
Structure and Properties of Reactively Extruded Opaque Post-Consumer Recycled PET

 , , , ,
, , , ,  , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Intrinsic Viscosity
2.3. Thermogravimetry Analysis (TGA)
2.4. Transmission Electron Microscopy (TEM)
2.5. DSC Analysis
2.6. Wide Angle X-ray Diffraction (WAXS)
2.7. Rheology
2.8. Injection Molding of Samples
2.9. Mechanical Properties
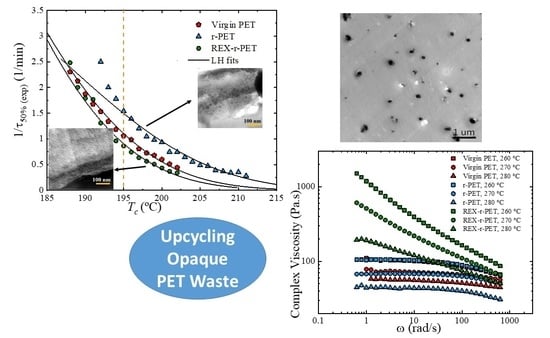
3. Results
3.1. Intrinsic Viscosity
3.2. Thermal Stability
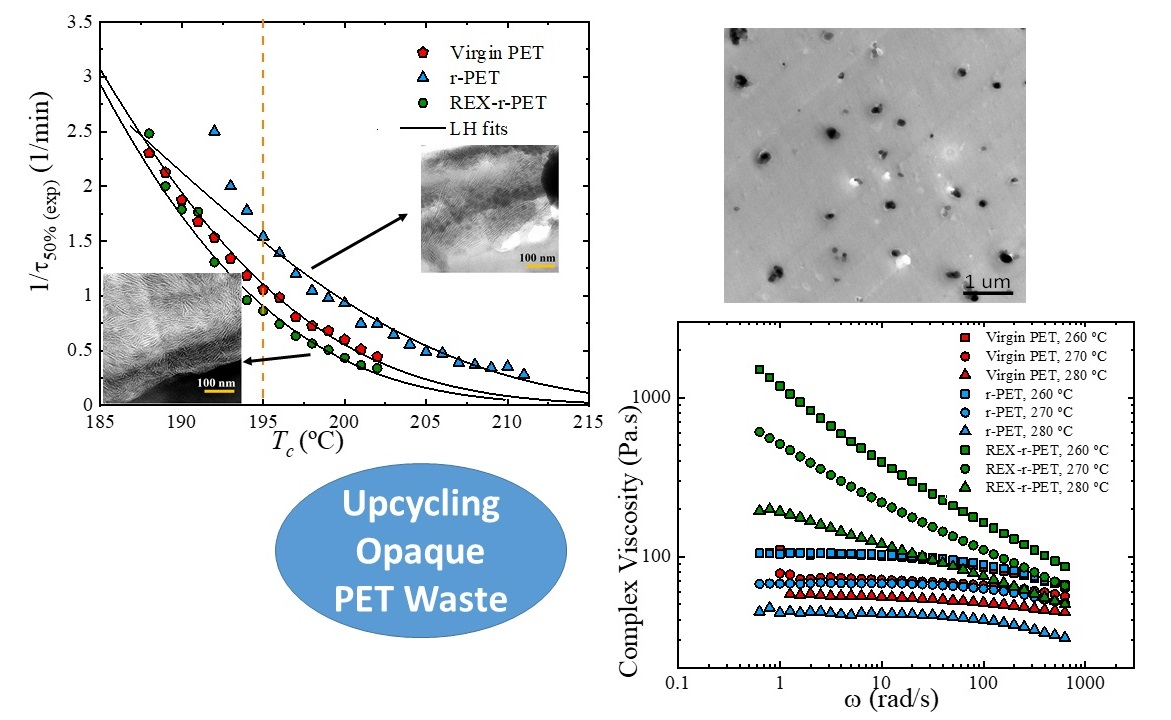
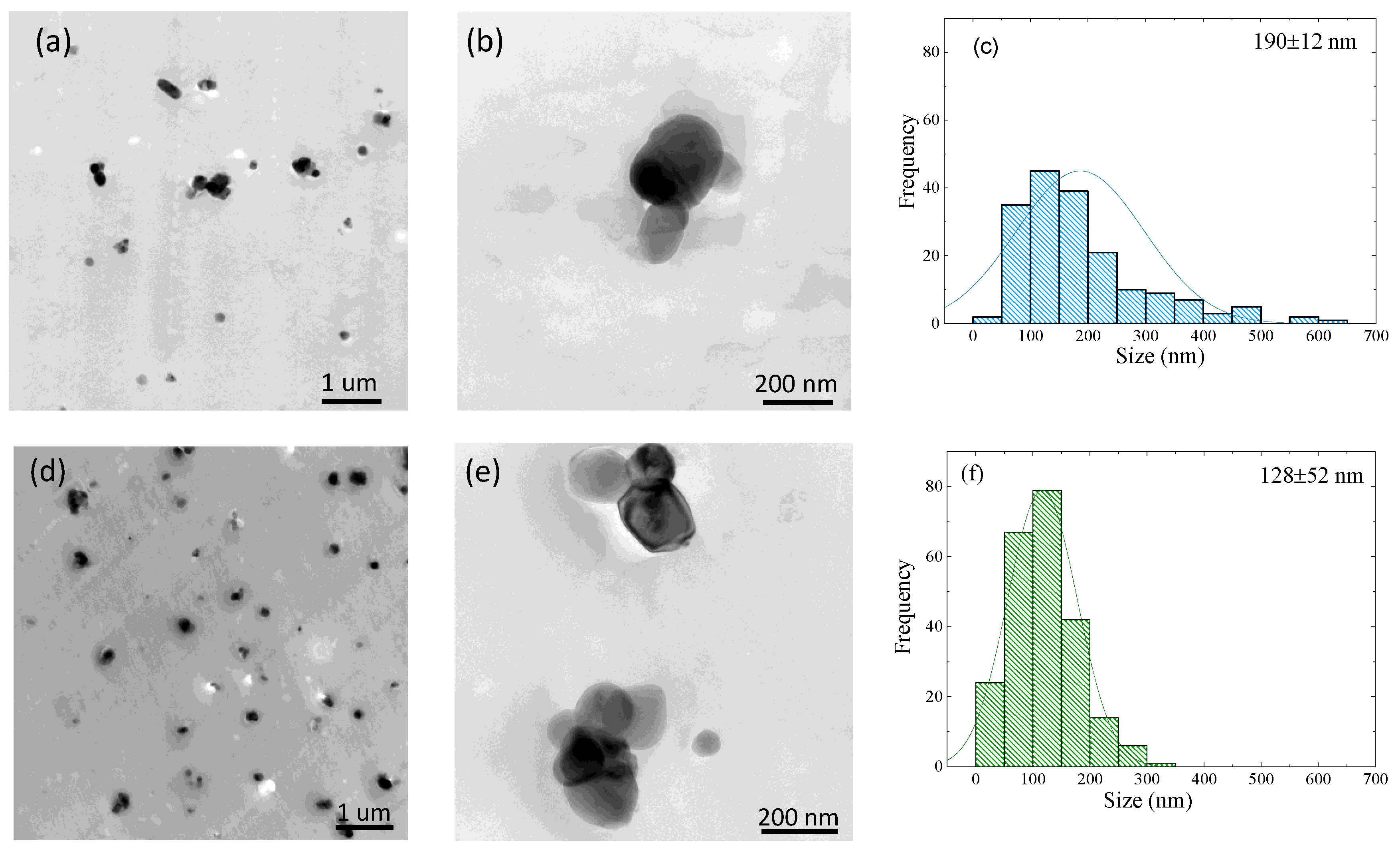
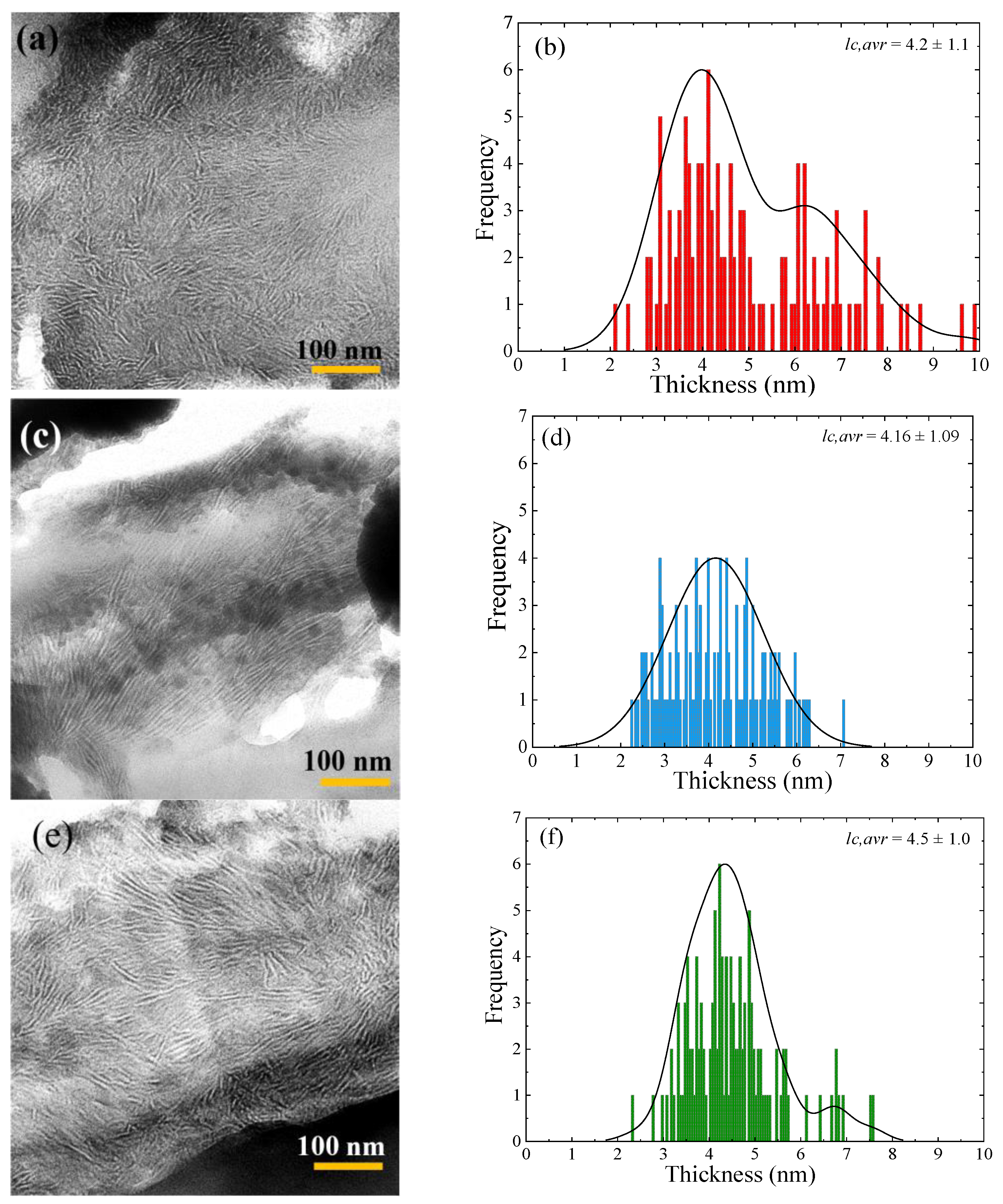
3.3. TEM Observations of TiO2 Nanoparticle Dispersion
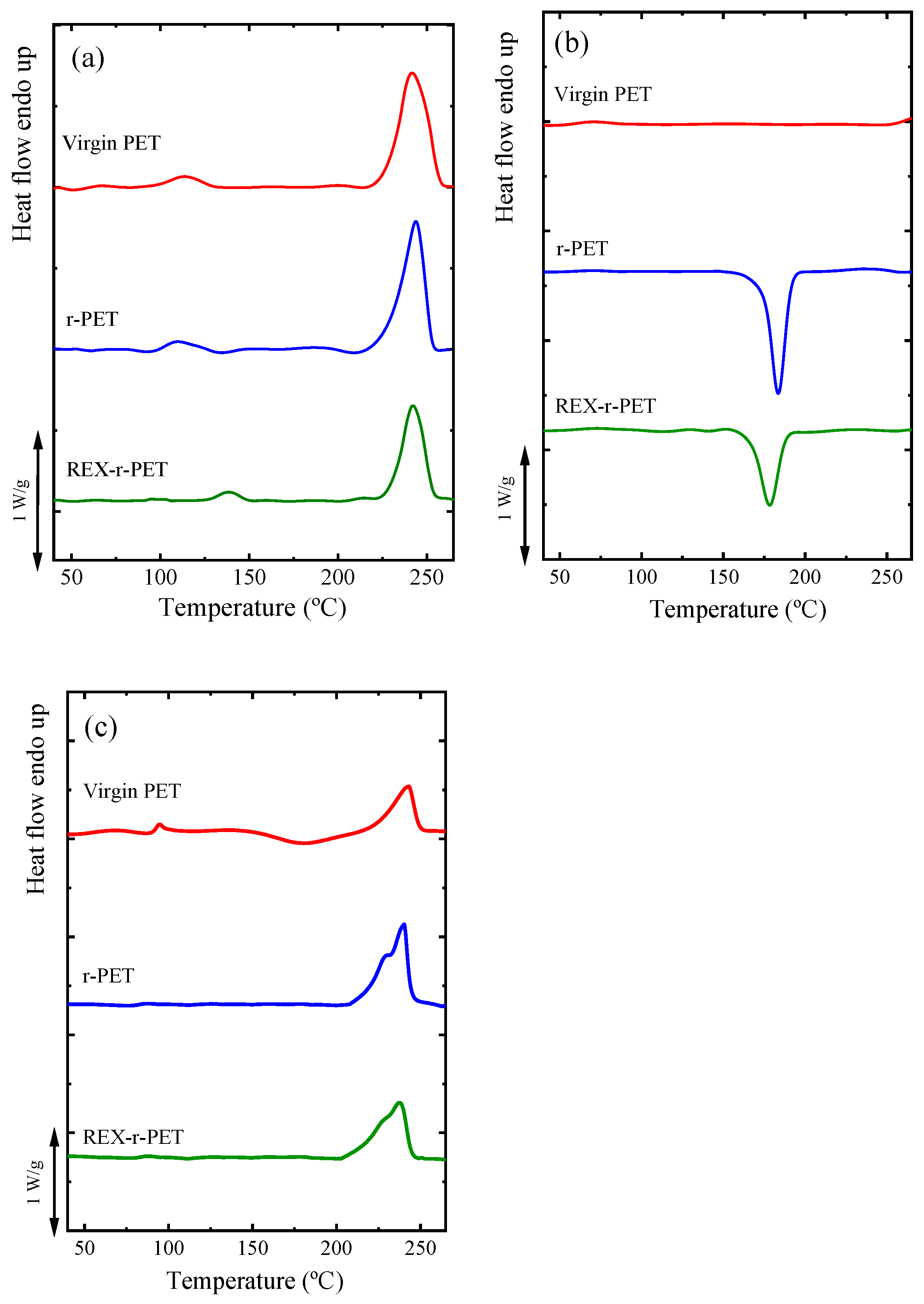
