A Robust and Sensitive Spectrophotometric Assay for the Enzymatic Activity of Bacterial Adenylate Cyclase Toxins




1
Biochemistry of Macromolecular Interactions Unit, Department of Structural Biology and Chemistry, Institut Pasteur, Université Paris Cité, CNRS UMR 3528, 75015 Paris, France
2
Université Paris Cité, 75014 Paris, France
3
Structural Bioinformatic Unit, Department of Structural Biology and Chemistry, Institut Pasteur, Université Paris Cité, CNRS UMR 3528, 75015 Paris, France
4
Université Paris Sorbonne, 75231 Paris, France
*
Author to whom correspondence should be addressed.
Toxins 2022, 14(10), 691; https://doi.org/10.3390/toxins14100691
Submission received: 12 September 2022
/
Revised: 27 September 2022
/
Accepted: 28 September 2022
/
Published: 8 October 2022
(This article belongs to the Special Issue Toxins: Mr Hyde or Dr Jekyll?)
Abstract
:Various bacterial pathogens are producing toxins that target the cyclic Nucleotide Monophosphate (cNMPs) signaling pathways in order to facilitate host colonization. Among them, several are exhibiting potent nucleotidyl cyclase activities that are activated by eukaryotic factors, such as the adenylate cyclase (AC) toxin, CyaA, from Bordetella pertussis or the edema factor, EF, from Bacillus anthracis. The characterization of these toxins frequently requires accurate measurements of their enzymatic activity in vitro, in particular for deciphering their structure-to-function relationships by protein engineering and site-directed mutagenesis. Here we describe a simple and robust in vitro assay for AC activity based on the spectrophotometric detection of cyclic AMP (cAMP) after chromatographic separation on aluminum oxide. This assay can accurately detect down to fmol amounts of B. pertussis CyaA and can even be used in complex media, such as cell extracts. The relative advantages and disadvantages of this assay in comparison with other currently available methods are briefly discussed.
Keywords:
adenylate cyclase toxin; Bordetella pertussis; cyclic nucleotide; cAMP; spectrophotometric enzymatic assayKey Contribution: A simple and robust in vitro spectrophotometric assay for adenylate cyclase activity is described and shown to be capable of detecting fmol amounts of Bordetella pertussis CyaA toxin.
1. Introduction
Cyclic Nucleotides Monophosphates (cNMPs) are key messengers in most cell types that have been implicated in the modulation of numerous physiological processes [1]. Many pathogens have evolved sophisticated strategies to disrupt cNMP metabolism in the hosts they attempt to colonize [2]. In particular, several bacterial pathogens produce toxins that are endowed with endogenous nucleotidyl cyclase activity [3]. These toxins are able to invade eukaryotic cells where they are stimulated by endogenous co-factors to produce large amounts of cNMP, thus disrupting cell signaling and altering cell physiology. To date, the best characterized types are the adenylate cyclase (AC) toxin, CyaA, from Bordetella pertussis [4,5] and the edema factor, EF, from Bacillus anthracis [6]. They are key virulence factors that target the immune cells to promote efficient bacterial colonization of the hosts [7,8,9]. Both CyaA and EF are potently activated by the eukaryotic protein, calmodulin (CaM), and have very high catalytic efficiency (with kcat > 1000 s−1). Their catalytic moieties share remarkable structural similarity, while these two toxins differ markedly in their sequences, their secretion mechanisms, and their modes of invasion of eukaryotic target cells [3]. A distinct sub-family of virulent factors with nucleotidyl cyclase activity [10,11] has been recently characterized as being activated by eukaryotic actin [12]. It includes the ExoY toxin that is a type-3 virulent effector produced by P. aeruginosa and various ExoY-like modules present in different MARTX (Multifunctional-Autoprocessing Repeats-in-Toxin) toxins produced by certain Vibrio species as well as by other Gram-negative bacteria [3,13].
The characterization of these virulent factors frequently requires the determination of their enzymatic activities, which is the conversion of adenosine triphosphate (ATP) into cyclic AMP (cAMP) and pyrophosphate (PPi). Indeed, numerous studies have been devoted to the identification of critical residues in the catalytic reaction and/or the binding of the toxins to their eukaryotic co-factors [2,3]. Simple and rapid assays for determining enzymatic activities of recombinant enzymes harboring specific mutations would greatly facilitate such investigations.
One of the most widely applied in vitro techniques uses a radioactive substrate [α-32P or α-33P]-ATP that is converted into a radioactive cAMP product that is subsequently isolated by chromatography on neutral alumina and quantified with a scintillation counter. A main drawback of this approach is that it requires specific equipment and laboratory premises to safely handle radioactivity. To avoid this major hurdle, a variety of alternative enzymatic assays have been set up, including the measurement of cAMP with ELISA, use of fluorescent ATP substrates, measurement of the pyrophosphate with colorimetric of fluorimetric coupled assays, or measurement of the reverse reaction (eg cAMP + PPi -> ATP) with colorimetric assays (see Table 1).
Here we describe a simple and robust in vitro assay for AC activity based on the spectrophotometric detection of cAMP after chromatographic separation on aluminum oxide. This assay is cost-effective and straightforward for routine monitoring of AC toxin activity and sensitive enough to detect down to fmol amounts of B. pertussis AC. Moreover, with proper controls, it can also be applied to quantify AC in complex mixtures, such as cell extracts. The relative advantages and disadvantages of this assay in comparison with other methods (Table 1) are presented in the Discussion section.
