
Characterization of Bio-Inspired Electro-Conductive Soy Protein Films

 ,
,  , , ,
, , ,  and
and 
Abstract
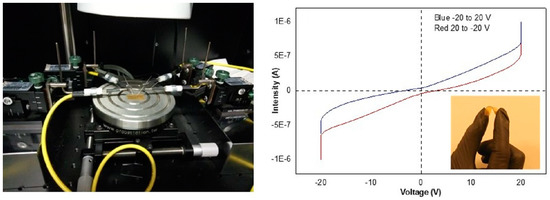
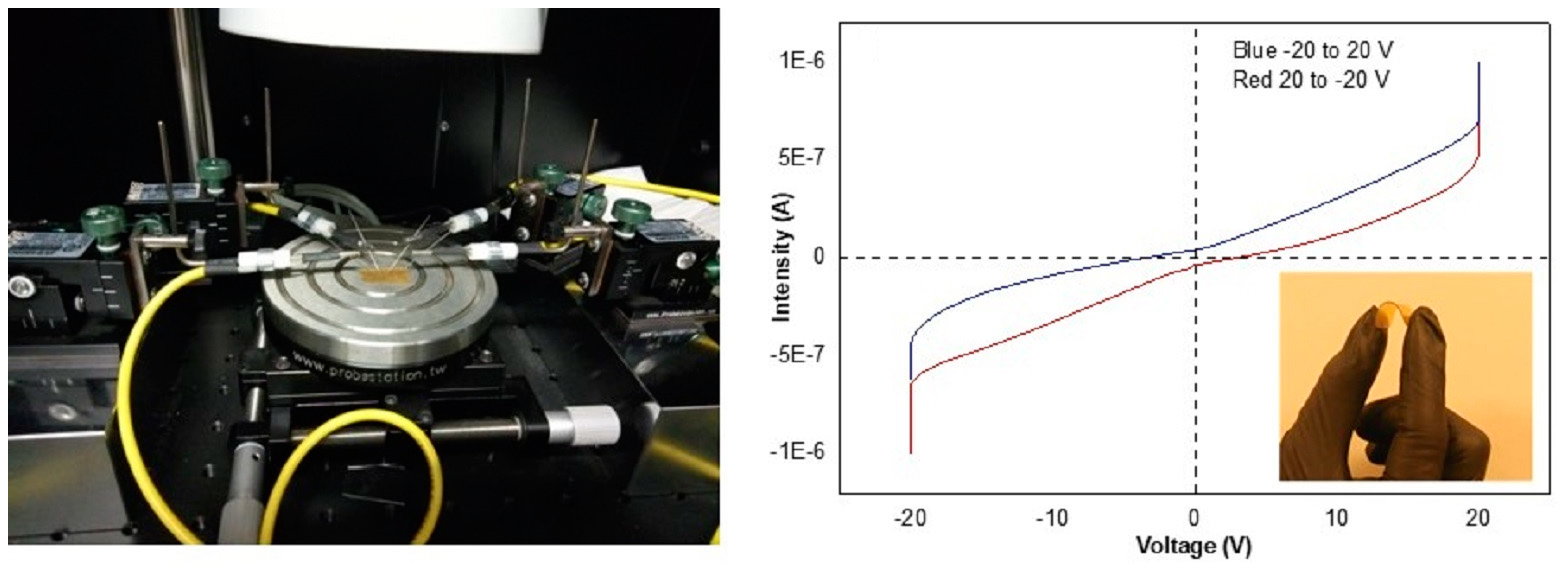
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Pellets Preparation
2.3. Film Preparation
2.4. Film Characterization
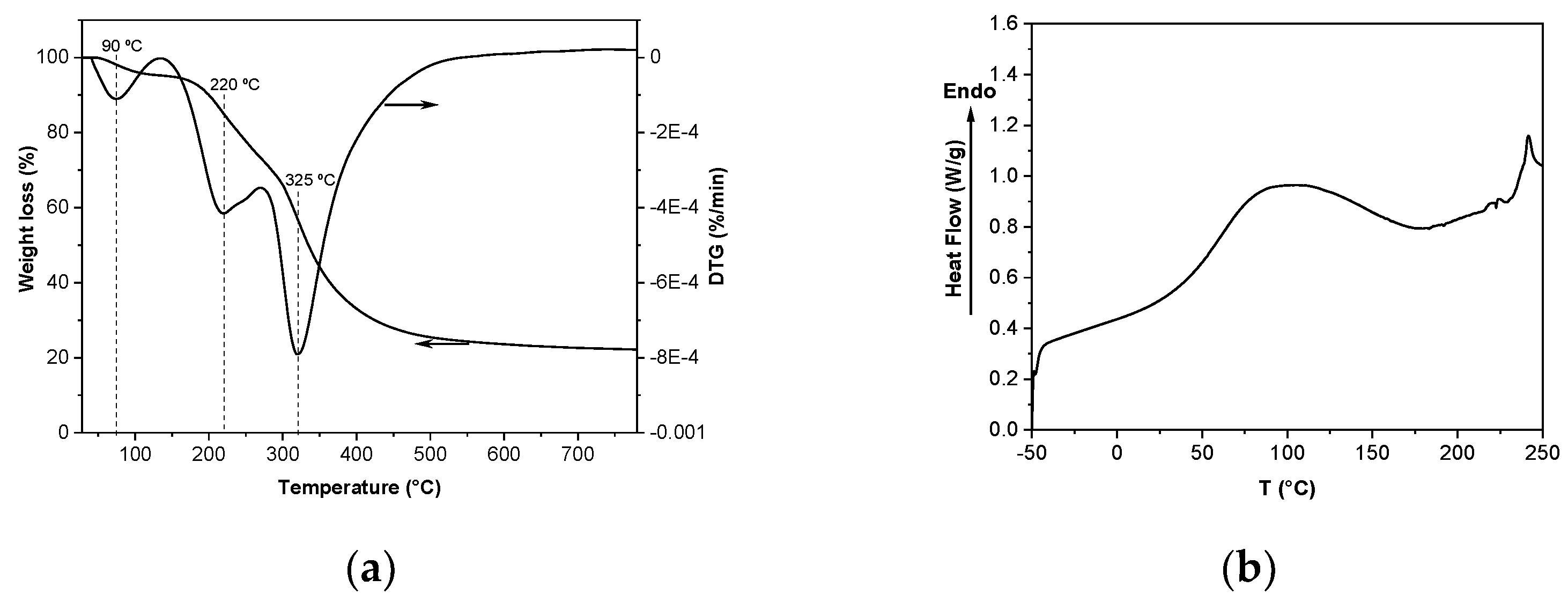
2.4.1. Differential Scanning Calorimetry (DSC) and Thermo-Gravimetric Analysis (TGA)
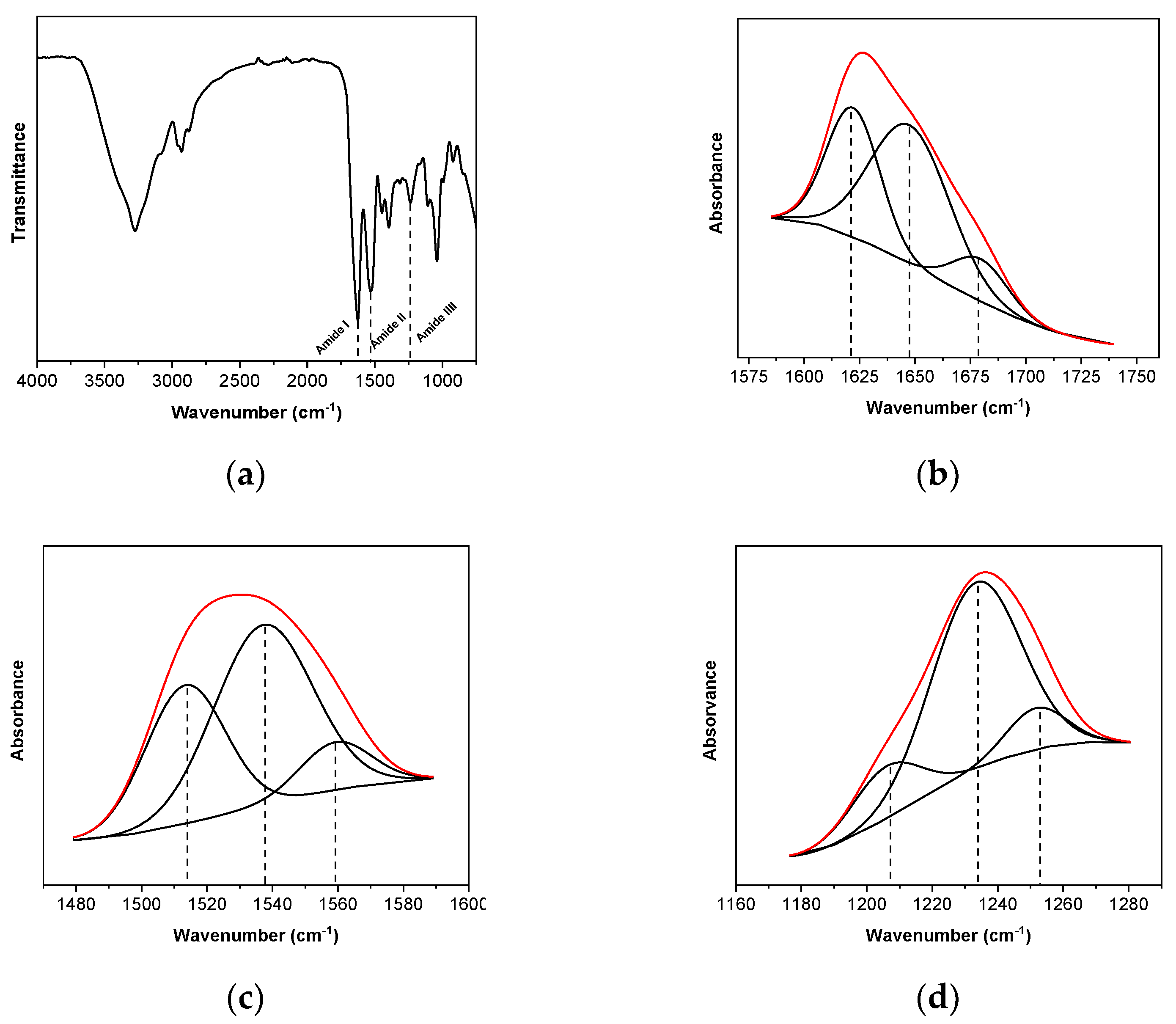
2.4.2. Fourier Transform Infrared (FTIR) Spectroscopy
2.4.3. Ultraviolet-Visible (UV-vis) and Fluorescence Spectroscopies
2.4.4. X-ray Diffraction (XRD) and X-ray Photoelectron Spectroscopy (XPS)
2.4.5. Scanning Electron Microscopy (SEM)
2.4.6. Mechanical Testing
2.4.7. Electrical Conductivity
2.4.8. Water Uptake
2.4.9. Water Contact Angle
2.4.10. Degradation Analyses
2.4.11. Cytotoxicity Studies
2.5. Statistical Analysis
3. Results and Discussion
3.1. Physicochemical Properties
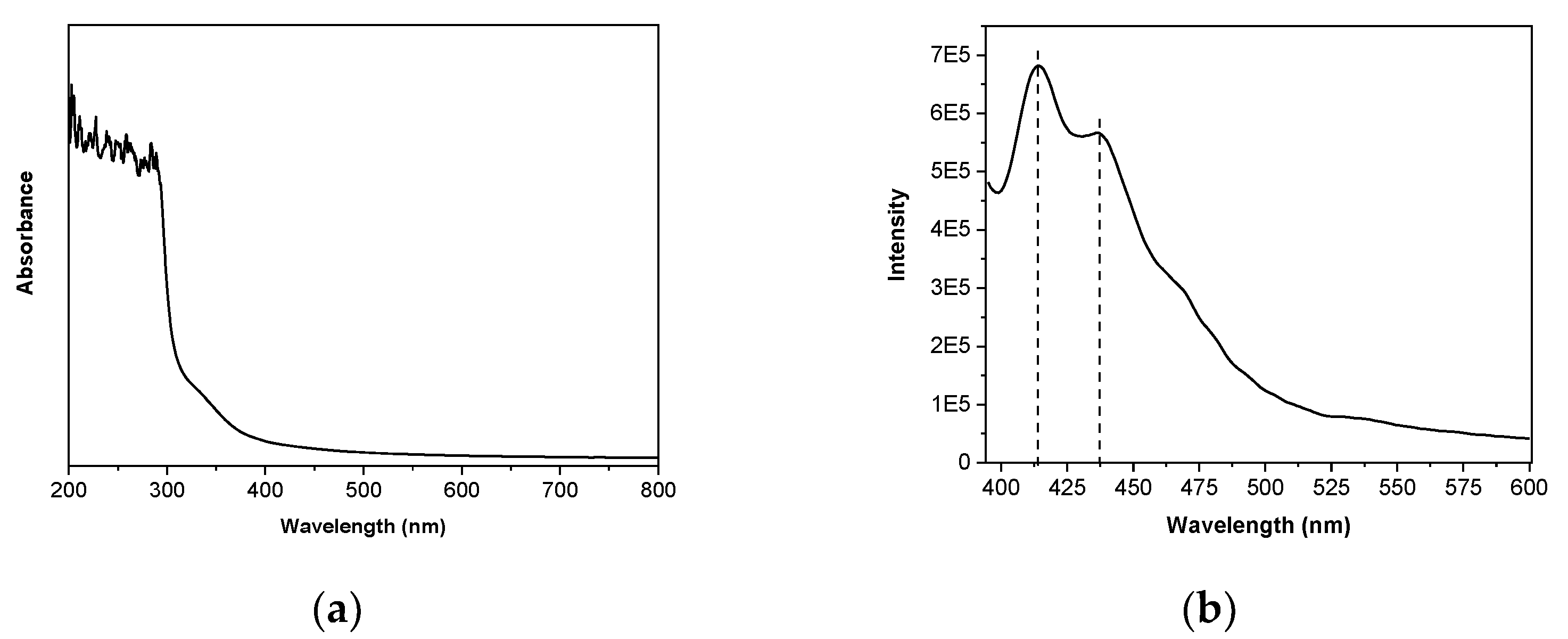
3.2. Optical Properties
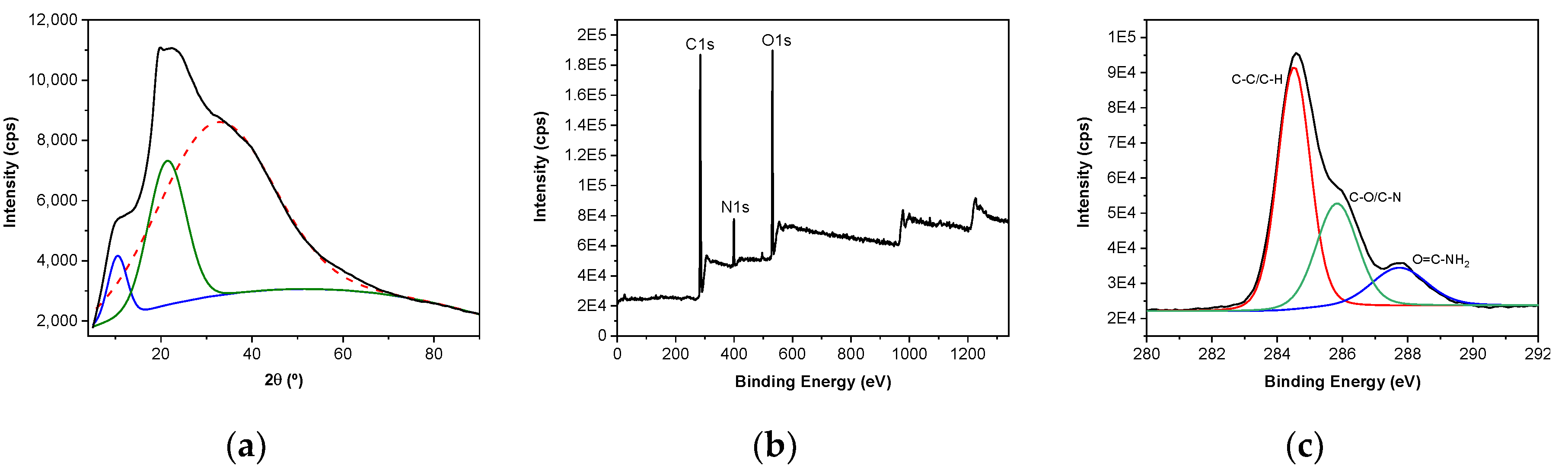
3.3. X-ray Diffraction (XRD) and X-ray Photoelectron Spectroscopy (XPS)
3.4. Film Microstructure and Mechanical and Electrical Properties
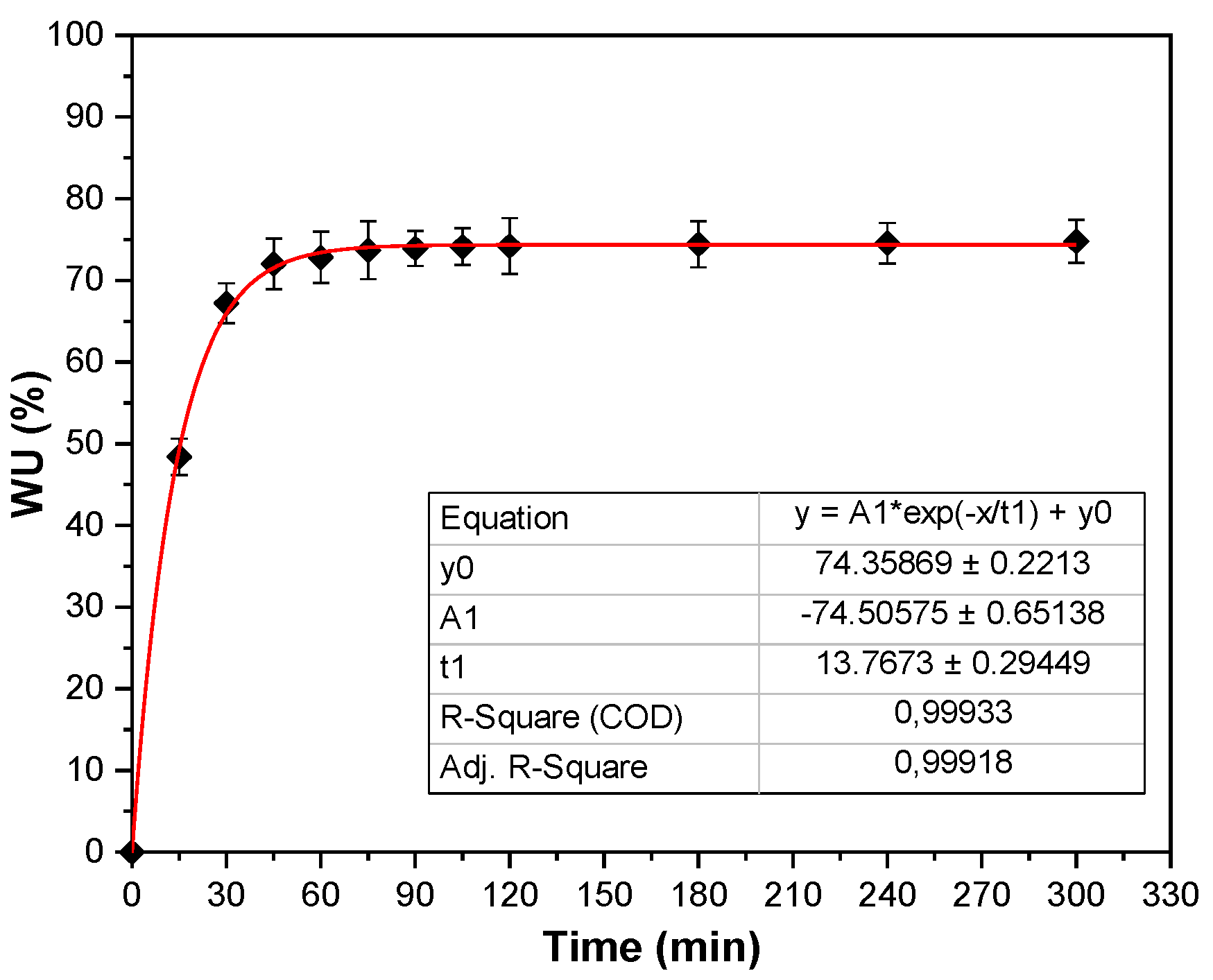
3.5. Water Uptake and Contact Angle
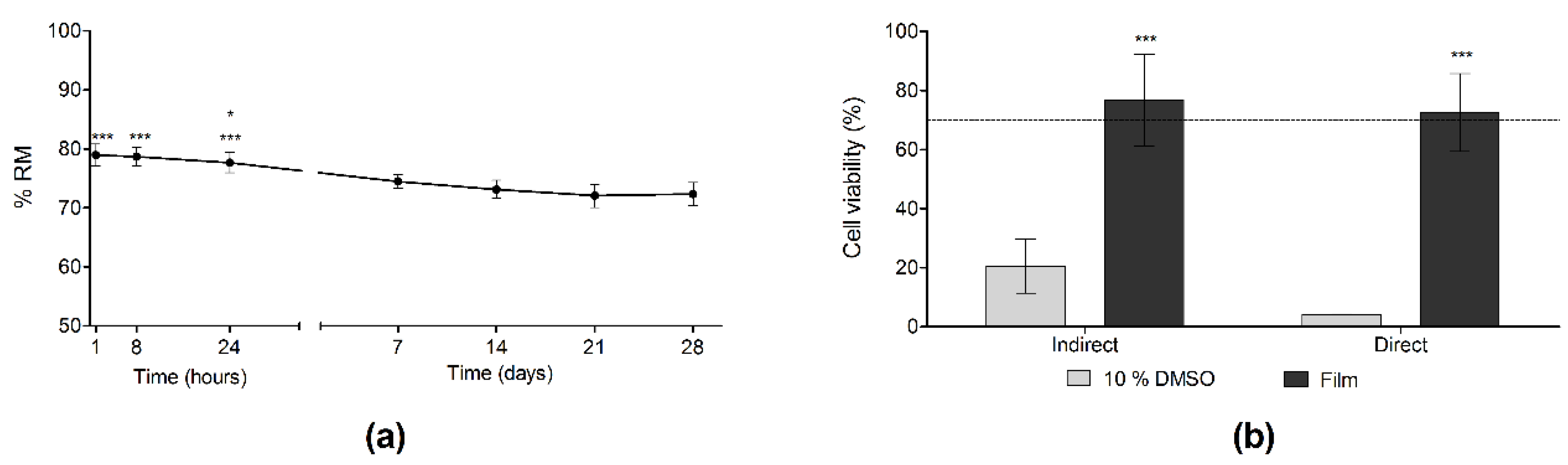