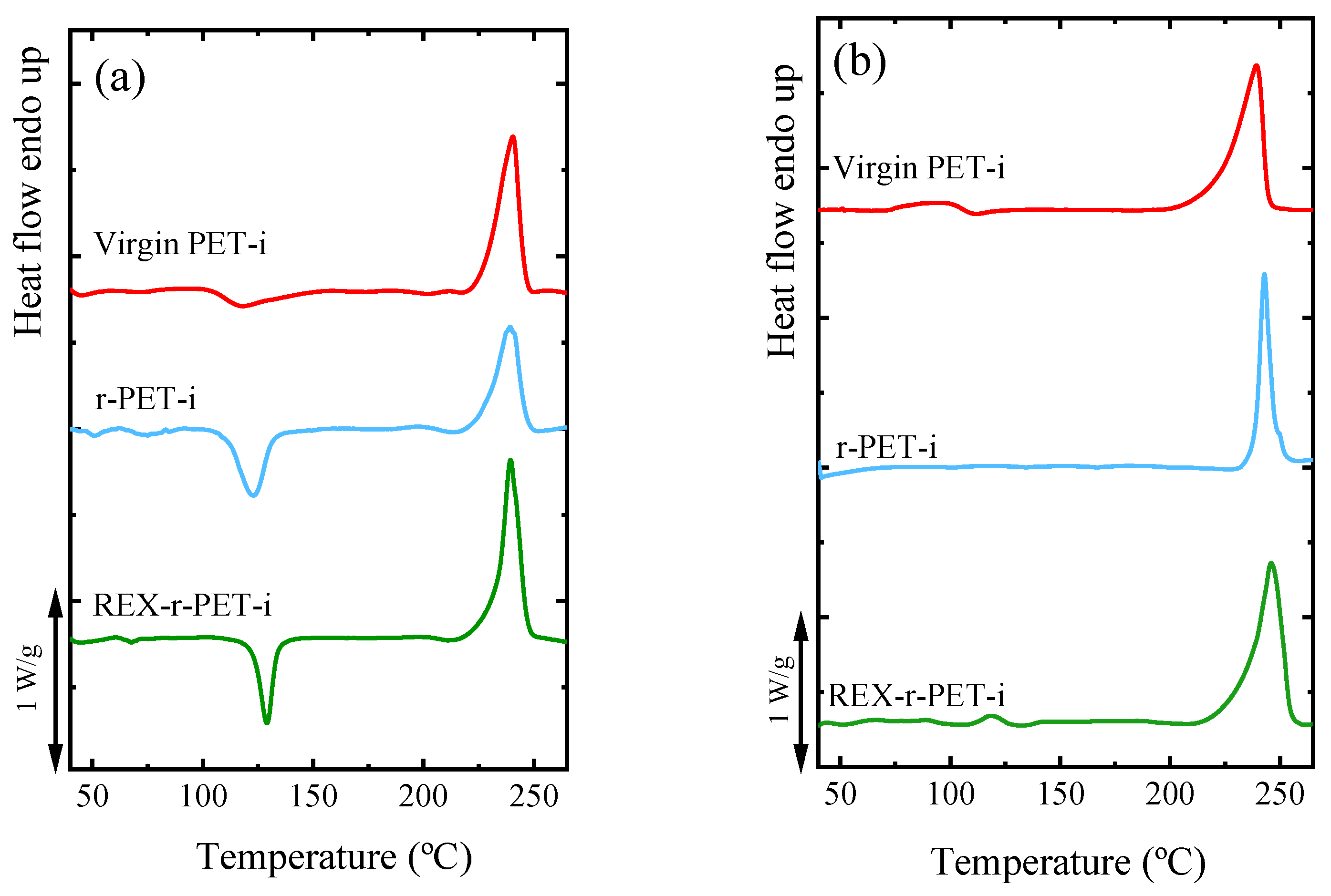
3.4. Nonisothermal Crystallization by DSC
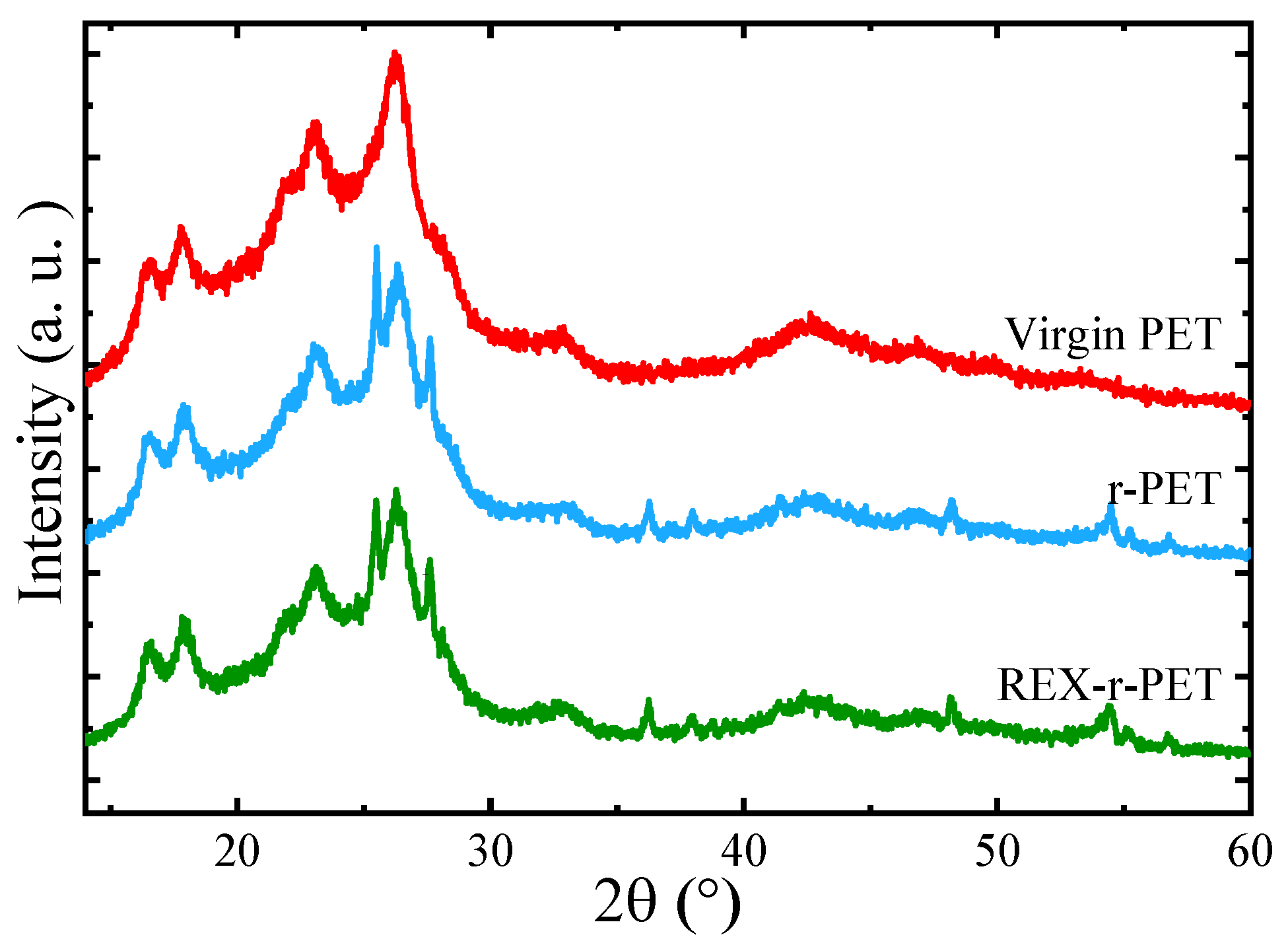
3.5. WAXS
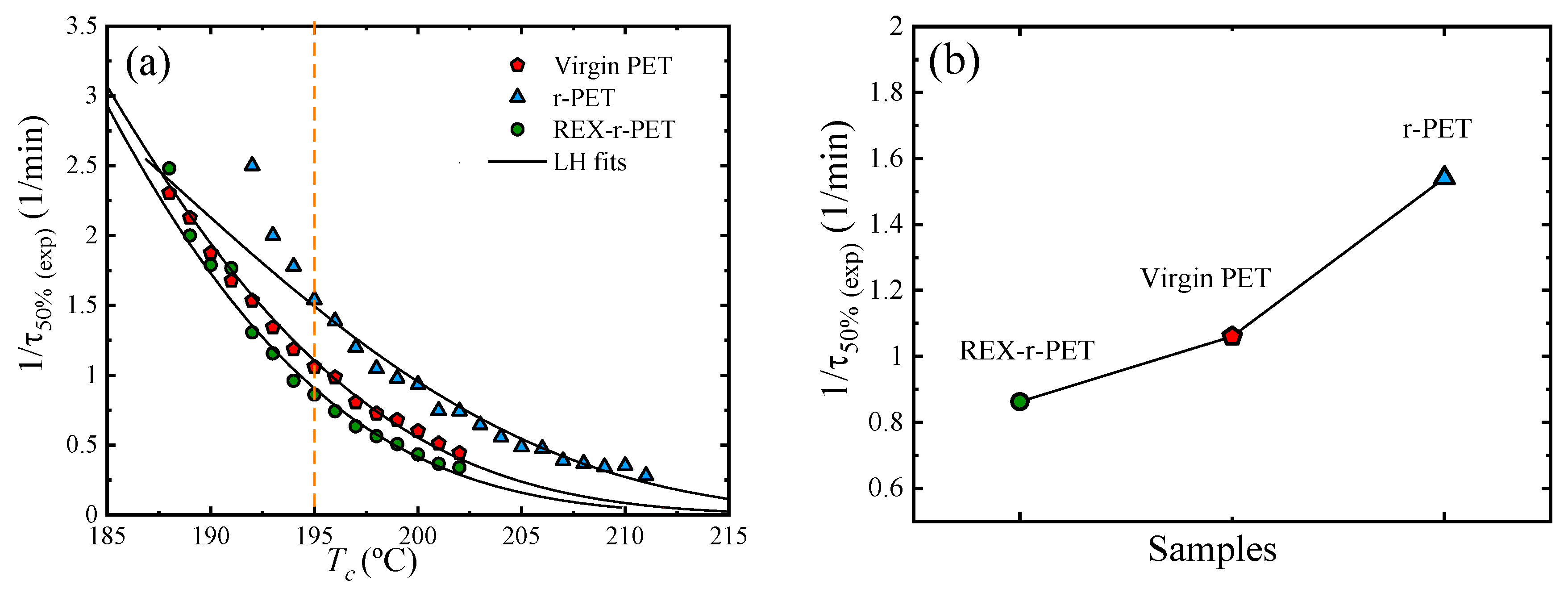
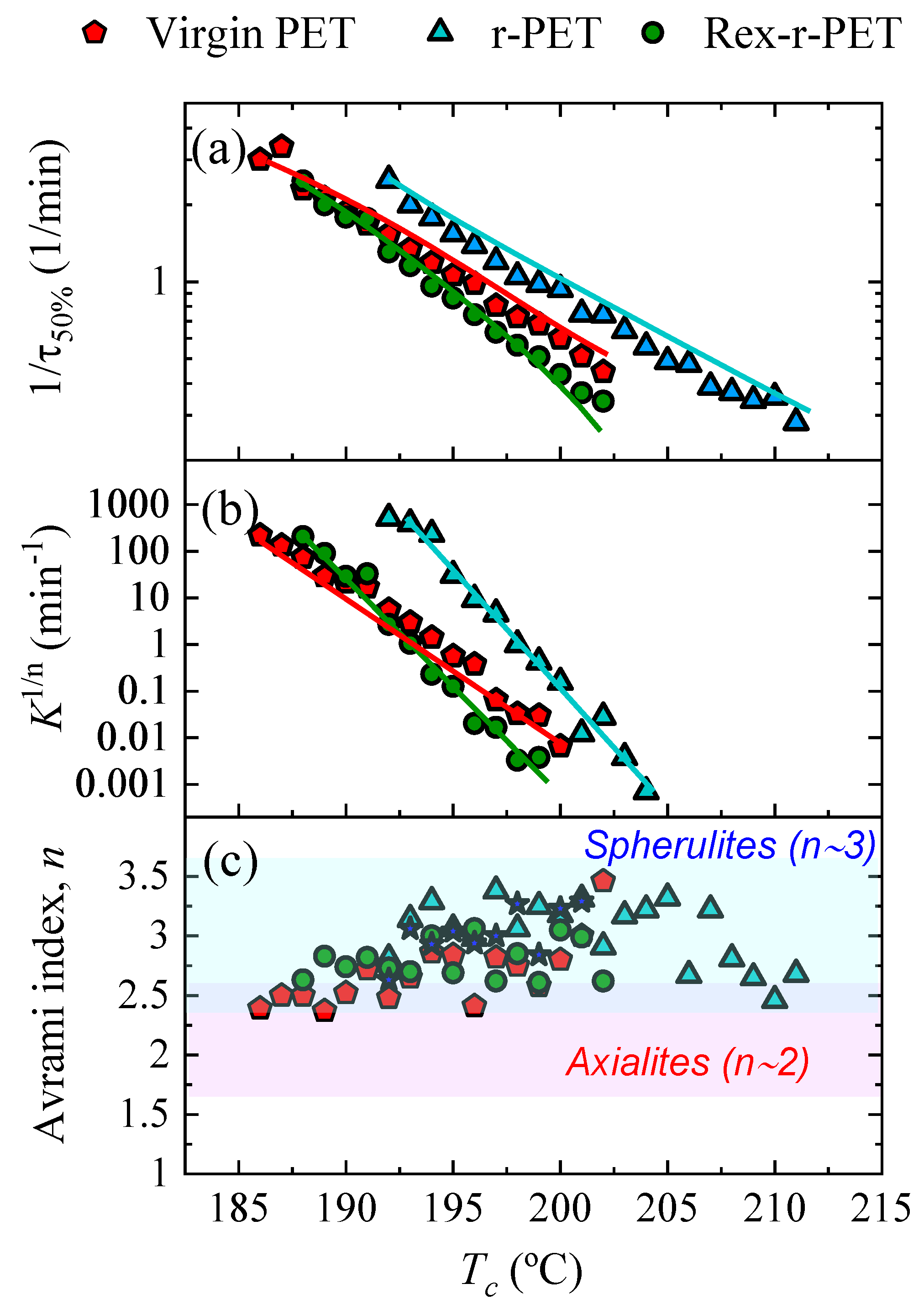
3.6. Overall Isothermal Crystallization by DSC
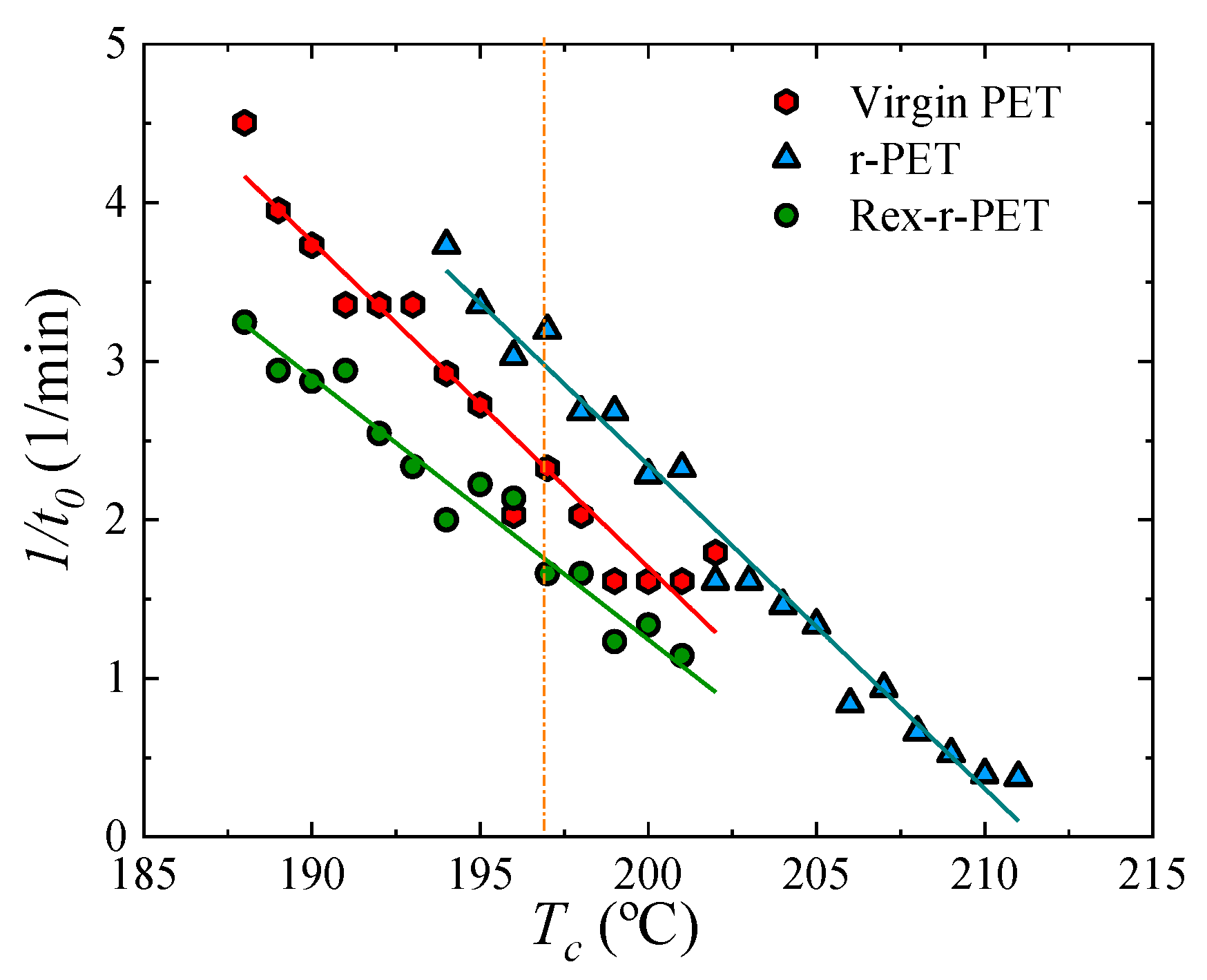
Fitting the DSC Isothermal Crystallization Data to the Avrami Theory
