
Differentially Expressed Conserved Plant Vegetative Phase-Change-Related microRNAs in Mature and Rejuvenated Silver Birch In Vitro Propagated Tissues

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification of Conserved miRNAs in Silver Birch
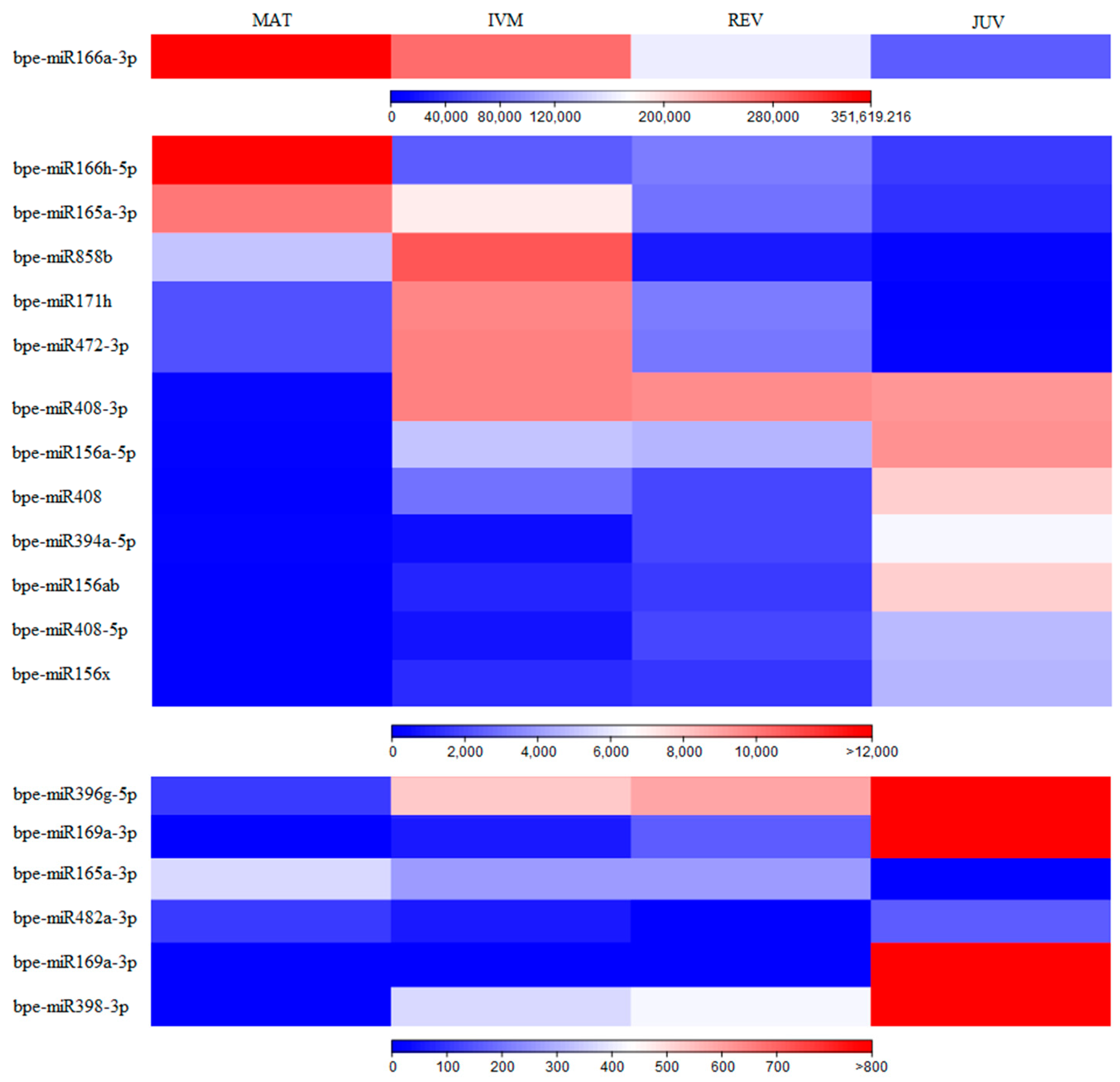
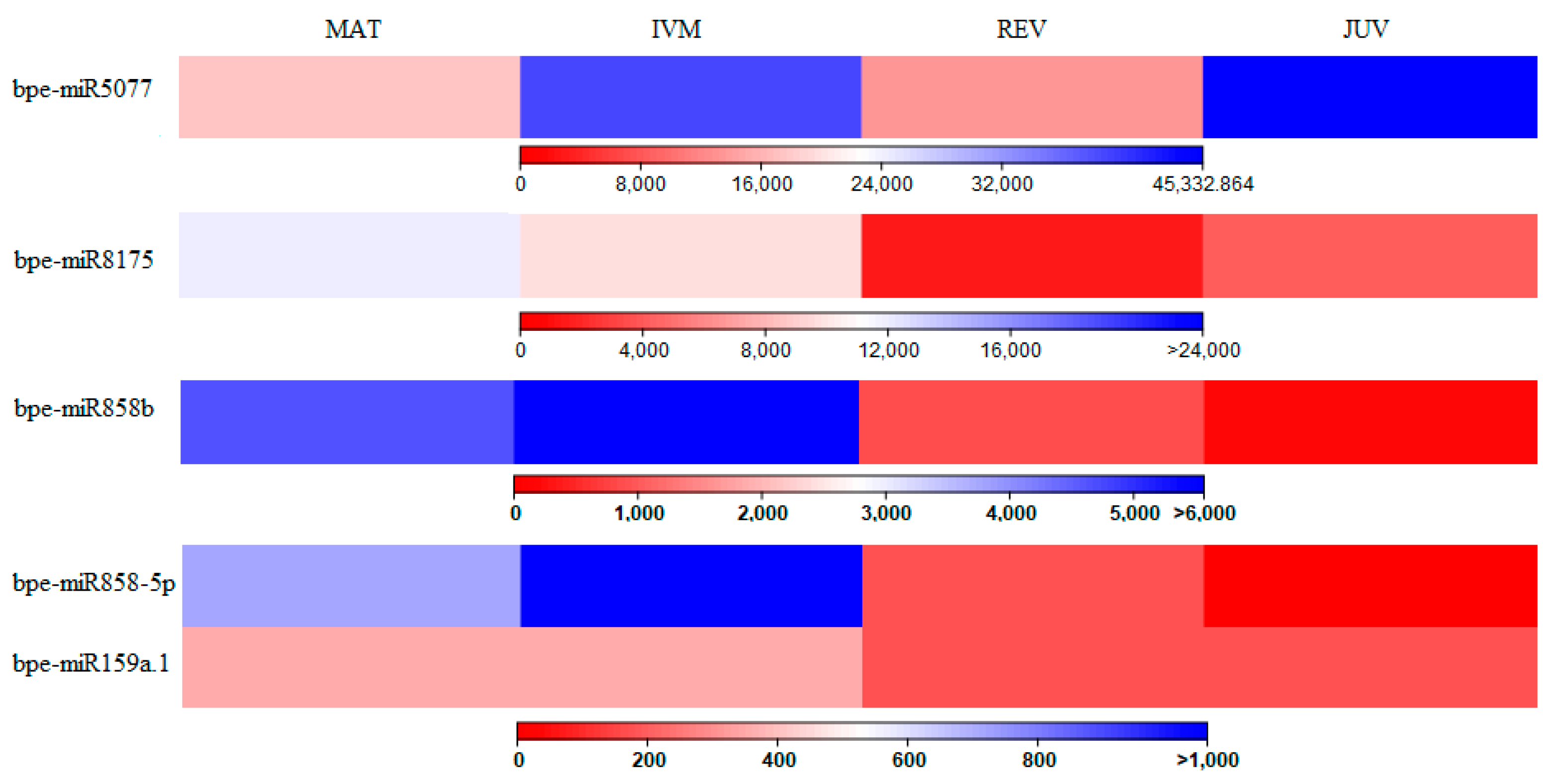
2.2. Differentially Expressed miRNAs
2.3. Identification of Potential miRNA Precursors for Differentially Expressed miRNAs
2.4. Expression Analysis of Precursor miRNAs Using qPCR
2.5. miRNA Target Gene Identification
3. Discussion
4. Materials and Methods
4.1. Plant Material and Extraction of RNA
4.2. RNA Quality Control
4.3. Small RNA Enrichment, Library Preparation and Sequencing Analysis
4.4. Conserved miRNA Identification
4.5. Identification of Differentially Expressed miRNAs
4.6. Target Gene Identification
4.7. Analysis of Expression of Precursor miRNAs Using qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Poethig, R.S. Vegetative Phase Change and Shoot Maturation in Plants. Curr. Top. Dev. Biol. 2013, 105, 125–152. [Google Scholar] [CrossRef]
- Poethig, R.S. The Past, Present, and Future of Vegetative Phase Change. Plant Physiol. 2010, 154, 541–544. [Google Scholar] [CrossRef]
- Werner, S.; Bartrina, I.; Schmülling, T. Cytokinin regulates vegetative phase change in Arabidopsis thaliana through the miR172/TOE1-TOE2 module. Nat. Commun. 2021, 12, 5816. [Google Scholar] [CrossRef]
- Yang, L.; Xu, M.; Koo, Y.; He, J.; Scott Poethig, R. Sugar promotes vegetative phase change in Arabidopsis thaliana by repressing the expression of MIR156A and MIR156C. eLife 2013, 2, e00260. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, C.; Zhou, C.M.; Zhang, T.Q.; Lian, H.; Sun, Y.; Wu, J.; Huang, J.; Wang, G.; Wang, J.W. Sugar is an endogenous cue for juvenile-to-adult phase transition in plants. eLife 2013, 2, e00269. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, Y.B.; Wang, H.S.; Feng, S.J.; Zhang, Y.T. Involvement of miR156 in the Regulation of Vegetative Phase Change in Plants. J. Am. Soc. Hortic. Sci. 2015, 140, 387–395. [Google Scholar] [CrossRef]