3.6. Degradation and Biocompatibility Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karan, S.K.; Mandal, D.; Khatua, B.B. Self-powered flexible Fe-doped RGO/PVDF nanocomposite: An excellent material for a piezoelectric energy harvester. Nanoscale 2015, 7, 10655–10666. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, J.; Wang, K.; Qian, X.; Sun, R. Highly thermostable, flexible, and conductive films prepared from cellulose, graphite, and polypyrrole nanoparticles. ACS Appl. Mater. Interfaces 2015, 7, 15641–15648. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Li, K.; Gao, Q.; Zhang, W.; Chen, H.; Li, J. Development of conductive protein-based film reinforced by cellulose nanofibril template-directed hyperbranched copolymer. Carbohydr. Polym. 2020, 237, 116141. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Zheng, R.; Chen, J.; Su, W.; Dong, B.; Lin, C.; Huang, B.; Lua, B. Microfibrillated cellulose enhancement to mechanical and conductive properties of biocompatible hydrogels. Carbohydr. Polym. 2019, 205, 244–254. [Google Scholar] [CrossRef]
- Tollefson, J. Power from the oceans: Blue energy. Nature 2014, 508, 302. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Shi, B.; Fan, F.; Wang, X.; Yan, L.; Yuan, W.; Wang, S.; Liu, H.; Li, Z.; Wang, Z.L. In vivo powering of pacemaker by breathing-driven implanted triboelectric nanogenerator. Adv. Mater. 2014, 26, 5851–5856. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, G.; Yang, R.; Wang, A.C.; Wang, Z.L. Muscle-driven in vivo nanogenerator. Adv. Mater. 2010, 22, 2534–2537. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Mandal, D. High-performance bio-piezoelectric nanogenerator made with fish scale. Appl. Phys. Lett. 2016, 109, 103701. [Google Scholar] [CrossRef]