2. Results
2.1. Spectrophotometric Detection of AC Activity
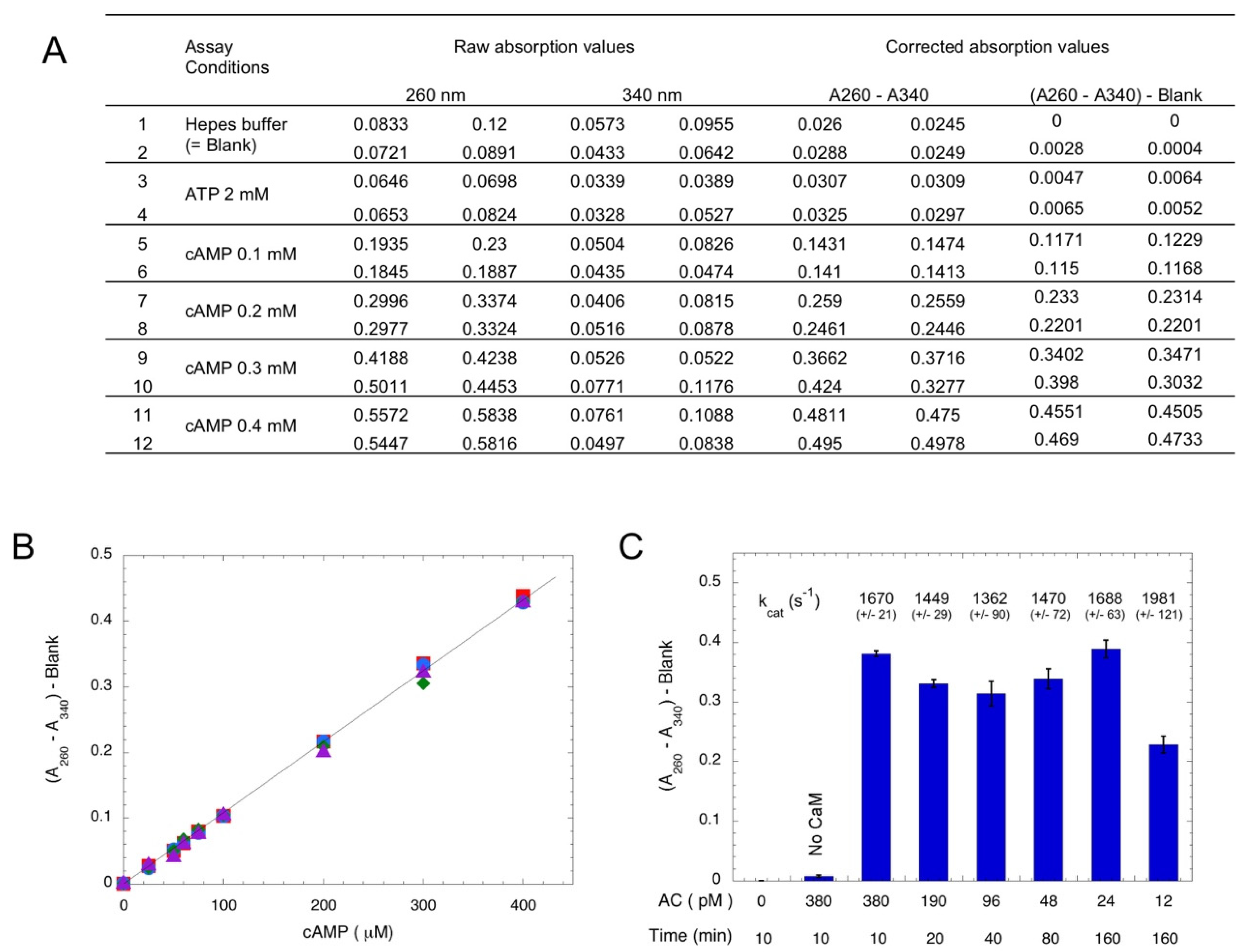
Chromatography over aluminum oxide has been widely used for the separation of cyclic nucleotides from other nucleotides (NTP, NDP, or NMP), as these latter nucleotides are selectively adsorbed on Al2O3 [26,27]. In the following assay procedure, ATP is converted into cAMP by AC in a first step, and in a second step, cAMP is separated from ATP by chromatography on Al2O3; at pH 7.2–7.6, deprotonated ATP (ATP4−, as well as ADP3− and AMP2−) is retained on the alumina, whereas cAMP− is not (provided the ionic strength of the elution buffer is higher than 0.1 M NaCl—data not shown). Thus, the alumina eluent exclusively contains cAMP, which can be easily determined by measuring the absorption at 260 nm, given the high absorption coefficient of cAMP (molar extinction coefficient, ε260nm = 15,000 M−1·cm−1). When using relatively pure AC preparations or highly-active ACs like bacterial AC toxins, there are no other contaminating molecules that absorb light at 260 nm, so the assay is very straightforward and robust. In standard conditions, the reaction is carried out at a 0.1 mL final volume with 2 mM ATP for appropriate time (from 5 min up to several hrs). Incubation is arrested by directly adding 0.3 g of Al2O3 dry powder to the reaction mixture (alternatively, an excess of EDTA can be added to chelate Mg2+, an essential co-factor, before transferring the reaction mixture into pre-weighted Al2O3 dry powder). After the addition of 0.9 mL of HBS buffer (50 mM Hepes, pH 7.5, 0.2 M NaCl), the mixture is incubated for a few minutes with agitation, the tube is centrifuged (5 min at 10–13,000× g), and the absorption at 260 nm (A260) of supernatant is recorded. When multiple assays are carried out in parallel, it is convenient to transfer supernatant (e.g., 0.3 mL) into a UV-transparent microplate to record the absorption with a microplate reader. Raw data are directly available in a spreadsheet file, which facilitates downstream calculations. In addition, the absorption of supernatant at another wavelength (e.g., at 340 nm, A340) is also recorded in order to correct for the potential light scattering that may arise from the contamination of the supernatant by Al2O3 particles, as illustrated in Figure 1A. Figure 1B shows the correspondence between the absorption at 260 nm minus the absorption at 340 nm (A260–A340) of the Al2O3 supernatant as a function of when the cAMP spiked into the standard assay medium. An excellent linearity is observed for cAMP, which varies from 25 to 400 μM. These values correspond to the conversion of 1.25% to 20% of the initial ATP substrate (2 mM), a range that is suitable for standard enzymatic assays. The time and concentration dependence of cAMP synthesis by the purified catalytic domain of CyaA (encoded by the first 384 residues of toxin, AC384 [28]) is shown Figure 1C. Given the high catalytic efficiency of AC384 (with kcat in the range of 1500 to 2000 s−1) and its excellent time stability, this simple assay allows for an accurate determination (i.e., with robust absorption values) of its AC activity down to fmol amounts of purified AC enzyme.
2.2. CaM-Dependent Activation of AC and EF Activities
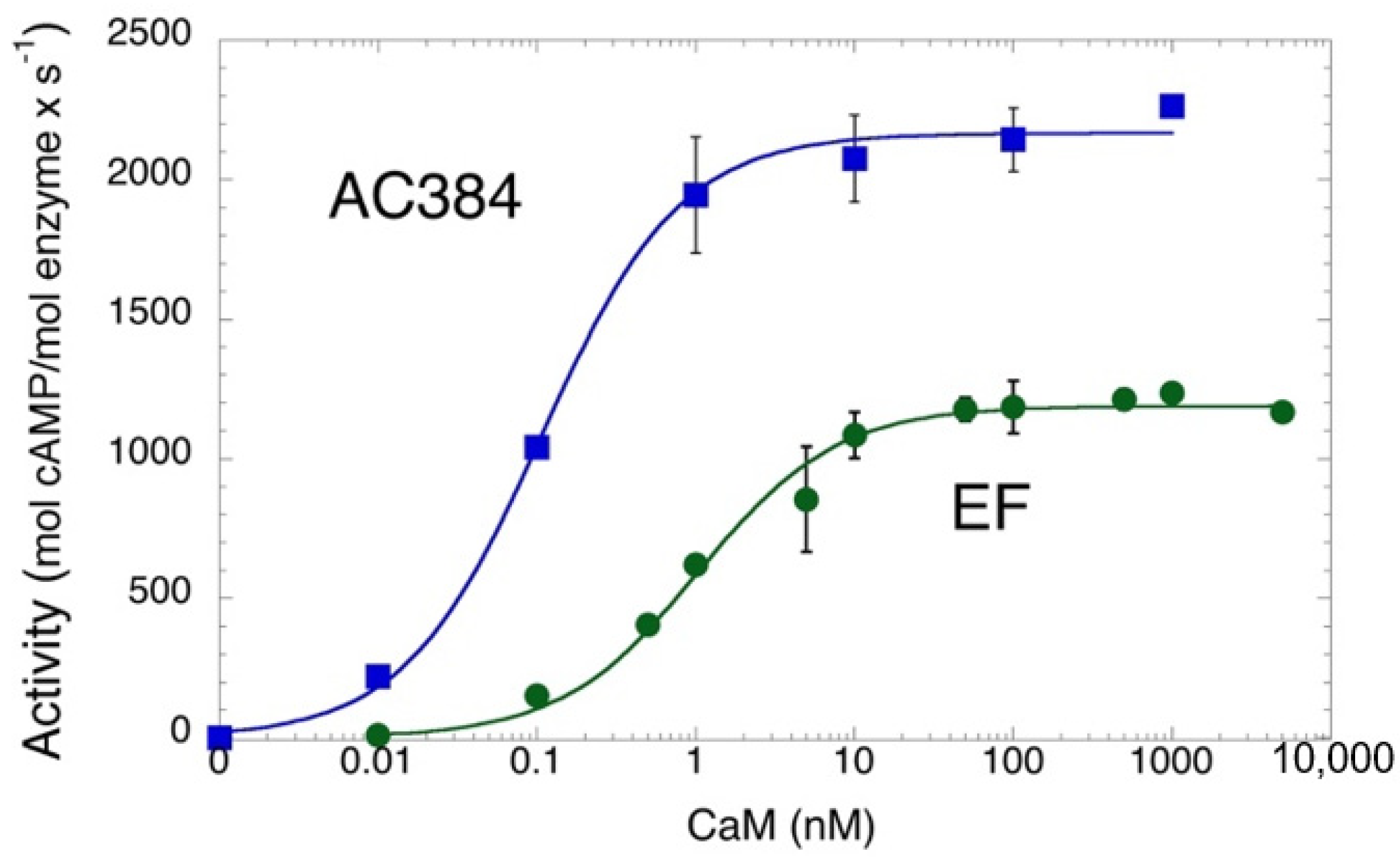
Figure 2 shows the CaM-dependence of the AC enzymatic activity of B. pertussis AC384 as well as that of a purified edema factor, EF, from the B. cereus (G9241 strain; this protein differs from the well-studied B. anthracis EF toxin by three amino acids, Pitard et al. manuscript in preparation). Both enzymes show high catalytic activities and high CaM-affinities in accordance with prior studies [25,29,30]. These data also indicate that the three amino acid replacements in B. cereus EF (I318T, V694A, and N789K, using amino acid numbers from B anthracis EF) have no impact on the catalytic efficiency or CaM affinity of the enzyme. This is consistent with the fact that they are located at the protein surface, away from the catalytic site and the CaM binding interface [31].