- Wang, J.W.; Park, M.Y.; Wang, L.J.; Koo, Y.; Chen, X.Y.; Weigel, D.; Poethig, R.S. MiRNA Control of Vegetative Phase Change in Trees. PLOS Genet. 2011, 7, e1002012. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.U.; Hayward, A.; Irihimovitch, V.; Fletcher, S.; Tanurdzic, M.; Pocock, A.; Beveridge, C.A.; Mitter, N. Juvenility and vegetative phase transition in tropical/subtropical tree crops. Front. Plant Sci. 2019, 10, 729. [Google Scholar] [CrossRef]
- Hiti-Bandaralage, J.; Hayward, A.; O’Brien, C.; Ahsan, U.; Gleeson, M.; Xue, Y.; Mitter, N. Phase Change Related microRNA Profiles in the Plant Regeneration Process of Avocado through Shoot-tip Culture. Ann. Adv. Agric. Sci. 2020, 4, 9–17. [Google Scholar] [CrossRef]
- Xing, L.; Zhang, D.; Li, Y.; Zhao, C.; Zhang, S.; Shen, Y.; An, N.; Han, M. Genome-wide identification of vegetative phase transition-associated microRNAs and target predictions using degradome sequencing in Malus hupehensis. BMC Genomics 2014, 15, 1–22. [Google Scholar] [CrossRef]
- Jia, X.L.; Chen, Y.K.; Xu, X.Z.; Shen, F.; Zheng, Q.B.; Du, Z.; Wang, Y.; Wu, T.; Xu, X.F.; Han, Z.H.; et al. miR156 switches on vegetative phase change under the regulation of redox signals in apple seedlings. Sci. Rep. 2017, 7, 14223. [Google Scholar] [CrossRef]
- Xing, S.; Salinas, M.; Höhmann, S.; Berndtgen, R.; Huijser, P. miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell 2010, 22, 3935–3950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, Y.; Li, Y. Plant rejuvenation: From phenotypes to mechanisms. Plant Cell Rep. 2020, 39, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Carrington, J.C.; Ambros, V. Role of MicroRNAs in Plant and Animal Development. Science 2003, 301, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Neutelings, G.; Fénart, S.; Lucau-Danila, A.; Hawkins, S. Identification and characterization of miRNAs and their potential targets in flax. J. Plant Physiol. 2012, 169, 1754–1766. [Google Scholar] [CrossRef]