- Rajala, S.; Siponkoski, T.; Sarlin, E.; Mettänen, M.; Vuoriluoto, M.; Pammo, A.; Juuti, J.; Rojas, O.J.; Franssila, S.; Tuukkanen, S. Cellulose nanofibril film as a piezoelectric sensor material. ACS Appl. Mater. Interfaces 2016, 8, 15607–15614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.M.; Pippel, E.; Moutanabbir, O.; Kim, J.-H.; Lee, H.-J.; Knez, M. In situ raman spectroscopic study of al-infiltrated spider dragline silk under tensile deformation. ACS Appl. Mater. Interfaces 2014, 6, 16827–16834. [Google Scholar] [CrossRef]
- Zubeldía, F.; Ansorena, M.R.; Marcovich, N.E. Wheat gluten films obtained by compression molding. Polym. Test. 2015, 43, 68–77. [Google Scholar] [CrossRef]
- Shi, W.; Dumont, M.J. Review: Bio-based films from zein, keratin, pea, and rapeseed protein feedstocks. J. Mater. Sci. 2014, 49, 1915–1930. [Google Scholar] [CrossRef]
- Las Heras, K.; Santos-Vizcaíno, E.; Garrido, T.; Gutiérrez, F.B.; Aguirre, J.J.; de la Caba, K.; Guerrero, P.; Igartua, M.; Hernandez, R.M. Soy protein and chitin sponge-like scaffolds: From natural by-products to cell delivery systems for biomedical applications. Green Chem. 2020, 22, 3445–3460. [Google Scholar] [CrossRef]
- Ahn, S.; Chantre, C.O.; Gannon, A.R.; Lind, J.U.; Campbell, P.H.; Grevesse, T.; O’Connor, B.B.; Parker, K.K. Soy protein/cellulose nanofiber scaffolds mimicking skin extracellular matrix for enhanced wound healing. Adv. Healthc. Mater. 2018, 7, 1701175. [Google Scholar] [CrossRef]