2.3. Enzymatic Assays in Complex Media
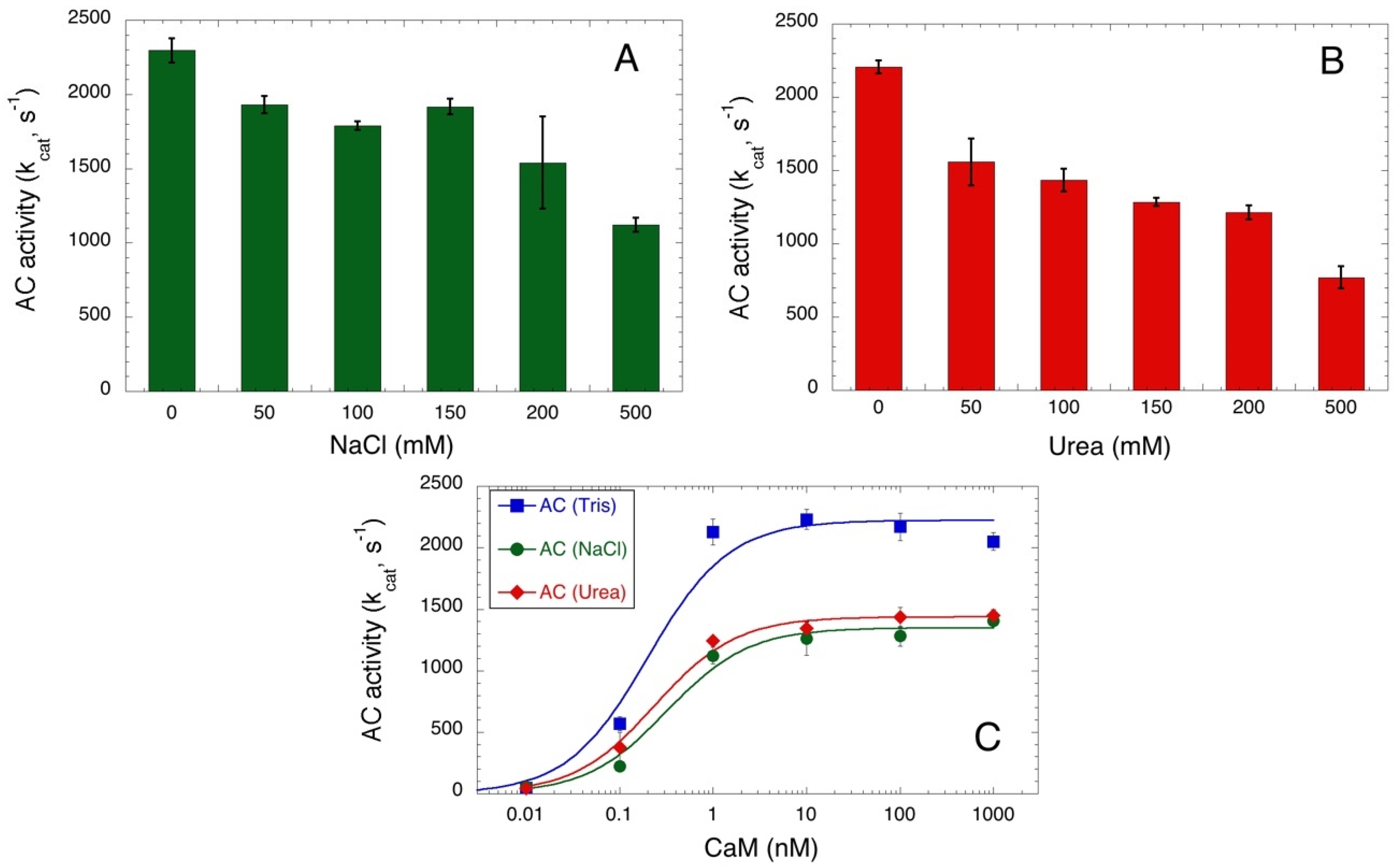
One advantage of this spectrophotometric assay is that it does not require any coupled enzyme to quantify the formation of the cAMP product. Therefore, it can be applied to measure AC activity in conditions that might affect the activity of many enzymes, like the presence of high salt or chaotropic agent concentrations. Figure 3A,B show the AC384 activity in the presence of an increasing concentration of NaCl or urea, respectively. A gradual decrease of activity is observed in both cases, although AC384 retains a significant catalytic efficiency (i.e., higher than 50% of its maximal kcat) even in the presence of up to 0.5 M NaCl or 0.2 M urea. We further examined the CaM-dependency of AC384 activity in the presence of 0.2 M NaCl or 0.2 M urea (Figure 3C) and found that although the maximal activity was reduced, the affinity for CaM was only marginally affected by these compounds.