3.7. Lamellar Thickness Distribution and TEM Observations
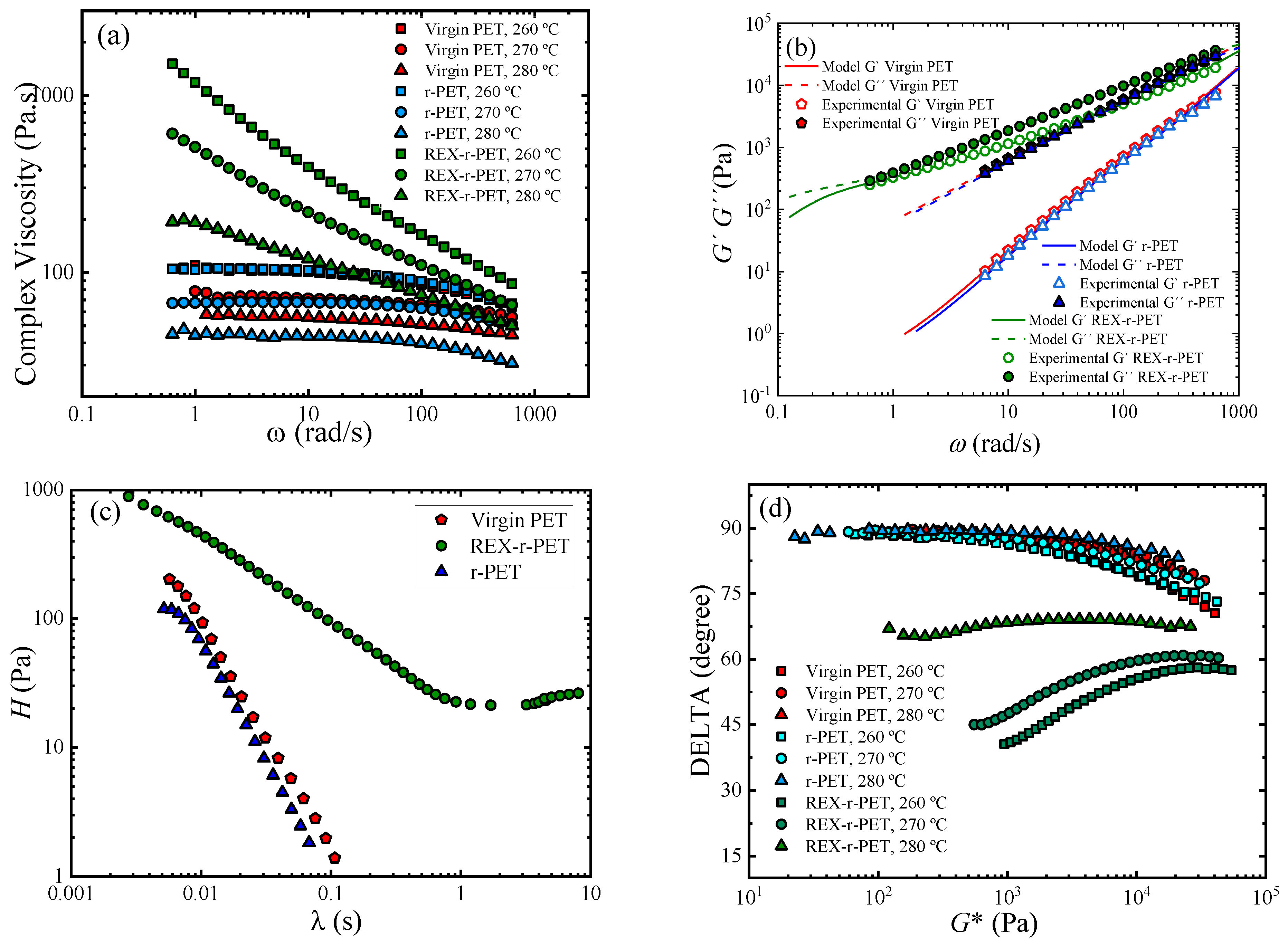
3.8. Rheology
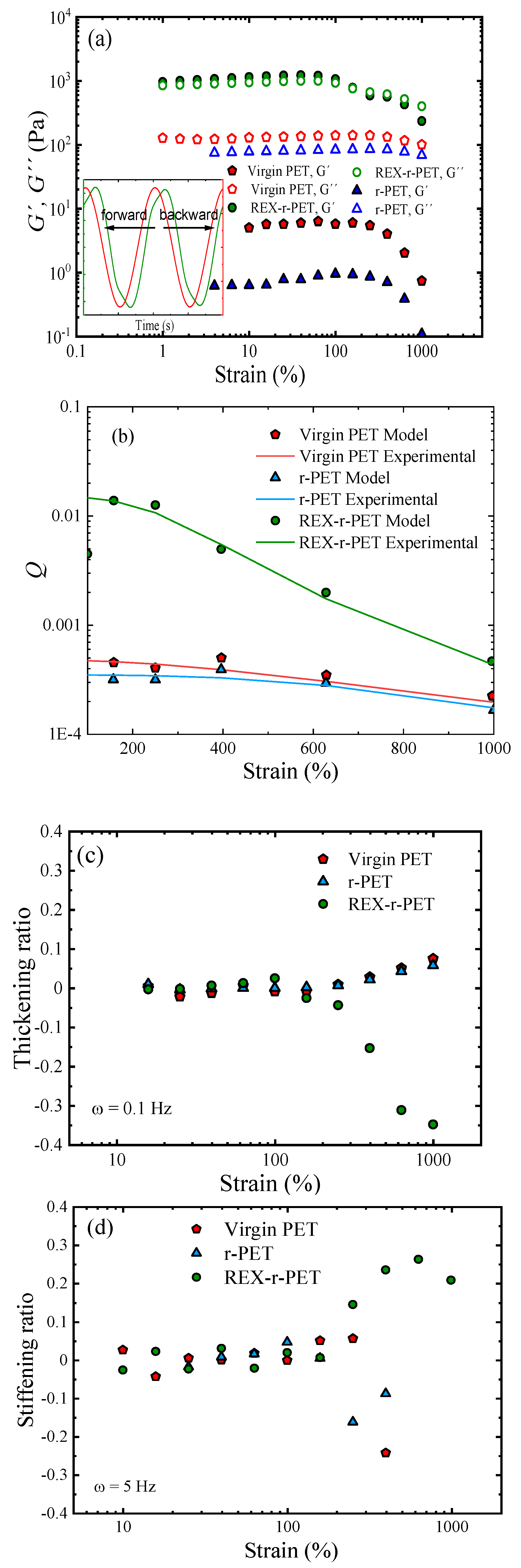
3.8.1. Linear Viscoelastic Data under SAOS (Small Amplitude Oscillatory Shear)
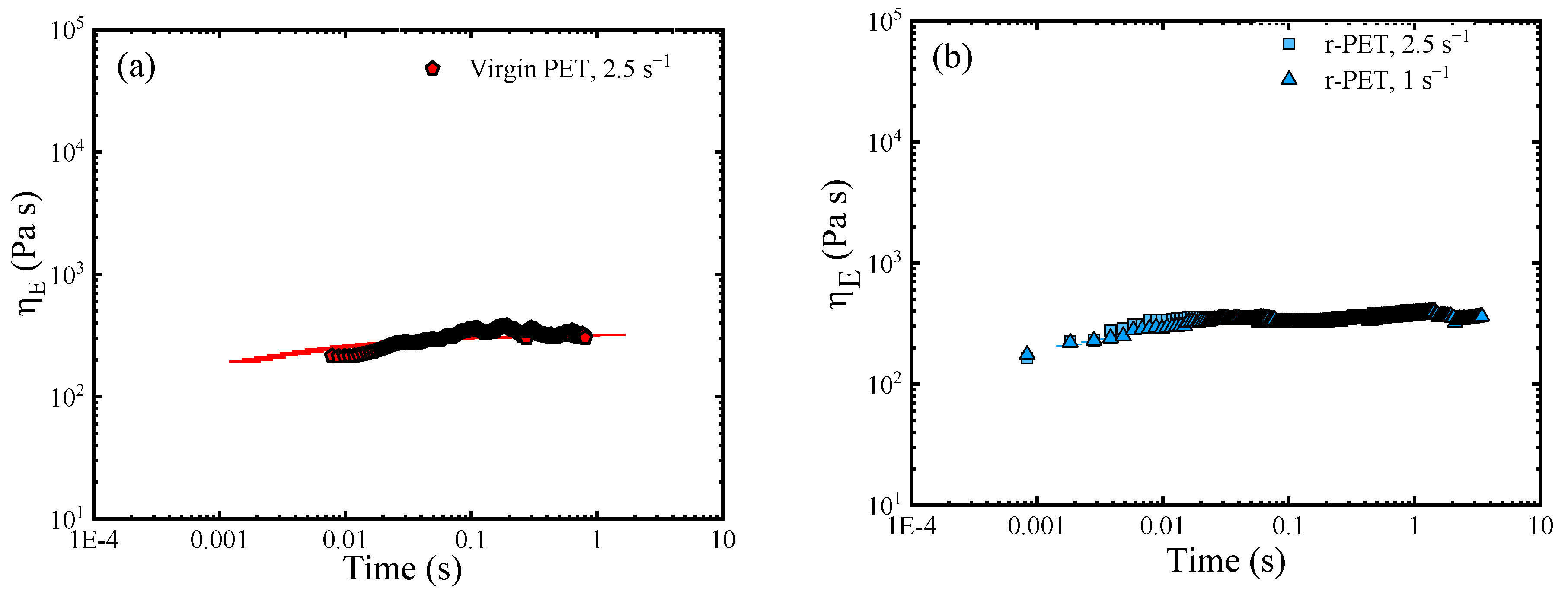
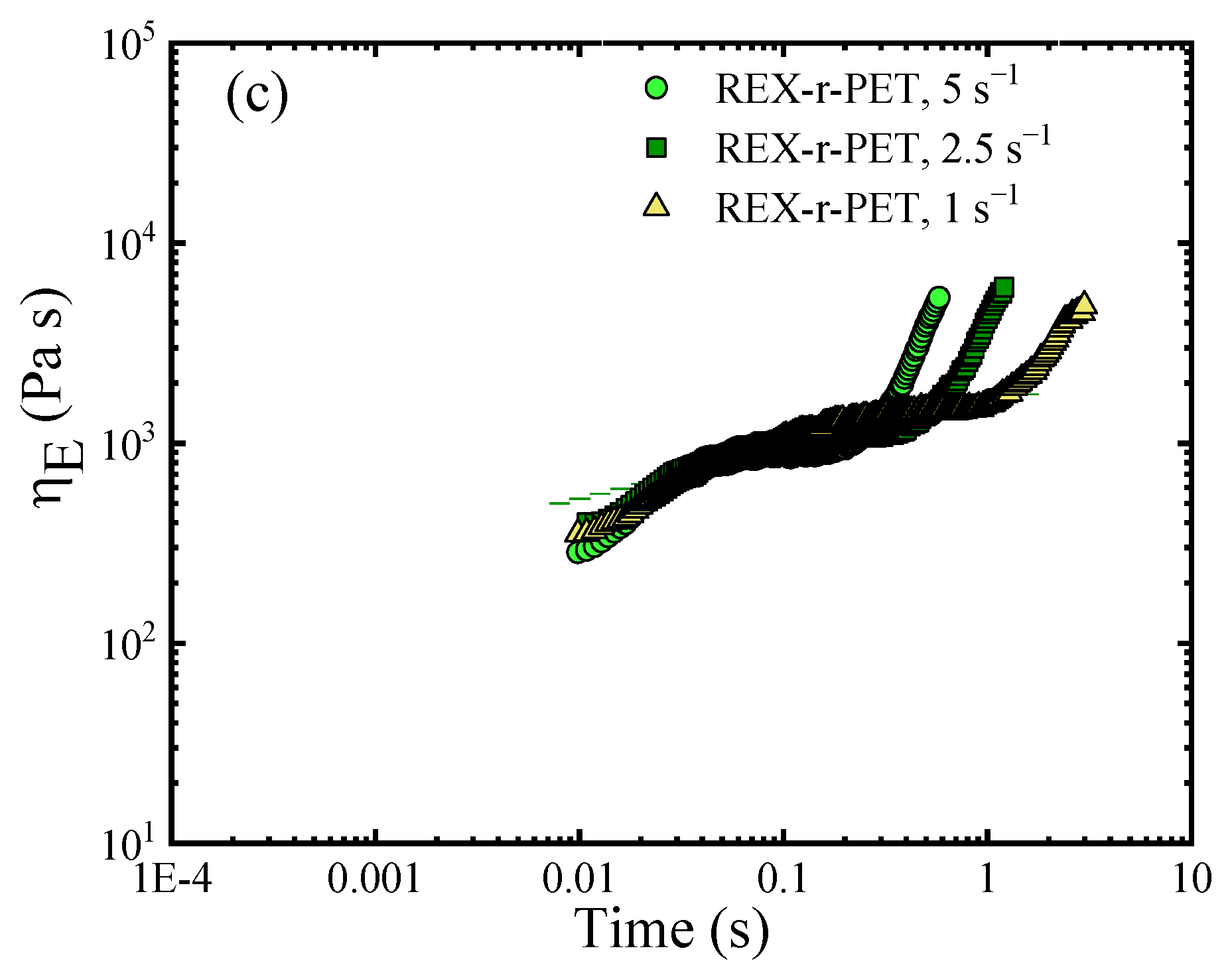
3.8.2. The Analysis of the Elongational Rheological Behavior