- Chatterjee, C.; Gleddie, S.; Xiao, C.-W. Soybean Bioactive Peptides and Their Functional Properties. Nutrients 2018, 10, 1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, N.; Yang, Z.; Liu, X.-Y.; Li, Y.; Xu, H.-Y. Structural origin of the strain-hardening of spider silk. Adv. Funct. Mater. 2011, 21, 772–778. [Google Scholar] [CrossRef]
- Andonegi, M.; las Heras, K.; Santos-Vizcaíno, E.; Igartua, M.; Hernández, R.M.; de la Caba, K.; Guerrero, P. Structure-properties relationship of chitosan/collagen films with potential for biomedical applications. Carbohydr. Polym. 2020, 237, 11615. [Google Scholar] [CrossRef]
- Guerrero, P.; Kerry, J.P.; de la Caba, K. FTIR characterization of protein-polysaccharide interactions in extruded blends. Carbohydr. Polym. 2014, 111, 598–605. [Google Scholar] [CrossRef]
- Sorgentini, D.A.; Wagner, J.R.; Añon, M.C. Effects of thermal treatment of soy protein isolate on the characteristics and structure-function relationship of soluble and insoluble fractions. J. Agric. Food Chem. 1995, 43, 2471–2479. [Google Scholar] [CrossRef]
- Yamauchi, F.; Yamagishi, T.; Iwabuchi, S. Molecular understanding of heat-induced phenomena of soybean protein. Food Rev. Int. 1991, 7, 721–729. [Google Scholar] [CrossRef]
- Guerrero, P.; Beatty, E.; Kerry, J.P.; de la Caba, K. Extrusion of soy protein with gelatin and sugars at low moisture content. J. Food Eng. 2012, 110, 53–59. [Google Scholar] [CrossRef]