2.4. Characterization of a Detoxified CyaAE5-OVA Vaccine
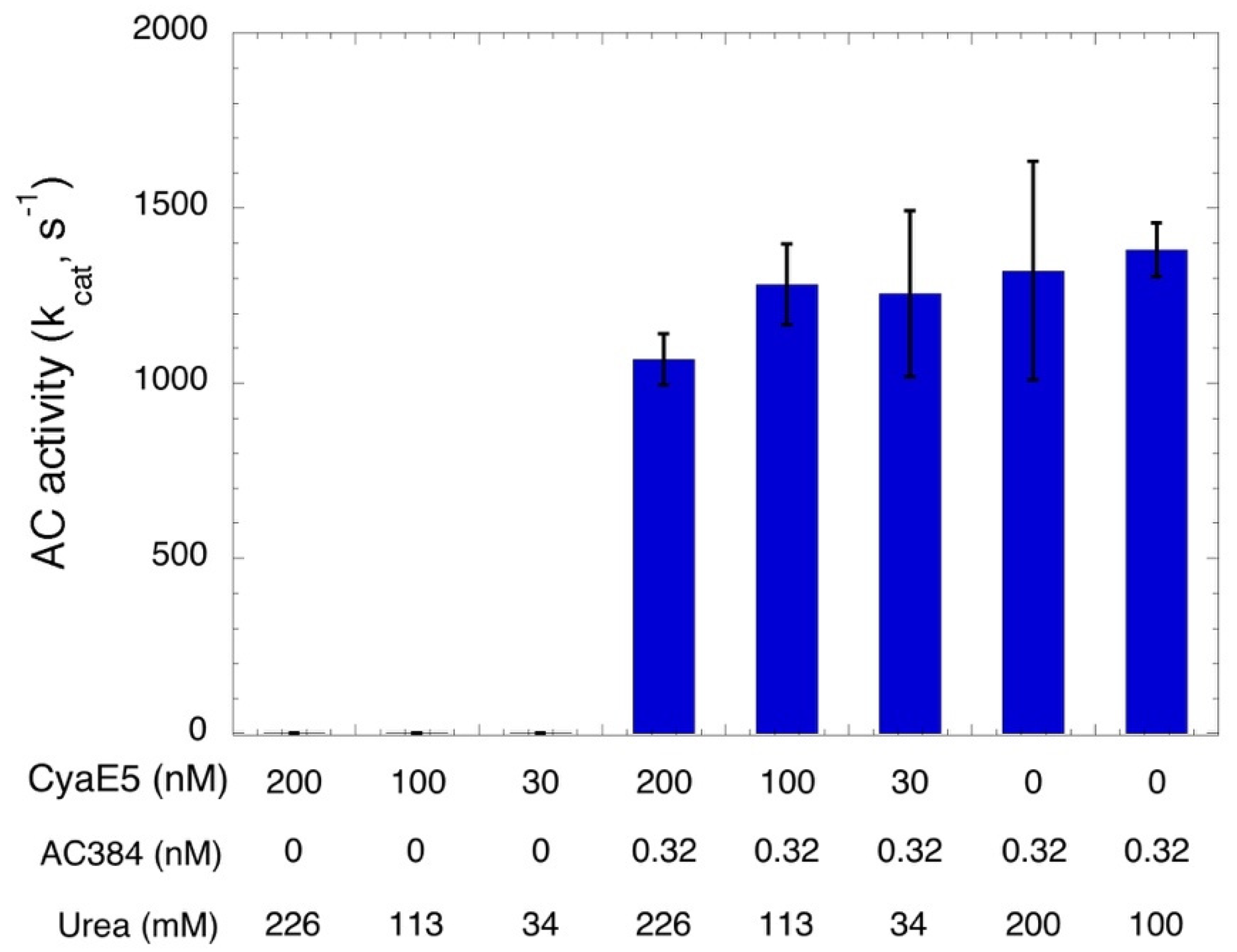
The non-radioactive AC assay can be useful for the characterization of CyaA-based vaccines. Indeed, earlier works have shown that detoxified recombinant CyaAs can be used as efficient antigen delivery vehicles. Various CyaA-based vaccines have been designed to trigger immune responses against infectious agents and/or cancer-specific antigens [32,33,34] (for a review [35]). The demonstration of the lack of AC activity (that is responsible for CyaA toxicity) of the detoxified CyaA vaccine is key in the process of GMP (Good Manufacturing Practice) preparation of protein lots for clinical evaluation. The spectrophotometric AC assay can be easily implemented for this, as illustrated in Figure 4, where a detoxified CyaA protein (CyaAE5, resulting from a specific mutation in catalytic site) carrying a model epitope (OVA, derived from ovalbumin), CyaAE5-OVA [36,37], was assayed at various concentrations. No traces of cAMP synthesis could be evidenced even with the highest concentration tested (200 nM CyaAE5-OVA, corresponding to 20 pmol per assay). Given that CyaAE5-OVA (as all CyaA proteins) was stored in a buffer containing ≈6.6 M urea to prevent the irreversible aggregation of the protein [5,38,39], we checked that the residual urea added to each assay (as indicated in Figure 4) did not affect the enzymatic detection by spiking sub-pmol amounts of active AC384. In all cases, high amounts of cAMP were easily detected. These experiments thus unambiguously demonstrate the total lack of AC activity of the purified CyaAE5-OVA preparation and indicate that this simple spectrophotometric AC assay could be easily implemented for control of GMP preparations of detoxified CyaA vaccines.
2.5. AC Assays in Crude Cell Extracts
To determine if the spectrophotometric assay could also be applied to unpurified or partially-purified AC preparations, or when the AC toxins would be diluted into complex cell extracts, we attempted to characterize CyaA binding to erythrocytes, which are frequently used as model target cells. Although these cells do not express the CyaA receptor (CD11b/CD18), CyaA can directly bind to the erythrocyte plasma membrane in a calcium- and temperature-dependent manner [40,41,42,43].
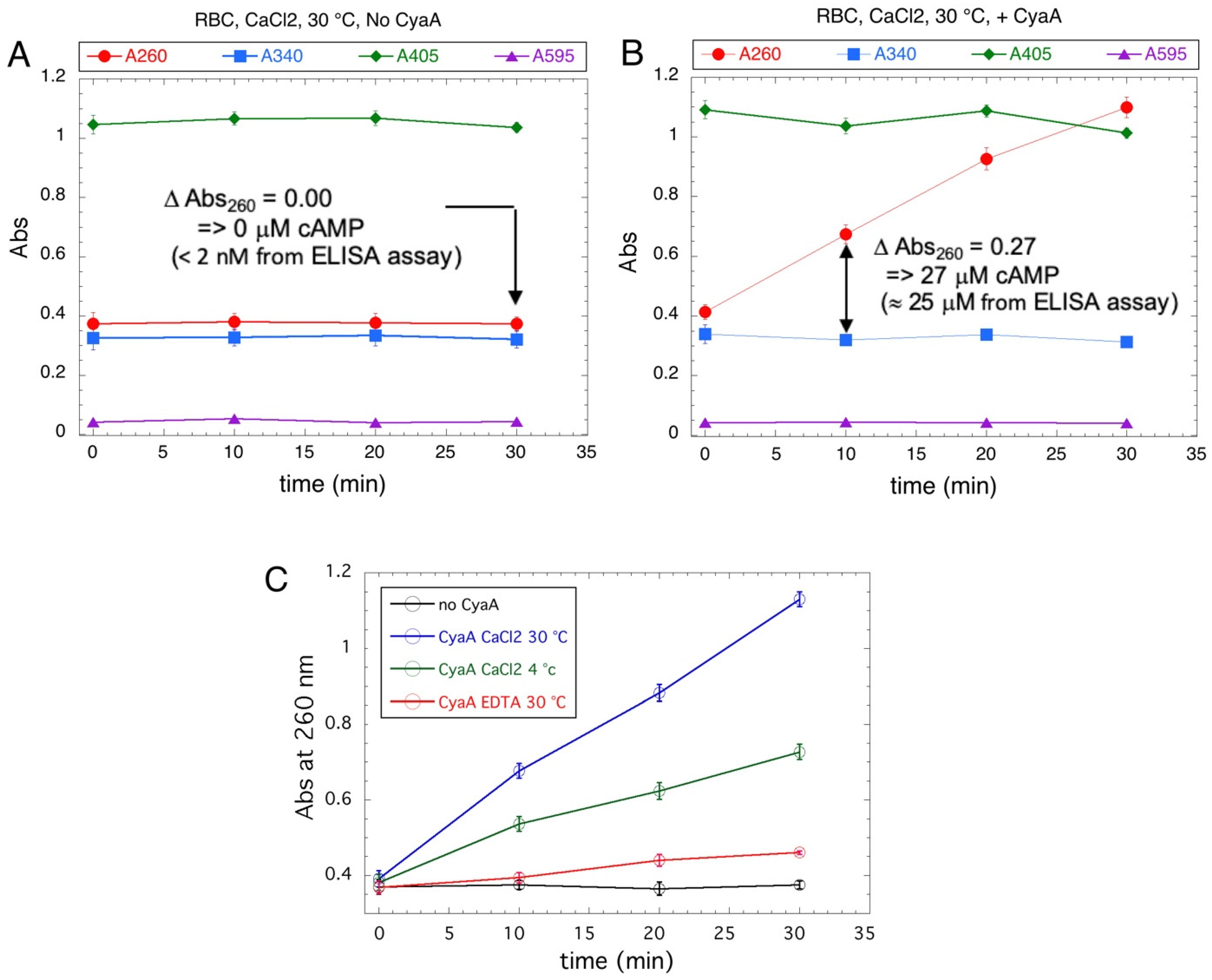
Purified CyaA was diluted in erythrocyte suspensions and incubated for 20 min at 30 °C in the presence of calcium (or in the presence of EDTA, or at 4 °C as controls). The cells were then extensively washed, resuspended in a small volume, and lysed with non-ionic detergent (e.g., Tween 20). The AC activities of the lysed samples were then assayed for various incubation times (0, 10, 20, and 30 min), and the absorption of the Al2O3 supernatants of all samples were recorded at different wavelengths (ie 260, 340, 405, and 595 nm). As anticipated, the cell lysates showed high absorption at 260, 340, or 405 nm (Figure 5). Yet, in control erythrocytes not incubated with CyaA, the absorption values remained constant over all the incubation periods (Figure 5A). In lysates of cells incubated with CyaA at 30 °C and in the presence of calcium (Figure 5B), the absorption at 260 nm (A260) increased linearly with time while the absorption at other wavelengths remained constant. By independently monitoring the cAMP content in the Al2O3 supernatants with an ELISA assay, we confirmed that the A260 increase precisely corresponded to the synthesis of cAMP due to the CyaA enzyme bound to cells (Figure 5B). The binding of CyaA to erythrocytes at 4 °C in the presence of calcium or at 30 °C in the absence of calcium (Figure 5C) was reduced in accordance with prior studies [41,42,43,44,45]. Hence, this spectrophotometric assay may be adapted to monitor AC toxin activity in complex cell extracts despite high absorption at useful wavelengths.
3. Discussion
We show here that the direct spectrophotometric detection of cAMP after chromatographic separation on aluminum oxide can be used to provide a robust in vitro assay for bacterial AC toxin activities. This simple assay allows for an accurate determination (i.e., with robust absorption values) of the AC activity of B. pertussis CyaA. Thanks to the high catalytic efficiency (kcat > 1000 s−1) and excellent time stability of this enzyme, we could detect down to the fmol amounts of purified AC. The assay is cost-effective and straightforward for routine monitoring of AC activity in safe conditions (i.e., without requiring manipulation of radioactivity). It can be easily set up in any laboratory without requiring expensive equipment or facilities (e.g., for manipulating radioactivity). It is perfectly appropriate for most standard assays of the bacterial nucleotidyl cyclase toxins that exhibit a high catalytic activity, such as the B. pertussis CyaA and B. anthracis EF—as shown here—and could be adapted as well for P. aeruginosa ExoY and other Gram- ExoY-like enzymes, which have turnover numbers in the range of 50 to 500 s−1 (depending on enzymes and/or substrates) [10,11,46,47]. Moreover, this spectrophotometric assay can also be applied to quantify the AC activity in complex mixtures, such as cell extracts, by shifting to a kinetic mode where the cAMP synthesis is measured via the specific increase of absorption at 260 nm (A260) as a function of the incubation time. Of course, proper controls should be performed to reliably establish that the A260 increase corresponds only to cAMP synthesis. It might also be adapted to characterize other less active nucleotidyl cyclases found in numerous bacterial or eukaryotic species—again provided that careful controls are applied.