- Bielewicz, D.; Dolata, J.; Zielezinski, A.; Alaba, S.; Szarzynska, B.; Szczesniak, M.W.; Jarmolowski, A.; Szweykowska-Kulinska, Z.; Karlowski, W.M. mirEX: A platform for comparative exploration of plant pri-miRNA expression data. Nucleic Acids Res. 2012, 40, D191–D197. [Google Scholar] [CrossRef]
- Tomasello, L.; Distefano, R.; Nigita, G.; Croce, C.M. The MicroRNA Family Gets Wider: The IsomiRs Classification and Role. Front. Cell Dev. Biol. 2021, 9, 668648. [Google Scholar] [CrossRef]
- Hynynen, J.; Niemistö, P.; Viherä-Aarnio, A.; Brunner, A.; Hein, S.; Velling, P. Silviculture of birch (Betula pendula Roth and Betula pubescens Ehrh.) in Northern Europe. Forestry 2010, 83, 103–119. [Google Scholar] [CrossRef]
- Gailis, A.; Karklina, A.; Purvinš, A.; Matisons, R.; Zeltinš, P.; Jansons, A. Effect of breeding on income at first commercial thinning in silver birch plantations. Forests 2020, 11, 327. [Google Scholar] [CrossRef]
- Ewald, D.; Naujoks, G.; Welander, M.; Zhu, L.H.; Hagqvist, R.; Salonen, M.; Harrison, A. Micropropagation and birch field trials. In Proceedings of the Workshop on High Quality Birch: Clonal Propagation and Wood Properties, Ronneby, Sweden, 27–28 August 2001; Swedish University of Agricultural Sciences, Department of Crop Science: Ronneby, Sweden, 2001; pp. 37–46. [Google Scholar]
- Zeltiņš, P.; Matisons, R.; Gailis, A.; Jansons, J.; Katrevičs, J.; Jansons, Ā. Genetic parameters of growth traits and stem quality of silver birch in a low-density clonal plantation. Forests 2018, 9, 52. [Google Scholar] [CrossRef]
- O’Dowd, N. The Improvement of Irish Birch. Phase 1: Selection of Individuals and Populations; COFORD: Dublin, Ireland, 2004. [Google Scholar]
- Salojärvi, J.; Smolander, O.P.; Nieminen, K.; Rajaraman, S.; Safronov, O.; Safdari, P.; Lamminmäki, A.; Immanen, J.; Lan, T.; Tanskanen, J.; et al. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat. Genet. 2017, 49, 904–912. [Google Scholar] [CrossRef]
- Potocki, L.; Karbarz, M.; Adamczyk-Grochala, J.; Kasprzyk, I.; Pawlina-Tyszko, K.; Lewinska, A.; Wnuk, M. Silver birch pollen-derived microRNAs promote NF-κB-mediated inflammation in human lung cells. Sci. Total Environ. 2021, 800, 149531. [Google Scholar] [CrossRef]