- ASTM D 638-03. Standard test method for tensile properties of plastics. In Annual Book of ASTM Standards; American Society of Testing and Materials: Philadelphia, PA, USA, 2003. [Google Scholar]
- ISO 10993-5. Biological Evaluation of Medical Devices-Part 5: Tests for In-Vitro Cytotoxicity; ISO: Geneve, Switzerland, 2009. [Google Scholar]
- Etxabide, A.; Vairo, C.; Santos-Vizcaino, E.; Guerrero, P.; Pedraz, J.L.; Igartua, M.; de la Caba, K.; Hernandez, R.M. Ultra thin hydro-films based on lactose-crosslinked fish gelatin for wound healing applications. Int. J. Pharm. 2017, 530, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, Y.; Dreisoerner, J.; Wei, Y. The effects of screw configuration on the screw fill degree and special mechanical energy in twin-screw extruder for high-moisture texturised defatted soybean meal. J. Food Eng. 2015, 157, 77–83. [Google Scholar] [CrossRef]
- Rhim, J.W.; Wang, L.F.; Hong, S.I. Preparation and characterization of agar/silver nanoparticles composite films with antimicrobial activity. Food Hydrocolloids 2013, 33, 327–335. [Google Scholar] [CrossRef]
- Wang, W.; Guo, Y.; Otaigbe, J.U. Synthesis, characterization and degradation of biodegradable thermoplastic elastomers from poly(ester urethane)s and renewable soy protein isolate biopolymer. Polymer 2010, 51, 5448–5455. [Google Scholar] [CrossRef]