Table 1 presents a comparison of the diverse assays that have been used to characterize bacterial AC toxins with their main advantages and drawbacks. The spectrometric assay described here lies in between the more sensitive and specific, but rather cumbersome, techniques that rely on cNMP detection (e.g., via radioactivity, ELISA, or LC/MS), and more high-throughput methods that might be less sensitive and more susceptible to interference by contaminating NTP- and/or pyrophosphate-metabolizing enzymes. Given its robustness and simplicity to set up, this in vitro spectrometric assay may thus become a method of choice for the routine characterization of the enzymatic activities of purified or partially-purified bacterial cyclase toxins.
4. Materials and Methods
4.1. Materials
Aluminum oxide 90 active neutral (activity stage I) for column chromatography was obtained from Merck (ref # 101077, 70–230 mesh ASTM, Al2O3). UV-transparent 96-well microplates (Nunc™ UV 96 well) were purchased from Thermo Scientific (Waltham, MA, USA). ATP, cAMP, Bovine Serum Albumin, and Tween 20 were from Sigma-Aldrich (Saint-Louis, MO, USA). All recombinant proteins were expressed in E. coli and purified as described previously: CyaA protein [39,42], CyaAE5-OVA [36,37], AC384 (catalytic domain of CyaA, corresponding to residues 1 to 384), and CaM [28]. The EF enzyme tested here was from the Bacillus cereus G9241 strain (kind gift from P. L. Goossens, Pitard et al. manuscript in preparation) and comprises the adenylyl cyclase domain without the protective antigen-binding domain (i.e., residues 291 to 798 of full-length EF). This protein differs from the wild-type EF toxin from B anthracis by three amino acid changes: I318T, V694A, N789K (aa numbering from B anthracis EF shown in 1XFV.pdb, [48]). It was expressed in E. coli and purified as described by Drum et al. [31].
4.2. Enzymatic AC Assay
The enzymatic reactions were carried out in 1.5 mL Eppendorf tubes by sequentially adding: 50 μL of AC×2 buffer (Adenylate Cyclase assay buffer 2-times concentrated: 100 mM Tris-HCl, pH 8.0, 15 mM MgCl2, 0.2 mM CaCl2, 1 mg/mL bovine serum albumin (BSA), 10 μL of CaM (calmodulin) diluted in buffer D (dilution buffer: 10 mM Tris-HCl, pH 8.0, 0.1% Tween 20, or other non-ionic detergent such NP40 or Triton ×100) to appropriate concentrations (final concentration of 2 μM for standard assays), 10 to 30 μL of enzyme samples to be tested, diluted in buffer D to appropriate concentrations. Buffer D was added to a complete volume of 90 μL. The tubes were equilibrated at 30 °C for 3–5 min in a Thermomixer (Eppendorf, Montesson, France), and the enzymatic reactions were initiated by adding 10 μL of 20 mM ATP (stock solution in H2O, adjusted to pH ≈ 7.5). The tubes were incubated at 30 °C for appropriate incubation times (usually 5–20 min, but it could be extended to several hrs when needed, see Figure 1). A blank assay containing no enzyme (or no CaM) was carried out in parallel. The enzymatic reactions were stopped by adding 0.3 g (conveniently measured with a 0.2 mL Eppendorf tube) of dried aluminum oxide 90 active powder (the reaction was stopped immediately as the ATP binds to alumina). Alternatively, the enzymatic reaction could be stopped by adding 100 μL of 50 mM HEPES pH 7.5 or 50 mM EDTA (stop solution) to chelate the Mg2+ essential co-factor. Then, 0.9 mL (or 0.8 mL if 100 μL of stop solution have been added) of 20 mM Hepes-Na, pH 7.5 or 0.15 M NaCl (Chromatography buffer) were added to elute cAMP from the alumina powder. The tubes were mixed by inversion a few times over the course of 3–5 min and then centrifuged for 5 min at 12–14,000 rpm (room temperature) to pellet the alumina powder. The supernatants were carefully collected and the absorption at 260 and 340 nm were recorded. The cAMP concentration was then determined using an absorption coefficient at 260 nm of 15.4 mM−1 after subtracting the background absorption of the blank tube. The measurement of the absorption at 340 nm (A340) is very convenient for monitoring the potential light diffusion that may happen due to the presence of traces contaminating alumina powder in the supernatant solution. In our routine procedure, the OD260–OD340 was used to measure the cAMP content in each fraction.