3.8.3. The study of Large Deformation Oscillatory Shear Measurements (LAOS)
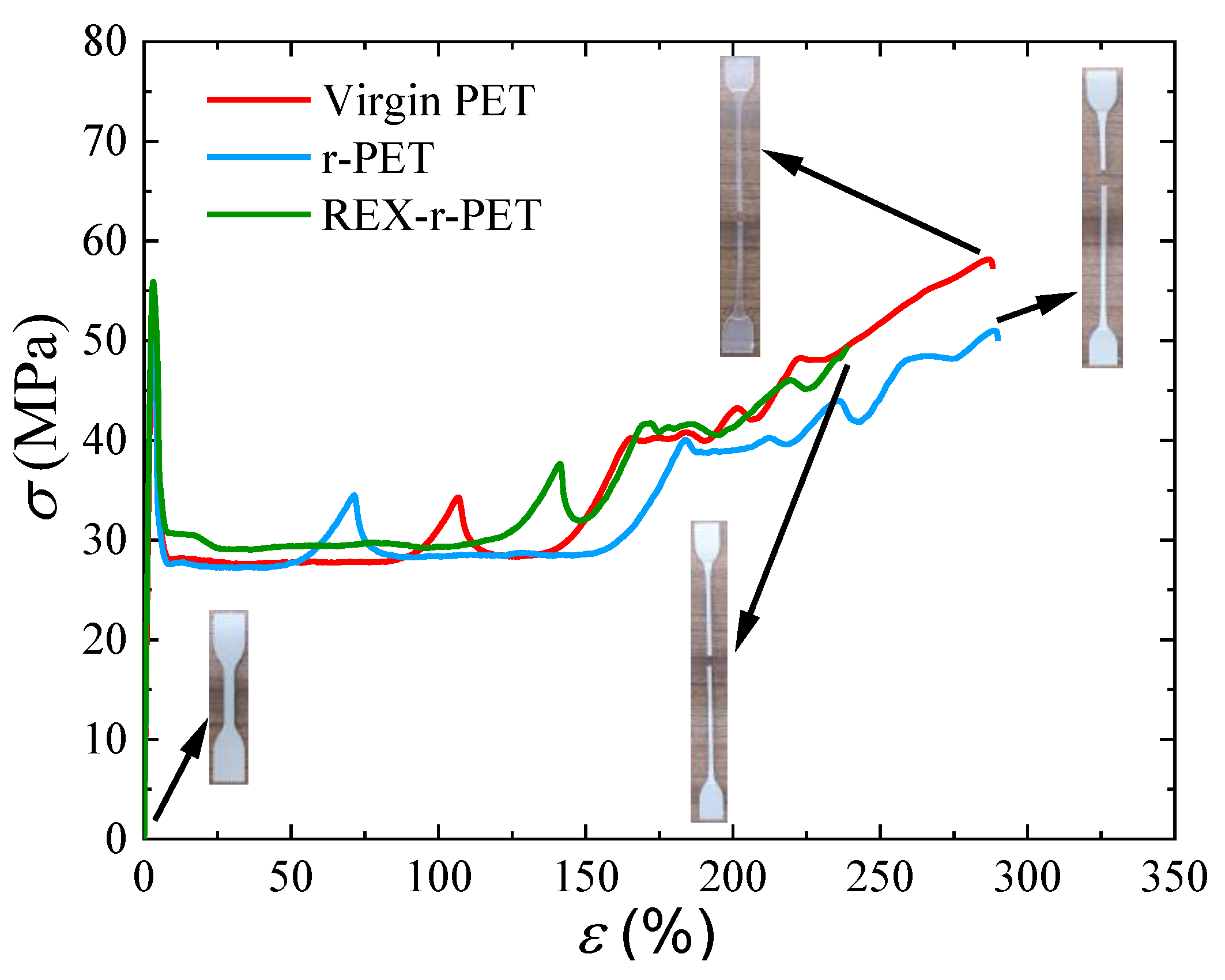
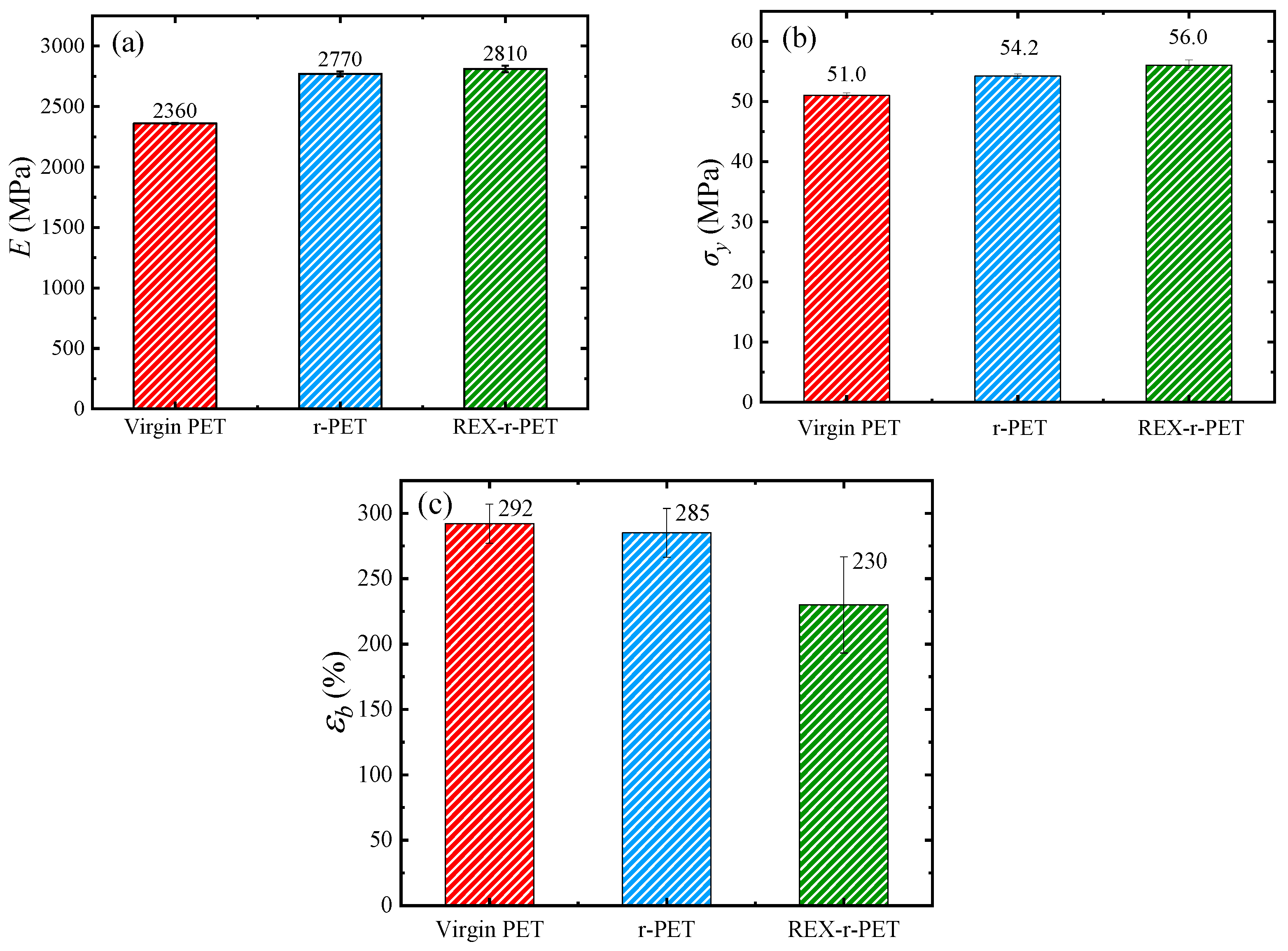
3.9. Mechanical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Plastics Europe. Plastics—the Facts 2020. Available online: https://www.plasticseurope.org/ (accessed on 10 August 2021).