- Kumar, R.; Choudhary, V.; Mishra, S.; Varma, I.K.; Mattiason, B. Adhesives and plastics based on soy protein products. Ind. Crop. Prod. 2002, 16, 155–172. [Google Scholar] [CrossRef]
- Steven, E.; Saleh, W.R.; Lebedev, V.; Acquah, S.F.A.; Laukhin, V.; Alamo, R.G.; Brooks, J.S. Carbon nanotubes on a spider silk scaffold. Nat. Commun. 2013, 4, 2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, J.; Baldwin, M.A.; Cohen, F.E.; Prusine, S.B. Protein peptides induce α-helix to β-sheet conformational transitions. Biochemistry 1995, 34, 4186–4192. [Google Scholar] [CrossRef]
- Bonwell, E.S.; Wetzel, D.L. Innovative FTIR imaging of protein film secondary structure before and after heat treatment. J. Agric. Food Chem. 2009, 57, 10067–10072. [Google Scholar] [CrossRef]
- Hu, X.; Kaplan, D.L.; Cebe, P. Dynamic protein−water relationships during β-sheet formation. Macromolecules 2008, 41, 3939–3948. [Google Scholar] [CrossRef]
- Barth, A. The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 2000, 74, 141–173. [Google Scholar] [CrossRef]
- Ling, S.; Qi, Z.; Knight, D.P.; Shao, Z.; Chen, X. Synchrotron FTIR microspectroscopy of single natural silk fibers. Biomacromolecules 2011, 12, 3344–3349. [Google Scholar] [CrossRef] [PubMed]
- Pal, G.K.; Nidheesh, T.; Suresh, P.V. Comparative study on characteristics and in vitro fibril formation ability of acid and pepsin soluble collagen from the skom pf catla (Catla catla) and rohu (Labeo rohita). Food Res. Int. 2015, 76, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, B.L.; Gao, L.Z.; Chen, H.L. Studies on bullfrog skin collagen. Food Chem. 2004, 84, 65–69. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, L. Structure and properties of soy protein plastics plasticized with acetamide. Macromol. Mater. Eng. 2006, 291, 820–828. [Google Scholar] [CrossRef]
- Eftink, M.R. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys. J. 1991, 66, 482–501. [Google Scholar] [CrossRef] [Green Version]
- Xiang, B.Y.; Ngadi, M.O.; Simpson, B.K.; Simpson, M.V. Pulsed electric field induced structural modification of soy protein isolate as studied by fluorescence spectroscopy. J. Food Process Preserv. 2011, 35, 563–570. [Google Scholar] [CrossRef]
- Karan, S.K.; Maiti, S.; Kwon, O.; Paria, V.; Maitra, A.; Si, S.K.; Kim, Y.; Kim, J.K.; Khatua, B.B. Nature driven spider silk as high energy conversion efficient biopiezoelectric nanogenerator. Nano Energy 2018, 49, 655–666. [Google Scholar] [CrossRef]
- Zhou, L.; Fu, P.; Cai, X.; Zhou, S.; Yuan, Y. Naturally derived carbon nanofibers as sustainable electrocatalysts for microbial energy harvesting: A new application of spider silk. Appl. Catal. B 2016, 188, 31–38. [Google Scholar] [CrossRef]
- Irimia-Vladu, M.; Głowacki, E.D.; Voss, G.; Bauer, S.; Sariciftci, N.S. Green and biodegradable electronics. Mater. Today 2012, 15, 340–346. [Google Scholar] [CrossRef]