A convenient format to carry out multiple assays is to transfer 0.3 mL aliquots of alumina supernatants into wells of a UV-transparent acrylic microplate, which allows for easy sequential measurements of the absorption at 260 and 340 nm (as well as other wavelengths see below) with direct data being recorded into an excel sheet. This greatly facilitates the downstream calculations.
4.3. Assay of CyaA Binding to Erythrocytes
CyaA toxin binding to sheep erythrocytes was essentially assayed as previously described [42,43]. Sheep erythrocytes (from Charles River Laboratories, Wilmington, MA, USA) were washed and resuspended (≈5% of dry pellet) in HBS buffer (20 mM Hepes-Na, pH 7.5, 150 mM NaCl) supplemented with 5 mM glucose. The purified CyaA toxin (stored at 1 mg/mL in 8 M urea, 20 mM Hepes-Na) was directly diluted to 11.2 nM (2 μg/mL final concentration) into 1 mL of erythrocyte suspension supplemented with either 2 mM CaCl2 or 2 mM EDTA. A 20 μL aliquot was removed to determine the total adenylate cyclase activity added to each sample. The mixtures were then incubated at 30 °C, or 4 °C, for 30 min. The cell suspensions were chilled on ice, centrifuged at 4 °C, and cell pellets were washed two times with 1 mL cold HBS buffer (transferring the resuspended cells into new tubes to eliminate potential non-specific absorption of CyaA to the tube wall). Finally, the pelleted erythrocytes were resuspended in 100 μL of 10 mM Tris-HCl, pH 8.0, containing 0.1% Triton X−100 in order to lyse the cells. A total of 20 μL of lysates were tested in standard AC assays as described above and incubated for 0, 10, 20, or 30 min. The supernatants of aluminum oxide were then collected in a microplate and the absorption at 260, 340, 405, and 595 nm (the build-in filters available in our microplate reader–other wavelengths could be used as well in addition to the 260 nm) were successively recorded. Independent quantification of cAMP was performed on Al2O3 supernatants with an ELISA assay using a cAMP-biotinylated-BSA conjugate coated on ELISA plates and detected with a specific rabbit anti-cAMP antiserum [49].
Author Contributions
Conceptualization: D.L.; Investigation: M.D., M.S., I.P., A.C. and D.L.; Writing: original draft preparation: D.L.; Writing: review and editing: M.D., M.S., I.P., A.C. and D.L.; Project administration: D.L.; Funding acquisition: A.C. and D.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Institut Pasteur, the Centre National de la Recherche Scientifique, CNRS UMR 3528 (Biologie Structurale et Agents Infectieux), and Agence Nationale de la Recherche (grant #: ANR 21-CE11–0014-01-TransCyaA). M.S. was supported by the Pasteur–Paris University (PPU) International PhD Program. I.P. was funded by DGA support 2017033 and the Ecole Doctorale Complexité du Vivant (ED515).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Acknowledgments
We thank P. L. Goossens, Institut Pasteur, and the Production and Purification of Recombinant Proteins Facility from Institut Pasteur for their assistance in producing the B. cereus EF enzyme.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Seifert, R.; Schneider, E.H.; Bähre, H. From Canonical to Non-Canonical Cyclic Nucleotides as Second Messengers: Pharmacological Implications. Pharmacol. Ther. 2015, 148, 154–184. [Google Scholar] [CrossRef]
- The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th ed.; Alouf, J.E.; Ladant, D.; Popoff, M.R. (Eds.) Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Teixeira-Nunes, M.; Retailleau, P.; Comisso, M.; Deruelle, V.; Mechold, U.; Renault, L. Bacterial Nucleotidyl Cyclases Activated by Calmodulin or Actin in Host Cells: Enzyme Specificities and Cytotoxicity Mechanisms Identified to Date. IJMS 2022, 23, 6743. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, E.L.; Urban, M.A.; Manclark, C.R.; Wolff, J. Extracytoplasmic Adenylate Cyclase of Bordetella pertussis. Proc. Natl. Acad. Sci. USA 1976, 73, 1926–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogel, A.; Schultz, J.E.; Brownlie, R.M.; Coote, J.G.; Parton, R.; Hanski, E. Bordetella pertussis Adenylate Cyclase: Purification and Characterization of the Toxic Form of the Enzyme. EMBO J. 1989, 8, 2755–2760. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S.H. Anthrax Toxin Edema Factor: A Bacterial Adenylate Cyclase That Increases Cyclic AMP Concentrations of Eukaryotic Cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masin, J.; Osicka, R.; Bumba, L.; Sebo, P. Bordetella Adenylate Cyclase Toxin: A Unique Combination of a Pore-Forming Moiety with a Cell-Invading Adenylate Cyclase Enzyme. Pathog. Dis. 2015, 73, ftv075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiso, N. Bordetella Adenylate Cyclase-Hemolysin Toxins. Toxins 2017, 9, 277. [Google Scholar] [CrossRef]
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax Pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef]
- Yahr, T.L.; Vallis, A.J.; Hancock, M.K.; Barbieri, J.T.; Frank, D.W. ExoY, an Adenylate Cyclase Secreted by the Pseudomonas Aeruginosa Type III System. Proc. Natl. Acad. Sci. USA 1998, 95, 13899–13904. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, C.D.; Alexeyev, M.; Pastukh, V.; Balczon, R.; Stevens, T. Pseudomonas Aeruginosa Exotoxin Y Is a Promiscuous Cyclase That Increases Endothelial Tau Phosphorylation and Permeability. J. Biol. Chem. 2012, 287, 25407–25418. [Google Scholar] [CrossRef]
- Belyy, A.; Raoux-Barbot, D.; Saveanu, C.; Namane, A.; Ogryzko, V.; Worpenberg, L.; David, V.; Henriot, V.; Fellous, S.; Merrifield, C.; et al. Actin Activates Pseudomonas Aeruginosa ExoY Nucleotidyl Cyclase Toxin and ExoY-like Effector Domains from MARTX Toxins. Nat. Commun. 2016, 7, 13582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziolo, K.J.; Jeong, H.-G.; Kwak, J.S.; Yang, S.; Lavker, R.M.; Satchell, K.J.F. Vibrio vulnificus Biotype 3 Multifunctional Autoprocessing RTX Toxin Is an Adenylate Cyclase Toxin Essential for Virulence in Mice. Infect. Immun. 2014, 82, 2148–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, A.A. Separation and Purification of Cyclic Nucleotides by Alumina Column Chromatography. Methods Enzymol. 1974, 38, 41–46. [Google Scholar] [CrossRef]
- Duriez, E.; Goossens, P.L.; Becher, F.; Ezan, E. Femtomolar Detection of the Anthrax Edema Factor in Human and Animal Plasma. Anal. Chem. 2009, 81, 5935–5941. [Google Scholar] [CrossRef]