- Lonca, G.; Lesage, P.; Majeau-Bettez, G.; Bernard, S.; Margni, M. Assessing scaling effects of circular economy strategies: A case study on plastic bottle closed-loop recycling in the USA PET market. Resour. Conserv. Recycl. 2020, 162, 105013. [Google Scholar] [CrossRef]
- Nisticò, R. Polyethylene terephthalate (PET) in the packaging industry. Polym. Test. 2020, 90, 106707. [Google Scholar] [CrossRef]
- Provin, A.P.; de Aguiar, A.R.; Aguiar, I.C.; Gouveia, S.; Vieira, A. Circular economy for fashion industry: Use of waste from the food industry for the production of biotextiles. Technol. Forecast. Soc. Chang. 2021, 169, 120858. [Google Scholar] [CrossRef]
- Meys, R.; Frick, F.; Westhues, S.; Sternberg, A.; Klankermayer, J.; Bardow, A. Towards a circular economy for plastic packaging wastes—The environmental potential of chemical recycling. Resour. Conserv. Recycl. 2020, 162, 105010. [Google Scholar] [CrossRef]
- Cladman, W.; Scheffer, S.; Goodrich, N.; Griffiths, M.W. Shelf-life of milkpackaged in plastic containers with and without treatment to reduce light transmission. Int. Dairy J. 1998, 8, 629–636. [Google Scholar] [CrossRef]
- Karaman, A.D.; Özer, B.; Pascall, M.A.; Álvarez, V. Recent Advances in Dairy Packaging. Food Rev. Int. 2015, 31, 295–318. [Google Scholar] [CrossRef]
- Welle, F. Twenty years of PET bottle to bottle recycling—An overview. Resour. Conserv. Recycl. 2011, 55, 865–875. [Google Scholar] [CrossRef]
- Awaja, F.; Pavel, D. Recycling of PET. Eur. Polym. J. 2005, 41, 1453–1477. [Google Scholar] [CrossRef]
- Miller, C. Polyethylene terephthalate. Waste Age 2002, 33, 102–106. [Google Scholar]
- Wu, H.; Lv, S.; He, Y.; Qu, J. The study of the thermomechanical degradation and mechanical properties of PET recycled by industrial-scale elongational processing. Polym. Test. 2018, 77, 105882. [Google Scholar] [CrossRef]
- Fann, D.M.; Huang, S.K.; Lee, L.Y. Kinetics and thermal crystallinity of recycled PET. II. Topographic study on thermal crystallinity of the injection-molded recycled PET. J. Appl. Polym. Sci. 1996, 61, 261–271. [Google Scholar] [CrossRef]
- Abu-isa, I.; Jaynes, C.B.; Ogara, J.F. High-impact-strength poly(ethylene terephthalate) (PET) from virgin and recycled resins. J. Appl. Polym. Sci. 1996, 59, 1957–2105. [Google Scholar] [CrossRef]
- Schyns, Z.; Shaver, M.P. Mechanical Recycling of Packaging Plastics: A Review. Macromol. Rapid Commun. 2020, 42, 2000415. [Google Scholar] [CrossRef]
- La Mantia, F.P.; Vinci, M. Recycling poly(ethyleneterephthalate). Polym. Degrad. Stab. 1994, 45, 121–125. [Google Scholar] [CrossRef]
- Alvarado Chacon, F.; Brouwer, M.T.; Thoden van Velzen, E.U. Effect of recycled content and rPET quality on the properties of PET bottles, part I: Optical and mechanical properties. Packaging Technology and Science. Am. J. Nano Res. Appl. 2020, 3, 11–16. [Google Scholar] [CrossRef]
- Elamri, A.; Abid, K.; Harzallah, O.; Lallam, A. Characterization of Recycled/ Virgin PET Polymers and their Composites. Am. J. Nano Res. Appl. Spec. Issue Nanocompos. Coat. Manuf. 2015, 3, 11–16. [Google Scholar] [CrossRef]
- Asensio, M.; Nuñez, K.; Guerrero, J.; Herrero, M.; Merino, J.C.; Pastor, J.M. Rheological modification of recycled poly(ethylene terephthalate): Blending and reactive extrusion. Polym. Degrad. Stab. 2020, 179, 109258. [Google Scholar] [CrossRef]
- Cavalcanti, N.; Teofilo, E.T.; Rabello, M.S.; Silva, S.M.L. Chain extension and degradation during reactive processing of PET in the presence of triphenyl phosphite. Polym. Eng. Sci. 2007, 47, 2155–2163. [Google Scholar] [CrossRef]
- Raffa, P.; Coltelli, M.B.; Savi, S.; Bianchi, S.; Castelvetro, V. Chain extension and branching of poly(ethylene terephthalate) (PET) with di- and multifunctional epoxy or isocyanate additives: An experimental and modelling study. React. Funct. Polym. 2012, 72, 50–60. [Google Scholar] [CrossRef]
- Benvenuta Tapia, J.J.; Hernández Valdez, M.; Cerna Cortez, J.; Díaz García, V.M. Ultraviolet-induced chain extension of poly(ethylene terephthalate) based on radical reaction with the aid of trimethylolpropane triacrylate and glycidyl methacrylate during extrusion. J. Polym. Environ. 2018, 26, 4221. [Google Scholar] [CrossRef]
- Wang, K.; Qian, J.; Lou, F.; Yan, W.; Wu, G.; Guo, W. The effects of two-step reactive processing on the properties of recycled poly(ethylene terephthalate). Polym. Bull. 2017, 74, 2479. [Google Scholar] [CrossRef]
- González, J.; Rosales, C.; Luis, A.; Candal, M.; Albano, C. Morphological, Thermal and Mechanical Behavior of Pet/Pp-G-Dem and Nylon 6/Pp-G-Dem Blends. In Proceedings of the ANTEC’98, Atlanta, GA, USA, 26–30 April 1998. [Google Scholar]
- Zhang, Z.; Wang, C.; Mai, K. Reinforcement of Recycled PET for Mechanical Properties of Isotactic Polypropylene. Adv. Ind. Eng. Polym. Res. 2019, 2, 69–76. [Google Scholar] [CrossRef]
- Ávila, A.F.; Duarte, M.V. A mechanical analysis on recycled PET/HDPE composites. Polym. Degrad. Stab. 2003, 80, 373–382. [Google Scholar] [CrossRef]
- Velásquez, E.J.; Garrido, L.; Guarda, A.; Galotto, M.J.; López de Dicastillo, C. Increasing the incorporation of recycled PET on polymeric blends through the reinforcement with commercial nanoclays. Appl. Clay Sci. 2019, 180, 105185. [Google Scholar] [CrossRef]
- Duarte, I.S.; Tavares, A.A.; Lima, P.S.; Andrade, D.L.; Carvalho, L.H.; Canedo, E.L.; Silva, S.M. Chain extension of virgin and recycled poly(ethylene terephthalate): Effect of processing conditions and reprocessing. Polym. Degrad. Stab. 2016, 124, 26–34. [Google Scholar] [CrossRef]
- Awaja, F.; Daver, F.; Kosior, E. Recycled poly (ethylene terephthalate) chain extension by a reactive extrusion process. Polym. Eng. Sci. 2004, 44, 1579–1587. [Google Scholar] [CrossRef]
- Incarnato, L.; Scarfato, P.; Di Maio, L.; Acierno, D. Structure and rheology of recycled PET modified by reactive extrusion. Polymer 2000, 41, 6825–6831. [Google Scholar] [CrossRef]
- Tramis, C.; Garnier, C.; Yus, S.; Irusta, F.; Chaberta, F. Enhancement of the fatigue life of recycled PP by incorporation of recycled opaque PET collected from household milk bottle wastes. Waste Manag. 2021, 125, 49–57. [Google Scholar] [CrossRef]
- American Society for Testing and Materials. ASTM D 4603: Standard Test Method for Determining Inherent Viscosity of Poly(Ethylene Terephthalate) (PET) by Glass Capillary Viscometer; American Society for Testing and Materials: West Conshohocken, PA, USA, 1993. [Google Scholar]
- Karayannidis, G.P.; Kokkalas, D.E.; Bikiaris, D.N. Solid-state polycondensation of poly(ethylene terephthalate) recycled from post-consumer soft-drink bottles. I. J. Appl. Polym. Sci. 1993, 50, 2135–2142. [Google Scholar] [CrossRef]
- Lorenzo, A.T.; Arnal, M.L.; Albuerne, J.; Müller, A.J. DSC isothermal polymer crystallization kinetics measurements and the use of the Avrami equation to fit the data: Guidelines to avoid common problems. Polym. Test. 2007, 26, 222–231. [Google Scholar] [CrossRef]
- Karsl, N.G. A study on the fracture, mechanical and thermal properties of chain extended recycled poly(ethylene terephthalate). J. Thermoplast. Compos. Mater. 2017, 30, 1157–1172. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Li, H.; Du, Z.; Li, C. Chain extension of poly (ethylene terephthalate) with bisphenol-A dicyanate. J. Appl. Polym. Sci. 2010, 117, 2003–2008. [Google Scholar] [CrossRef]
- Tavares, E.; Silva, A.A.; Lima, D.F.A.; Andrade, P.S.; Silva, D.L.A.C.S.; Canedo, S.M.L. Chain extension of virgin and recycled polyethylene terephthalate. Polym. Test. 2016, 50, 26–32. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, J.; Ma, Y.; Agarwal, U.S. Study on mechanical properties, thermal stability and crystallization behavior of PET/MMT nanocomposites. Compos. Part B Eng. 2006, 37, 399–407. [Google Scholar] [CrossRef]
- Hu, G.; Zhao, C.; Zhang, S.; Yang, M.; Wang, Z. Low percolation thresholds of electrical conductivity and rheology in poly(ethylene terephthalate) through the networks of multi-walled carbon nanotubes. Polymer 2006, 47, 480–488. [Google Scholar] [CrossRef]
- Das, P.; Tiwari, P. Thermal degradation study of waste polyethylene terephthalate (PET) under inert and oxidative environments. Thermochim. Acta 2019, 679, 178340. [Google Scholar] [CrossRef]
- Liu, H.; Wang, R.; Xu, X. Thermal stability and flame retardancy of PET/magnesium salt composites. Polym. Degrad. Stab. 2010, 95, 1466–1470. [Google Scholar] [CrossRef]
- Alongi, J.; Ciobanu, M.; Tata, J.; Carosio, F.; Malucelli, G. Thermal stability and flame retardancy of polyester, cotton, and relative blend textile fabrics subjected to sol–gel treatments. J. Appl. Polym. Sci. 2011, 119, 1961–1969. [Google Scholar] [CrossRef]