- Aradhya, S.V.; Venkataraman, L. Single-molecule junctions beyond electronic transport. Nat. Nanotechnol. 2013, 8, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B. Electrical conductivity of proteins. Nature 1962, 193, 364–365. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Prabhakaran, M.P.; Kai, D.; Annamalai, S.K.; Arunachalam, K.D.; Ramakrishna, S. Tissue engineered plant extracts as nanofibrous wound dressing. Biomaterials 2013, 34, 724–734. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FTIR Band | Amide I | Amide II | Amide III | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Wavenumber (cm−1) | 1622 | 1649 | 1678 | 1513 | 1537 | 1559 | 1206 | 1233 | 1252 |
| Area (%) | 33.90 | 54.50 | 11.60 | 34.25 | 55.23 | 10.52 | 15.83 | 73.31 | 10.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerrero, P.; Garrido, T.; Garcia-Orue, I.; Santos-Vizcaino, E.; Igartua, M.; Hernandez, R.M.; de la Caba, K. Characterization of Bio-Inspired Electro-Conductive Soy Protein Films. Polymers 2021, 13, 416. https://doi.org/10.3390/polym13030416
Guerrero P, Garrido T, Garcia-Orue I, Santos-Vizcaino E, Igartua M, Hernandez RM, de la Caba K. Characterization of Bio-Inspired Electro-Conductive Soy Protein Films. Polymers. 2021; 13(3):416. https://doi.org/10.3390/polym13030416
Chicago/Turabian StyleGuerrero, Pedro, Tania Garrido, Itxaso Garcia-Orue, Edorta Santos-Vizcaino, Manoli Igartua, Rosa Maria Hernandez, and Koro de la Caba. 2021. "Characterization of Bio-Inspired Electro-Conductive Soy Protein Films" Polymers 13, no. 3: 416. https://doi.org/10.3390/polym13030416