- Feng, S.; Xu, Y.; Guo, C.; Zheng, J.; Zhou, B.; Zhang, Y.; Ding, Y.; Zhang, L.; Zhu, Z.; Wang, H.; et al. Modulation of miR156 to identify traits associated with vegetative phase change in tobacco (Nicotiana tabacum). J. Exp. Bot. 2016, 67, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Guo, C.; Zhou, B.; Li, C.; Wang, H.; Zheng, B.; Ding, H.; Zhu, Z.; Peragine, A.; Cui, Y.; et al. Regulation of Vegetative Phase Change by SWI2/SNF2 Chromatin Remodeling ATPase BRAHMA. Plant Physiol. 2016, 172, 2416. [Google Scholar] [CrossRef] [PubMed]
- Krivmane, B.; Šņepste, I.; Škipars, V.; Yakovlev, I.; Fossdal, C.G.; Vivian-Smith, A.; Ruņgis, D. Identification and in Silico Characterization of Novel and Conserved MicroRNAs in Methyl Jasmonate-Stimulated Scots Pine (Pinus sylvestris L.) Needles. Forest 2020, 11, 384. [Google Scholar] [CrossRef]
- Krivmane, B.; Girgžde, E.; Samsone, I.; Ruņģis, D. Expression of juvenility related microRNAs and target genes during micropropagation of silver birch (Betula pendula Roth.). Plant Cell Tissue Organ Cult. 2022, 152, 455–469. [Google Scholar] [CrossRef]
- Tang, M.; Bai, X.; Niu, L.J.; Chai, X.; Chen, M.S.; Xu, Z.F. Mir172 Regulates Both Vegetative and Reproductive Development in the Perennial Woody Plant Jatropha Curcas. Plant Cell Physiol. 2018, 59, 2549–2563. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Szwerdszarf, D.; Abu-Abied, M.; Mordehaev, I.; Yaniv, Y.; Riov, J.; Arazi, T.; Sadot, E. Profiling microRNAs in Eucalyptus grandis reveals no mutual relationship between alterations in miR156 and miR172 expression and adventitious root induction during development. BMC Genomics 2014, 15, 524. [Google Scholar] [CrossRef]
- Hu, J.; Jin, J.; Qian, Q.; Huang, K.; Ding, Y. Small RNA and degradome profiling reveals miRNA regulation in the seed germination of ancient eudicot Nelumbo nucifera. BMC Genomics 2016, 17, 684. [Google Scholar] [CrossRef]
- Fouracre, J.P.; Poethig, R.S. The role of small RNAs in vegetative shoot development. Curr. Opin. Plant Biol. 2016, 29, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, Y.; Ponso, M.A.; Belén, F.; Vegetti, A.C.; Dotto, M.C. MicroRNA miR394 regulates flowering time in Arabidopsis thaliana. Plant Cell Rep. 2022, 41, 1375–1388. [Google Scholar] [CrossRef]
