLoS ONE 2013, 8, e69787. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Jin, J.Y.; Alejandro, S.; Martinoia, E.; Lee, Y. Overexpression of AtABCG36 improves drought and salt stress resistance in Arabidopsis. Physiol. Plant. 2010, 139, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Niño-González, M.; Novo-Uzal, E.; Richardson, D.N.; Barros, P.M.; Duque, P. More transporters, more substrates: The Arabidopsis Major Facilitator Superfamily revisited. Mol. Plant 2019, 12, 1182–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinders, A.; Ward, J.M. Functional characterization of the alpha-glucoside transporter Sut1p from Schizosaccharomyces pombe, the first fungal homologue of plant sucrose transporters. Mol. Microbiol. 2001, 39, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Beyhl, D.; Marten, I.; Wormit, A.; Neuhaus, E.; Poschet, G.; Büttner, M.; Schneider, S.; Sauer, N.; Hedrich, R. Proton-driven sucrose symport and antiport are provided by the vacuolar transporters SUC4 and TMT1/2. Plant J. 2011, 68, 129–136. [Google Scholar] [CrossRef]
- Gong, X.; Liu, M.; Zhang, L.; Ruan, Y.; Ding, R.; Ji, Y.; Zhang, N.; Zhang, S.; Farmer, J.; Wang, C. Arabidopsis AtSUC2 and AtSUC4, encoding sucrose transporters, are required for abiotic stress tolerance in an ABA-dependent pathway. Physiol. Plant. 2015, 153, 119–136. [Google Scholar] [CrossRef]
- Blumwald, E.; Aharon, G.S.; Apse, M.P. Sodium transport in plant cells. Biochim. Biophys. Acta 2000, 1465, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Lu, C.; Guo, J.; Qiao, Z.; Sui, N.; Qiu, N.; Wang, B. C2H2 zinc finger proteins: Master regulators of abiotic stress responses in plants. Front. Plant Sci. 2020, 11, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ji, X.; Nie, X.; Qu, M.; Zheng, L.; Tan, Z.; Zhao, H.; Huo, L.; Liu, S.; Zhang, B.; et al. Arabidopsis AtbHLH112 regulates the expression of genes involved in abiotic stress tolerance by binding to their E-box and GCG-box motifs. New Phytol. 2015, 207, 692–709. [Google Scholar] [CrossRef]
- Qian, Y.; Zhang, T.; Yu, Y.; Gou, L.; Yang, J.; Xu, J.; Pi, E. Regulatory mechanisms of bHLH transcription factors in plant adaptive responses to various abiotic stresses. Front. Plant Sci. 2021, 12, 677611. [Google Scholar] [CrossRef]
- Han, G.; Qiao, Z.; Li, Y.; Wang, C.; Wang, B. The roles of CCCH zinc-finger proteins in plant abiotic stress tolerance. Int. J. Mol. Sci. 2021, 22, 8327. [Google Scholar] [CrossRef]
- Li, J.; Han, G.; Sun, C.; Sui, N. Research advances of MYB transcription factors in plant stress resistance and breeding. Plant Signal. Behav. 2019, 14, 1613131. [Google Scholar] [CrossRef]
- Thouly, C.; Le Masson, M.; Lai, X.; Carles, C.C.; Vachon, G. Unwinding BRAHMA functions in plants. Genes 2020, 11, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Hong, Y.; Ren, Z.; Tang, K.; Zhang, H.; Zhu, J.-K.; Zhao, C. A role for PICKLE in the regulation of cold and salt stress tolerance in Arabidopsis. Front. Plant Sci. 2019, 10, 900. [Google Scholar] [CrossRef] [PubMed]
- Zou, B.; Sun, Q.; Zhang, W.; Ding, Y.; Yang, D.L.; Shi, Z.; Hua, J. The Arabidopsis chromatin-remodeling factor CHR5 regulates plant immune responses and nucleosome occupancy. Plant Cell Physiol. 2017, 58, 2202–2216. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zhang, L.; Yang, Z.; Wei, Y.; Dong, T. Identification and functional characterization of plant miRNA under salt stress shed light on salinity resistance improvement through miRNA manipulation in crops. Front. Plant Sci. 2021, 12, 665439. [Google Scholar] [CrossRef]
- Chen, T.; Cui, P.; Xiong, L. The RNA-binding protein HOS5 and serine/arginine-rich proteins RS40 and RS41 participate in miRNA biogenesis in Arabidopsis. Nucleic Acids Res. 2015, 43, 8283–8298. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Moon, S.-J.; Park, S.R.; Kim, B.-G.; Byun, M.-O. Elongation factor 1α from A. thaliana functions as molecular chaperone and confers resistance to salt stress in yeast and plants. Plant Sci. 2009, 177, 156–160. [Google Scholar] [CrossRef]
- Synthesis of complementary DNA. Nat. Methods 2005, 2, 151–152. [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable venn and euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rodriguez, P.; Riano-Pachon, D.M.; Correa, L.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, D822–D827. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, P.S.; Kaur, N.; Randhawa, G.S. Identification of reference genes for qRT-PCR gene expression studies during seed development and under abiotic stresses in Cyamopsis tetragonoloba. Crop Sci. 2019, 59, 252–265. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Groups | DEGs | Upregulated | Downregulated |
|---|---|---|---|---|
| Treatment vs. Control | T03L vs. C03L T03R vs. C03R T22L vs. C22L T22R vs. C22R | 853 8263 1070 1006 | 357 1805 644 511 | 496 6478 426 495 |
| Tolerant vs. Sensitive | C03L vs. C22L C03R vs. C22R T03L vs. T22L T03R vs. T22R | 1049 1355 1031 1727 | 541 583 430 538 | 508 772 601 1189 |
| Leaf vs. Root | C03L vs. C03R C22L vs. C22R T03L vs. T03R T22L vs. T22R | 41,540 35,936 21,383 27,383 | 10,259 8978 8168 8702 | 31,281 26,958 13,215 18,681 |
| ECiw | Ion Concentration in Mmolc L−1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | dS m−1 | NO3− | SO42− | Cl− | PO43− | CO3H− | Ca2+ | Mg2+ | Na+ | K+ |
| Control | 1.46 | 5.4 | 1.44 | 1.41 | 1.5 | 4.2 | 3.35 | 2.1 | 1.88 | 6.6 |
| Saline | 13.65 | 5.4 | 27.32 | 128.4 | 1.5 | 3.5 | 29.6 | 23 | 106.9 | 6.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acharya, B.R.; Sandhu, D.; Dueñas, C.; Ferreira, J.F.S.; Grover, K.K. Deciphering Molecular Mechanisms Involved in Salinity Tolerance in Guar (Cyamopsis tetragonoloba (L.) Taub.) Using Transcriptome Analyses. Plants 2022, 11, 291. https://doi.org/10.3390/plants11030291
Acharya BR, Sandhu D, Dueñas C, Ferreira JFS, Grover KK. Deciphering Molecular Mechanisms Involved in Salinity Tolerance in Guar (Cyamopsis tetragonoloba (L.) Taub.) Using Transcriptome Analyses. Plants. 2022; 11(3):291. https://doi.org/10.3390/plants11030291
Chicago/Turabian StyleAcharya, Biswa R., Devinder Sandhu, Christian Dueñas, Jorge F. S. Ferreira, and Kulbhushan K. Grover. 2022. "Deciphering Molecular Mechanisms Involved in Salinity Tolerance in Guar (Cyamopsis tetragonoloba (L.) Taub.) Using Transcriptome Analyses" Plants 11, no. 3: 291. https://doi.org/10.3390/plants11030291