- Gottle, M.; Dove, S.; Kees, F.; Schlossmann, J.; Geduhn, J.; Konig, B.; Shen, Y.; Tang, W.-J.; Kaever, V.; Seifert, R. Cytidylyl and Uridylyl Cyclase Activity of Bacillus anthracis Edema Factor and Bordetella pertussis CyaA. Biochemistry 2010, 49, 5494–5503. [Google Scholar] [CrossRef] [Green Version]
- Beste, K.Y.; Spangler, C.M.; Burhenne, H.; Koch, K.-W.; Shen, Y.; Tang, W.-J.; Kaever, V.; Seifert, R. Nucleotidyl Cyclase Activity of Particulate Guanylyl Cyclase A: Comparison with Particulate Guanylyl Cyclases E and F, Soluble Guanylyl Cyclase and Bacterial Adenylyl Cyclases CyaA and Edema Factor. PLoS ONE 2013, 8, e70223. [Google Scholar] [CrossRef] [Green Version]
- Laine, E.; Goncalves, C.; Karst, J.C.; Lesnard, A.; Rault, S.; Tang, W.-J.; Malliavin, T.E.; Ladant, D.; Blondel, A. Use of Allostery to Identify Inhibitors of Calmodulin-Induced Activation of Bacillus anthracis Edema Factor. Proc. Natl. Acad. Sci. USA 2010, 107, 11277–11282. [Google Scholar] [CrossRef] [Green Version]
- Selwa, E.; Davi, M.; Chenal, A.; Sotomayor-Perez, A.-C.; Ladant, D.; Malliavin, T.E. Allosteric Activation of Bordetella pertussis Adenylyl Cyclase by Calmodulin: Molecular Dynamics and Mutagenesis Studies. J. Biol. Chem. 2014, 289, 21131–21141. [Google Scholar] [CrossRef] [Green Version]
- Stein, R.L. Kinetic Studies of the Activation of Bordetella pertussis Adenylate Cyclase by Calmodulin. Biochemistry 2022, 61, 554–562. [Google Scholar] [CrossRef]
- Lawrence, A.J.; Coote, J.G.; Kazi, Y.F.; Lawrence, P.D.; MacDonald-Fyall, J.; Orr, B.M.; Parton, R.; Riehle, M.; Sinclair, J.; Young, J.; et al. A Direct Pyrophosphatase-Coupled Assay Provides New Insights into the Activation of the Secreted Adenylate Cyclase from Bordetella pertussis by Calmodulin. J. Biol. Chem. 2002, 277, 22289–22296. [Google Scholar] [CrossRef]
- Spangler, C.M.; Spangler, C.; Göttle, M.; Shen, Y.; Tang, W.-J.; Seifert, R.; Schäferling, M. A Fluorimetric Assay for Real-Time Monitoring of Adenylyl Cyclase Activity Based on Terbium Norfloxacin. Anal. Biochem. 2008, 381, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Israeli, M.; Rotem, S.; Elia, U.; Bar-Haim, E.; Cohen, O.; Chitlaru, T. A Simple Luminescent Adenylate-Cyclase Functional Assay for Evaluation of Bacillus anthracis Edema Factor Activity. Toxins 2016, 8, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munier, H.; Bouhss, A.; Krin, E.; Danchin, A.; Gilles, A.M.; Glaser, P.; Barzu, O. The Role of Histidine 63 in the Catalytic Mechanism of Bordetella pertussis Adenylate Cyclase. J. Biol. Chem. 1992, 267, 9816–9820. [Google Scholar] [CrossRef]
- Guo, Q.; Shen, Y.; Zhukovskaya, N.L.; Florián, J.; Tang, W.-J. Structural and Kinetic Analyses of the Interaction of Anthrax Adenylyl Cyclase Toxin with Reaction Products CAMP and Pyrophosphate. J. Biol. Chem. 2004, 279, 29427–29435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, A.A.; Zenser, T.V. Separation of Cyclic 3′,5′-Nucleoside Monophosphates from Other Nucleotides on Aluminum Oxide Columns. Application to the Assay of Adenyl Cyclase and Guanyl Cyclase. Anal. Biochem. 1971, 41, 372–396. [Google Scholar] [CrossRef]
- Ramachandran, J. A New Simple Method for Separation of Adenosine 3′,5′-Cyclic Monophosphate from Other Nucleotides and Its Use in the Assay of Adenyl Cyclase. Anal. Biochem. 1971, 43, 227–239. [Google Scholar] [CrossRef]
- Vougier, S.; Mary, J.; Dautin, N.; Vinh, J.; Friguet, B.; Ladant, D. Essential Role of Methionine Residues in Calmodulin Binding to Bordetella pertussis Adenylate Cyclase, as Probed by Selective Oxidation and Repair by the Peptide Methionine Sulfoxide Reductases. J. Biol. Chem. 2004, 279, 30210–30218. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Lee, Y.-S.; Soelaiman, S.; Bergson, P.; Lu, D.; Chen, A.; Beckingham, K.; Grabarek, Z.; Mrksich, M.; Tang, W.-J. Physiological Calcium Concentrations Regulate Calmodulin Binding and Catalysis of Adenylyl Cyclase Exotoxins. EMBO J. 2002, 21, 6721–6732. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, D.P.; Durand, D.; Voegele, A.; Hourdel, V.; Davi, M.; Chamot-Rooke, J.; Vachette, P.; Brier, S.; Ladant, D.; Chenal, A. Calmodulin Fishing with a Structurally Disordered Bait Triggers CyaA Catalysis. PLoS Biol. 2017, 15, e2004486. [Google Scholar] [CrossRef] [Green Version]
- Drum, C.L.; Yan, S.-Z.; Bard, J.; Shen, Y.-Q.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W.-J. Structural Basis for the Activation of Anthrax Adenylyl Cyclase Exotoxin by Calmodulin. Nature 2002, 415, 396–402. [Google Scholar] [CrossRef]
- Saron, M.F.; Fayolle, C.; Sebo, P.; Ladant, D.; Ullmann, A.; Leclerc, C. Anti-Viral Protection Conferred by Recombinant Adenylate Cyclase Toxins from Bordetella pertussis Carrying a CD8+ T Cell Epitope from Lymphocytic Choriomeningitis Virus. Proc. Natl. Acad. Sci. USA 1997, 94, 3314–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guermonprez, P.; Fayolle, C.; Rojas, M.-J.; Rescigno, M.; Ladant, D.; Leclerc, C. In Vivo Receptor-Mediated Delivery of a Recombinant Invasive Bacterial Toxoid to CD11c + CD8 Alpha -CD11bhigh Dendritic Cells. Eur. J. Immunol. 2002, 32, 3071–3081. [Google Scholar] [CrossRef]
- Preville, X.; Ladant, D.; Timmerman, B.; Leclerc, C. Eradication of Established Tumors by Vaccination with Recombinant Bordetella pertussis Adenylate Cyclase Carrying the Human Papillomavirus 16 E7 Oncoprotein. Cancer Res. 2005, 65, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Ladant, D. Bioengineering of Bordetella pertussis Adenylate Cyclase Toxin for Vaccine Development and Other Biotechnological Purposes. Toxins 2021, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Ladant, D.; Karimova, G.; Ullmann, A.; Leclerc, C. Direct Delivery of the Bordetella pertussis Adenylate Cyclase Toxin to the MHC Class I Antigen Presentation Pathway. J. Immunol. 1999, 162, 1910–1916. [Google Scholar]
- Guermonprez, P.; Fayolle, C.; Karimova, G.; Ullmann, A.; Leclerc, C.; Ladant, D. Bordetella pertussis Adenylate Cyclase Toxin: A Vehicle to Deliver CD8-Positive T-Cell Epitopes into Antigen Presenting Cells. Methods Enzymol. 2000, 326, 527–542. [Google Scholar] [PubMed]
- Hewlett, E.L.; Gordon, V.M.; McCaffery, J.D.; Sutherland, W.M.; Gray, M.C. Adenylate Cyclase Toxin from Bordetella pertussis. Identification and Purification of the Holotoxin Molecule. J. Biol. Chem. 1989, 264, 19379–19384. [Google Scholar] [CrossRef]