- Liebsch, D.; Palatnik, J.F. MicroRNA miR396, GRF transcription factors and GIF co-regulators: A conserved plant growth regulatory module with potential for breeding and biotechnology. Curr. Opin. Plant Biol. 2020, 53, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, Q.; Zuo, Z.-F.; Liu, L. MicroRNA398: A Master Regulator of Plant Development and Stress Responses. Int. J. Mol. Sci. 2022, 23, 10803. [Google Scholar] [CrossRef]
- Sakaguchi, J.; Watanabe, Y. miR165/166 and the development of land plants. Dev. Growth Differ. 2012, 54, 93–99. [Google Scholar] [CrossRef]
- Emery, J.F.; Floyd, S.K.; Alvarez, J.; Eshed, Y.; Hawker, N.P.; Izhaki, A.; Baum, S.F.; Bowman, J.L. Radial Patterning of Arabidopsis Shoots by Class III HD-ZIP and KANADI Genes. Curr. Biol. 2003, 13, 1768–1774. [Google Scholar] [CrossRef]
- Han, H.; Zhou, Y. Function and Regulation of microRNA171 in Plant Stem Cell Homeostasis and Developmental Programing. Int. J. Mol. Sci. 2022, 23, 2544. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, B.; Gao, C.; Zhang, H.; Wen, F.; Tao, L.; Fu, G.; Xiong, J. The Evolution and Functional Roles of miR408 and Its Targets in Plants. Int. J. Mol. Sci. 2022, 23, 530. [Google Scholar] [CrossRef] [PubMed]
- Solofoharivelo, M.C.; van der Walt, A.P.; Stephan, D.; Burger, J.T.; Murray, S.L. MicroRNAs in fruit trees: Discovery, diversity and future research directions. Plant Biol. 2014, 16, 856–865. [Google Scholar] [CrossRef]
- Liao, L.; Xie, B.; Guan, P.; Jiang, N.; Cui, J. New insight into the molecular mechanism of miR482/2118 during plant resistance to pathogens. Front. Plant Sci. 2022, 13, 1026762. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Upadhyaya, H. Aluminium Toxicity and Its Tolerance in Plant: A Review. J. Plant Biol. 2021, 64, 101–121. [Google Scholar] [CrossRef]