- Antoniadis, G.; Paraskevopoulos, K.M.; Vassiliou, A.A.; Papageorgiou, G.Z.; Bikiaris, D.; Chrissafis, K. Nonisothermal melt-crystallization kinetics for in situ prepared poly (ethylene terephthalate)/monmorilonite (PET/OMMT). Thermochim. Acta 2011, 521, 161–169. [Google Scholar] [CrossRef]
- Antoniadis, G.; Paraskevopoulos, K.M.; Bikiaris, D.; Chrissafis, K. Non-isothermal crystallization kinetic of poly (ethylene terephthalate)/fumed silica (PET/SiO2) prepared by in situ polymerization. Thermochim. Acta 2010, 510, 103–112. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A. Bloch Polymer Handbook, 4th ed.; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Varma, P.; Lofgren, E.A.; Jabarin, S. A Properties and kinetics of thermally crystallized orientated poly(ethylene terephthalate) (PET) I: Kinetics of crystallization. Polym. Eng. Sci. 1998, 38, 237–244. [Google Scholar] [CrossRef]
- Wang, Z.G.; Hsiao, B.S.; Fu, B.X.; Liu, L.; Yeh, F.; Sauer, B.B.; Chang, H.; Schultz, J.M. Correct determination of crystal lamellar thickness in semi crystalline poly (ethylene terephthalate) by small-angle X-ray scattering. Polymer 2000, 41, 1791–1797. [Google Scholar] [CrossRef]
- Daubeny, R.D.P.; Bunn, C.W.; Brown, C.J. The crystal structure of polyethylene terephthalate. Proc. R. Soc. 1956, A226, 531. [Google Scholar] [CrossRef]
- Lu, X.F.; Hay, J.N. Isothermal crystallization kinetics and melting behaviour of poly (ethylene terephthalate). Polymer 2001, 42, 9423–9431. [Google Scholar] [CrossRef]
- Müller, A.J.; Michell, R.M.; Lorenzo, A.T. Chapter 11. Isothermal Crystallization Kinetics of Polymers. In Polymer Morphology: Principles, Characterization, and Processing; Guo, Q., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 181–203. [Google Scholar] [CrossRef]
- Lorenzo, A.T.; Müller, A.J. Estimation of the nucleation and crystal growth contributions to the overall crystallization energy barrier. J. Polym. Sci. Part B Polym. Phys. 2008, 46, 1478–1487. [Google Scholar] [CrossRef]
- Gaonkar, A.A.; Murudkar, V.V.; Deshpande, V.D. Comparison of crystallization kinetics of polyethylene terephthalate (PET) and reorganized PET. Thermochim. Acta 2020, 683, 178472. [Google Scholar] [CrossRef]
- Sorrentino, L.; Iannace, S.; Di Maio, E.; Acierno, D. Isothermal crystallization kinetics of chain-extended PET. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 1966–1972. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Stone, J.; Sbirrazzuoli, N. Hoffman-Lauritzen parameters for non-isothermal crystallization of poly (ethylene terephthalate) and poly (ethylene oxide) melts. J. Therm. Anal. Calorim. 2005, 80, 177–180. [Google Scholar] [CrossRef]
- Jiang, X.L.; Luo, S.J.; Sun, K.; Chen, X.D. Effect of nucleating agents on crystallization kinetics of PET. Express Polym. Lett. 2007, 1, 245–251. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers; Wiley: New York, NY, USA, 1980. [Google Scholar]
- Tschoegl, N.W. The Phenomenological Theory of Linear Viscoelastic Behavior: An Introduction; Springer: New York, NY, USA, 1989. [Google Scholar]
- Bird, R.B.; Armstrong, R.C.; Hassager, O. Dynamics of Polymeric Liquids; Wiley: New York, NY, USA, 1987; Volume 1. [Google Scholar]
- Kruse, M.; Wagner, M. Rheological and molecular characterization of long-chaing branched poly(ethylene terphthalate). Rheol. Acta 2017, 56, 887–904. [Google Scholar] [CrossRef]
- Kil, S.B.; Augros, T.; Leterrier, Y.; Manson, J.A.E.; Christel, A.; Borer, C. Rheological properties of hyperbranched polymer/poly(ethylene terephthalate) reactive blends. Polym. Eng. Sci. 2003, 43, 329–343. [Google Scholar] [CrossRef]
- Ge, Y.; Yao, S.; Xu, M.; Gao, L.; Fang, Z.; Zhao, L.; Liu, T. Improvement of Poly(ethylene terephthalate) Melt-Foamability by Long-Chain Branching with the Combination of Pyromellitic Dianhydride and Triglycidyl Isocyanurate. Ind. Eng. Chem. Res. 2019, 58, 3666–3678. [Google Scholar] [CrossRef]
- Keßner, U.; Kaschta, J.; Münstedt, H. Determination of method-invariant activation energies of long-chain branched low-density polyethylenes. J. Rheol. 2009, 53, 1001–1016. [Google Scholar] [CrossRef]
- Van Gurp, M.; Palmen, J. Time-temperature superposition for polymeric blends. Rheol. Bull 1998, 67, 5–8. [Google Scholar]
- Lohse, D.J.; Milner, S.T.; Fetters, L.J.; Xenidou, M.; Hadjichristidis, N.; Mendelson, R.A.; García-Franco, C.A.; Lyon, M.K. Well-defined model long chain branched polyethylene. 2. Melt rheological behavior. Macromolecules 2002, 35, 3066–3075. [Google Scholar] [CrossRef]
- Wood-Adams, P.; Costeux, S. Thermorheological Behavior of Polyethylene: Effects of Microstructure and Long Chain Branching. Macromolecules 2001, 34, 6281–6290. [Google Scholar] [CrossRef]
- Trinkle, S.; Walter, P.; Friedrich, C. Van Gurp-Palmen plot II—Classification of long chain branched polymers by their topology. Rheol. Acta 2002, 41, 103–113. [Google Scholar] [CrossRef]
- Stadler, F.J.; Kaschta, J.; Munstedt, H. Thermorheological behavior of various long-chain branched polyethylene. Macromolecules 2008, 41, 1328–1333. [Google Scholar] [CrossRef]
- Abbasi, M.; Faust, L.; Riazi, K.; Wilhelm, M. Linear and Extensional Rheology of Model Branched Polystyrenes: From Loosely Grafted Combs to Bottlebrushes. Macromolecules 2017, 50, 5964–5977. [Google Scholar] [CrossRef]
- Lee, L.H.; Orfanou, K.; Driva, P.; Iatrou, H.; Hadjichristidis, N.; Lohse, D.J. Linear and Nonlinear Rheology of Dendritic Star Polymers: Experiment. Macromolecules 2008, 41, 9165–9178. [Google Scholar] [CrossRef]
- Nielsen, J.K.; Rasmussen, H.K.; Denberg, M.; Almdal, K.; Hassager, O. Nonlinear Branch-Point Dynamics of Multiarm Polystyrene. Macromolecules 2006, 39, 8844–8853. [Google Scholar] [CrossRef]
- Chen, X.; Rahman, M.S.; Lee, H.; Mays, J.; Chang, T.; Larson, R. Combined Synthesis, TGIC Characterization, and Rheological Measurement and Prediction of Symmetric H Polybutadienes and Their Blends with Linear and Star-Shaped Polybutadienes. Macromolecules 2011, 44, 7799–7809. [Google Scholar] [CrossRef]
- Kapnistos, M.; Vlassopoulos, D.; Roovers, J.; Leal, L.G. Linear Rheology of Architecturally Complex Macromolecules: Comb Polymers with Linear Backbones. Macromolecules 2005, 38, 7852–7862. [Google Scholar] [CrossRef]
- Lentzakis, H.; Vlassopoulos, D.; Read, D.J.; Lee, H.; Chang, T.; Driva, P.; Hadjichristidis, N. Uniaxial extensional rheology of well characterized comb polymers. J. Rheol. 2013, 57, 605–625. [Google Scholar] [CrossRef] [Green Version]
- Van Ruymbeke, E.; Kapnistos, M.; Vlassopoulos, D.; Huang, T.; Knauss, D.M. Linear Melt Rheology of Pom-Pom Polystyrenes with Unentangled Branches. Macromolecules 2007, 40, 1713–1719. [Google Scholar] [CrossRef]
- Lentzakis, H.; Das, C.; Vlassopoulos, D.; Read, D.J. Pom-pom like constitutive equations for comb polymers. J. Rheol. 2014, 58, 1855–1875. [Google Scholar] [CrossRef] [Green Version]
- Kempf, M.; Barroso, V.C.; Wilhelm, M. Anionic Synthesis and Rheological Characterization of Poly(p-methylstyrene) Model Comb Architectures with a Defined and Very Low Degree of Long Chain Branching. Macromol. Rapid Commun. 2010, 31, 2140–2145. [Google Scholar] [CrossRef]
- Liu, G.; Ma, H.; Lee, H.; De Xua, H.; Cheng, S.; Sun, H.; Chang, T.; Quirk, R.P.; Wang, S. Long-chain branched polymers to prolong homogeneous stretching and to resist melt breakup. Polymer 2013, 54, 6608–6616. [Google Scholar] [CrossRef]
- Dealy, J.M.; Larson, R.G. Structure and Rheology of Molten Polymers: From Structure to Flow Behavior and Back Again, 1st ed.; Carl Hanser Verlag: Munich, Germany, 2006. [Google Scholar]
- Hyun, K.; Kim, W. A new non-linear parameter Q from FT-Rheology under non-linear dynamic oscillatory shear for polymer melts system. Korea Aust. Rheol. J. 2011, 23, 227–235. [Google Scholar] [CrossRef]
- Hoyle, D.M.; Auhl, D.; Harlen, O.G.; Barroso, V.C.; Wilhelm, M.; McLeish, T.C.B. Large amplitude oscillatory shear and Fourier transform rheology analysis of branched polymer melts. J. Rheol. 2014, 58, 969–997. [Google Scholar] [CrossRef] [Green Version]
- Hyun, K.; Kim, W.; Park, S.; Wilhelm, M. Numerical simulation results of the non-linear coefficient Q from ft-rheology using a single mode pompom model. J. Rheol. 2013, 57, 1–25. [Google Scholar] [CrossRef]
- Reinheimer, K.; Grosso, M.; Hetzel, F.; Kübel, J.; Wilhelm, M. Fourier Transform Rheology as an innovative morphological characterization technique for the emulsion volume average radius and its distribution. J. Colloid Interface Sci. 2012, 380, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Salehiyan, R.; Song, H.Y.; Hyun, K. Non-linear behavior of PP / PS blends with and without clay under large amplitude oscillatory shear (LAOS) flow. Korea-Aust. Rheol. J. 2015, 27, 95–103. [Google Scholar] [CrossRef]