- Wu, J.; Wang, D.; Liu, Y.; Wang, L.; Qiao, X.; Zhang, S. Identification of miRNAs involved in pear fruit development and quality. BMC Genomics 2014, 15, 953. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Shen, C.-H.; Lin, W.-D.; Chu, H.-A.; Huang, B.-L.; Kuo, C.-I.; Yeh, K.-W.; Huang, L.-C.; Chang, I.-F. Small RNAs of Sequoia sempervirens during rejuvenation and phase change. Plant Biol. 2013, 15, 27–36. [Google Scholar] [CrossRef]
- Rubio-Piña, J.A.; Zapata-Pérez, O. Isolation of total RNA from tissues rich in polyphenols and polysaccharides of mangrove plants. Electron. J. Biotechnol. 2011, 14, 11. [Google Scholar] [CrossRef]
- Kulju, K.K.M.; Pekkinen, M.; Varvio, S. Twenty-three microsatellite primer pairs for Betula pendula (Betulaceae). Mol. Ecol. Notes 2004, 4, 471–473. [Google Scholar] [CrossRef]
- Dai, X.; Zhao, P.X. PsRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ruonala, R.; Rinne, P.L.H.; Baghour, M.; Moritz, T.; Tuominen, H.; Kangasjärvi, J. Transitions in the functioning of the shoot apical meristem in birch (Betula pendula) involve ethylene. Plant J. 2006, 46, 628–640. [Google Scholar] [CrossRef]
- Žiarovská, J.; Labajová, M.; Ražná, K.; Bežo, M.; Štefúnová, V.; Shevtsova, T.; Garkava, K.; Brindza, J. Changes in expression of BetV1 allergen of silver birch pollen in urbanized area of Ukraine. J. Environ. Sci. Health 2013, 48, 1479–1484. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krivmane, B.; Ruņģe, K.S.; Samsone, I.; Ruņģis, D.E. Differentially Expressed Conserved Plant Vegetative Phase-Change-Related microRNAs in Mature and Rejuvenated Silver Birch In Vitro Propagated Tissues. Plants 2023, 12, 1993. https://doi.org/10.3390/plants12101993
Krivmane B, Ruņģe KS, Samsone I, Ruņģis DE. Differentially Expressed Conserved Plant Vegetative Phase-Change-Related microRNAs in Mature and Rejuvenated Silver Birch In Vitro Propagated Tissues. Plants. 2023; 12(10):1993. https://doi.org/10.3390/plants12101993
Chicago/Turabian StyleKrivmane, Baiba, Kaiva Solvita Ruņģe, Ineta Samsone, and Dainis Edgars Ruņģis. 2023. "Differentially Expressed Conserved Plant Vegetative Phase-Change-Related microRNAs in Mature and Rejuvenated Silver Birch In Vitro Propagated Tissues" Plants 12, no. 10: 1993. https://doi.org/10.3390/plants12101993