- Sangroniz, L.; Palacios, J.K.; Fernández, M.; Ignacio, J.; Santamaria, A.; Müller, A.J. Linear and non-linear rheological behavior of polypropylene/polyamide blends modified with a compatibilizer agent and nanosilica and its relationship with the morphology. Eur. Polym. J. 2016, 83, 10–21. [Google Scholar] [CrossRef]
- Cho, K.; Hyun, K.; Kyung, L.; Seung, A. Geometrical interpretation of large amplitude oscillatory shear response. J. Rheol. 2005, 45, 747–758. [Google Scholar] [CrossRef]
- Ewoldt, R.H.; Hosoi, A.E.; McKinley, G.H. New measures for characterizing non-linear viscoelasticity in large amplitude oscillatory shear. J. Rheol. 2008, 52, 1427–1458. [Google Scholar] [CrossRef] [Green Version]
- Hyun, K.; Wilhelm, M.; Klein, C.O.; Soo Cho, K.; Gun Nam, J.; Hyun Ahn, H.; Jong Lee, S.; Ewoldt, R.H.; McKinley, G.H. A review of non-linear oscillatory shear tests: Analysis and application of large amplitude oscillatory shear (LAOS). Prog. Polym. Sci. 2011, 36, 1697–1753. [Google Scholar] [CrossRef]
- Sugimoto, M.; Suzuki, Y.; Hyun, K.; Ahn, K.H.; Ushioda, T.; Nishioka, A.; Taniguchi, T.; Koyama, K. Melt rheology of long-chain-branched polypropylenes. Rheol. Acta 2006, 46, 33–44. [Google Scholar] [CrossRef]
- Fan, X.; Xu, X.; Wu, C.; Song, Y.; Zheng, Q. Influences of chemical crosslinking, physical associating, and filler filling on non-linear rheological responses of polyisoprene. J. Rheol. 2020, 64, 775–784. [Google Scholar] [CrossRef]
- Hyun, K.; Baik, E.S.; Ahn, K.H.; Lee, S.J.; Sugimoto, M.; Koyama, K. Fourier-transform rheology under medium amplitude oscillatory shear for linear and branched polymer melts. J. Rheol. 2007, 51, 1319–1342. [Google Scholar] [CrossRef]
- Hyun, K.; Wilhelm, M. Establishing a new mechanical non-linear coefficient Q from FT-rheology: First investigation of entangled linear and comb polymer model systems. Macromolecules 2009, 42, 411–422. [Google Scholar] [CrossRef]
- Ahirwal, D.; Filipe, S.; Neuhaus, I.; Busch, M.; Schlatter, G.; Wilhelm, M. Large amplitude oscillatory shear and uniaxial extensional rheology of blends from linear and long-chain branched polyethylene and polypropylene. J. Rheol. 2014, 58, 635–658. [Google Scholar] [CrossRef]
- Botta, L.; Scaffaro, R.; Sutera, F.; Mistretta, M.C. Reprocessing of PLA/graphene nanoplatelets nanocomposites. Polymers 2018, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Selvin, T.P.; Kuruvilla, J.; Sabú, T. Mechanical properties of titanium dioxide-filled polystyrene microcomposites. Mater. Lett. 2004, 58, 281–289. [Google Scholar] [CrossRef]
- Aoyama, S.; Ismail, I.; Tae Park, Y.; Yoshida, Y.; Macosko, C.Y.; Ougizawa, T. Applied Nano Materials Polyethylene Terephthalate/Trimellitic Anhydride Modified Graphene Nanocomposites. ACS Appl. Nano Mater. 2018, 1, 6301–6311. [Google Scholar] [CrossRef]
- Fernandez-Menéndez, T.; García-López, D.; Argüelles, A.; Fernandez, A.; Viña, J. Industrially produced PET nanocomposites with enhanced properties for food packaging applications. Polym. Test. 2020, 90, 106729. [Google Scholar] [CrossRef]
- Nikam, P.N.; Deshpande, V.D. Thermal and tensile properties of alumina filled PET nanocomposites. AIP Conf. Proc. 2018, 1953, 090058. [Google Scholar]
- Kim, J.Y.; Park, H.S.; Kim, S.H. Multiwall-carbon-nanotube-reinforced poly(ethylene terephthalate) nanocomposites by melt compounding. J. Appl. Polym. Sci. 2007, 103, 1450–1457. [Google Scholar] [CrossRef]
- Rodríguez, J.L.; Equiazabal, J.I.; Nazaba, J. Phase Behavior and Interchange Reactions in Poly(butylene terephthalate)/Poly(ester-carbonate) Blends. Polym. J. 1996, 28, 501–506. [Google Scholar] [CrossRef] [Green Version]
- Vallejo, F.J.; Eguiazabal, J.L.; Nazabal, J. Solid-state features and mechanical properties of PEI/PBT blends. J. Appl. Polym. Sci. 2001, 80, 885–892. [Google Scholar] [CrossRef]
- Brostow, W.; Corneliussen, R.D. Failure of Plastics. Hanser Pub.: Munich, Germany, 1986. [Google Scholar]
- Yeh, C.C.; Chen, C.N.; Li, Y.T.; Chang, C.W.; Cheng, M.Y.; Chang, H.I. The Effect of Polymer Molecular Weight and UV Radiation on Physical Properties and Bioactivities of PCL Films. Cell. Polym. 2011, 30, 261–276. [Google Scholar] [CrossRef]
- Andrady, A.L.; Pegram, J.E.; Trpsha, Y. Changes in carbonyl index and average molecular weight on embrittlement of enhanced-photodegradable polyethylenes. J. Environ. Polym. Degrad. 1993, 1, 171–179. [Google Scholar] [CrossRef]
- Spinacé, M.A.S.; De Paoli, M.A. Characterization of poly(ethylene terephtalate) after multiple processing cycles. J. Appl. Polym. Sci. 2001, 80, 20–25. [Google Scholar] [CrossRef]
- Bao, R.Y. Balanced strength and ductility improvement of in situ crosslinked polylactide/poly(ethylene terephthalate glycol) blends. RSC Adv. 2015, 5, 34821–34830. [Google Scholar] [CrossRef]
- Lozano-González, M.J.; Rodriguez-Hernandez, M.T.; Gonzalez-De Los Santos, E.A.; Villalpando-Olmos, J. Physical-mechanical properties and morphological study on nylon-6 recycling by injection molding. J. Appl. Polym. Sci. 2000, 76, 851–858. [Google Scholar] [CrossRef]
- Ronkay, F.; Molnár, B.; Szalay, F.; Nagy, D.; Bodsay, B.; Sajó, I.; Bocz, K. Development of Flame-Retarded Nanocomposites from Recycled PET Bottles for the Electronics Industry. Polymers 2019, 11, 233. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Schlarb, A.K.; Evstatiev, M. Study of PET/PP/TiO2 microfibrillar-structured composites, Part 2: Morphology and mechanical properties. J. Appl. Polym. Sci. 2009, 113, 3300–3306. [Google Scholar] [CrossRef]
- Ronkay, F.; Molnar, B.; Gere, D.; Czigany, T. Plastic waste from marine environment: Demonstration of possible routes for recycling by different manufacturing technologies. Waste Manag. 2021, 119, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Nielsen, L.E. The notch sensitivity of polymeric materials. J. Appl. Polym. Sci. 1976, 20, 2193–2207. [Google Scholar] [CrossRef]
- Ogazi-Onyemaechi, B.C.; Leong, Y.W.; Hamada, H. Crack propagation behavior and toughness of V-notched polyethylene terephthalate injection moldings. J. Appl. Polym. Sci. 2010, 116, 132–141. [Google Scholar] [CrossRef]
- Chukwuemeka, B.; Wei, Y.; Hamada, H. Dependence of polyethylene terephthalate crack-tip temperature on stress intensity and notch sensitivity. Polym. J. 2010, 42, 592–599. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | [η] * (dL/g) | Mw (kDa) | Mn (kDa) |
|---|---|---|---|
| Virgin PET | 0.702 | 45.0 | 22.5 |
| r-PET | 0.526 | 27.1 | 13.6 |
| REX-r-PET | 0.626 | 37.3 | 18.7 |
| Sample | Td,2% | Td1 (°C) | Td2 (°C) | Residue at 800 °C (%) |
|---|---|---|---|---|
| Virgin PET | 326 | 432 | 560.9 | 0 |
| r-PET | 311 | 430 | 561.0 | 2.40 ± 0.08 |
| REX-r-PET | 324 | 432 | 561.0 | 2.62 ± 0.21 |
| First Heating | Cooling | Second Heating | ||||||
|---|---|---|---|---|---|---|---|---|
| Tm (°C) | ΔHm (J/g) | Xc (%) | Tc (°C) | ΔHc (J/g) | Tm (°C) | ΔHm (J/g) | Xc (%) | |
| Virgin PET | 241.0 | 34 | 24 | - | - | 243.5 179.8 (Tcc) | 31 14 (ΔHcc) | 11 |
| r-PET | 242.9 | 41 | 30 | 183.6 | 35 | 239.9 | 36 | 27 |
| REX-r-PET | 241.5 | 40 | 29 | 178.3 | 30 | 237.9 | 32 | 23 |
| Sample Name | (K2) | σ (erg/cm2) | σe (erg/cm2) | q (erg) | R2 |
|---|---|---|---|---|---|
| Virgin PET | 3.81E+05 | 9.03 | 275 | 1.01E−12 | 0.992 |
| r-PET | 3.84E+05 | 9.03 | 276 | 1.02E−12 | 0.978 |
| REX-r-PET | 5.91E+05 | 9.03 | 426 | 1.57E−12 | 0.991 |
| Surface | Core | |||||||
|---|---|---|---|---|---|---|---|---|
| Materials | Tm (°C) | ΔHm (J/g) | Tcc (°C) | ΔHcc (J/g) | Xc (%) | Tm (°C) | ΔHm (J/g) | Xc (%) |
| Virgin PET-i | 240.9 | 29 | 117.1 | 3.5 | 18 | 240.2 | 39 | 29 |
| r-PET-i | 239.4 | 24 | 123.0 | 9.4 | 11 | 244.2 | 32 | 23 |
| REX-r-PET-i | 239.2 | 27 | 129.1 | 10.3 | 12 | 245.9 | 41 | 30 |
| Sample | Impact Strength (J/m) |
|---|---|
| Virgin PET | 27.6 ± 0.7 |
| r-PET | 21.5 ± 3.1 |
| REX-r-PET | 24.3 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Candal, M.V.; Safari, M.; Fernández, M.; Otaegi, I.; Múgica, A.; Zubitur, M.; Gerrica-echevarria, G.; Sebastián, V.; Irusta, S.; Loaeza, D.; et al. Structure and Properties of Reactively Extruded Opaque Post-Consumer Recycled PET. Polymers 2021, 13, 3531. https://doi.org/10.3390/polym13203531
Candal MV, Safari M, Fernández M, Otaegi I, Múgica A, Zubitur M, Gerrica-echevarria G, Sebastián V, Irusta S, Loaeza D, et al. Structure and Properties of Reactively Extruded Opaque Post-Consumer Recycled PET. Polymers. 2021; 13(20):3531. https://doi.org/10.3390/polym13203531
Chicago/Turabian StyleCandal, María Virginia, Maryam Safari, Mercedes Fernández, Itziar Otaegi, Agurtzane Múgica, Manuela Zubitur, Gonzalo Gerrica-echevarria, Víctor Sebastián, Silvia Irusta, David Loaeza, and et al. 2021. "Structure and Properties of Reactively Extruded Opaque Post-Consumer Recycled PET" Polymers 13, no. 20: 3531. https://doi.org/10.3390/polym13203531