- Karst, J.C.; Ntsogo Enguene, V.Y.; Cannella, S.E.; Subrini, O.; Hessel, A.; Debard, S.; Ladant, D.; Chenal, A. Calcium, Acylation, and Molecular Confinement Favor Folding of Bordetella pertussis Adenylate Cyclase CyaA Toxin into a Monomeric and Cytotoxic Form. J. Biol. Chem. 2014, 289, 30702–30716. [Google Scholar] [CrossRef] [Green Version]
- Hanski, E.; Farfel, Z. Bordetella pertussis Invasive Adenylate Cyclase. Partial Resolution and Properties of Its Cellular Penetration. J. Biol. Chem. 1985, 260, 5526–5532. [Google Scholar] [CrossRef]
- Rogel, A.; Hanski, E. Distinct Steps in the Penetration of Adenylate Cyclase Toxin of Bordetella pertussis into Sheep Erythrocytes. Translocation of the Toxin across the Membrane. J. Biol. Chem. 1992, 267, 22599–22605. [Google Scholar] [CrossRef]
- Karimova, G.; Fayolle, C.; Gmira, S.; Ullmann, A.; Leclerc, C.; Ladant, D. Charge-Dependent Translocation of Bordetella pertussis Adenylate Cyclase Toxin into Eukaryotic Cells: Implication for the in Vivo Delivery of CD8(+) T Cell Epitopes into Antigen-Presenting Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 12532–12537. [Google Scholar] [CrossRef]
- Voegele, A.; Sadi, M.; O’Brien, D.P.; Gehan, P.; Raoux-Barbot, D.; Davi, M.; Hoos, S.; Brûlé, S.; Raynal, B.; Weber, P.; et al. A High-Affinity Calmodulin-Binding Site in the CyaA Toxin Translocation Domain Is Essential for Invasion of Eukaryotic Cells. Adv. Sci. 2021, 8, 2003630. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, E.L.; Gray, L.; Allietta, M.; Ehrmann, I.; Gordon, V.M.; Gray, M.C. Adenylate Cyclase Toxin from Bordetella pertussis. Conformational Change Associated with Toxin Activity. J. Biol. Chem. 1991, 266, 17503–17508. [Google Scholar] [CrossRef]
- Masin, J.; Osickova, A.; Sukova, A.; Fiser, R.; Halada, P.; Bumba, L.; Linhartova, I.; Osicka, R.; Sebo, P. Negatively Charged Residues of the Segment Linking the Enzyme and Cytolysin Moieties Restrict the Membrane-Permeabilizing Capacity of Adenylate Cyclase Toxin. Sci. Rep. 2016, 6, 29137. [Google Scholar] [CrossRef] [Green Version]
- Beckert, U.; Wolter, S.; Hartwig, C.; Bahre, H.; Kaever, V.; Ladant, D.; Frank, D.W.; Seifert, R. ExoY from Pseudomonas Aeruginosa Is a Nucleotidyl Cyclase with Preference for cGMP and cUMP Formation. Biochem. Biophys. Res. Commun. 2014, 450, 870–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raoux-Barbot, D.; Belyy, A.; Worpenberg, L.; Montluc, S.; Deville, C.; Henriot, V.; Velours, C.; Ladant, D.; Renault, L.; Mechold, U. Differential Regulation of Actin-Activated Nucleotidyl Cyclase Virulence Factors by Filamentous and Globular Actin. PLoS ONE 2018, 13, e0206133. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhukovskaya, N.L.; Guo, Q.; Florián, J.; Tang, W.-J. Calcium-Independent Calmodulin Binding and Two-Metal-Ion Catalytic Mechanism of Anthrax Edema Factor. EMBO J. 2005, 24, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Karimova, G.; Pidoux, J.; Ullmann, A.; Ladant, D. A Bacterial Two-Hybrid System Based on a Reconstituted Signal Transduction Pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 5752–5756. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Photometric detection of AC. Panel (A): Spectrophotometric measurements of cAMP. Each line corresponds to an independent assay with the indicated nucleotide addition. Two 0.3 mL samples taken from the Al2O3 supernatant for each assay were transferred to a microplate and the absorption at 260 and 340 nm were recorded. The absorption at 340 nm provides a good estimate of the light scattering that arises from contaminating Al2O3 particles that are inadvertently aspirated in the 0.3 mL supernatant samples. Subtracting the absorption at 340 nm from the absorption at 260 nm yields a very reliable quantification of the cAMP content. Panel (B): Correspondence between the absorption at 260 nm minus absorption at 340 nm (A260–A340) of the Al2O3 supernatant as a function of cAMP (4 independent data points for each concentration) when it is added to the standard assay medium containing 2 mM ATP. Subtraction of the absorption at 340 nm allows for the correction of the potential light scattering arising from residual Al2O3 particles in the supernatant (see Material and methods for detail). Panel (C): Time and concentration dependence of B. pertussis AC activity. Reactions were carried out for the indicated times with the indicated final concentrations of AC384 in the presence of 2 mM ATP and 1 μM CaM (except in bar labeled “No CaM”). The calculated kcat is indicated above the bar (mean of 4 independent experiments).
Figure 1.
Photometric detection of AC. Panel (A): Spectrophotometric measurements of cAMP. Each line corresponds to an independent assay with the indicated nucleotide addition. Two 0.3 mL samples taken from the Al2O3 supernatant for each assay were transferred to a microplate and the absorption at 260 and 340 nm were recorded. The absorption at 340 nm provides a good estimate of the light scattering that arises from contaminating Al2O3 particles that are inadvertently aspirated in the 0.3 mL supernatant samples. Subtracting the absorption at 340 nm from the absorption at 260 nm yields a very reliable quantification of the cAMP content. Panel (B): Correspondence between the absorption at 260 nm minus absorption at 340 nm (A260–A340) of the Al2O3 supernatant as a function of cAMP (4 independent data points for each concentration) when it is added to the standard assay medium containing 2 mM ATP. Subtraction of the absorption at 340 nm allows for the correction of the potential light scattering arising from residual Al2O3 particles in the supernatant (see Material and methods for detail). Panel (C): Time and concentration dependence of B. pertussis AC activity. Reactions were carried out for the indicated times with the indicated final concentrations of AC384 in the presence of 2 mM ATP and 1 μM CaM (except in bar labeled “No CaM”). The calculated kcat is indicated above the bar (mean of 4 independent experiments).

Figure 2.
CaM-dependent AC activity of B. pertussis AC384 (blue) and B. cereus EF enzymes (green). AC activities (expressed in mol of cAMP produced per mol of enzyme per sec) at the indicated CaM concentrations were fitted to equation: A = (AMax × [CaM])/(KD + [CaM]). Maximal activities (AMax) of 2165 ± 30 and 1183 ± 22 s−1 were calculated for AC384 and EF, respectively, while the CaM concentration at half-maximal activation (≈KD) were 0.11 ± 0.01 and 1.04 ± 0.13 nM for AC384 and EF, respectively.
Figure 2.
CaM-dependent AC activity of B. pertussis AC384 (blue) and B. cereus EF enzymes (green). AC activities (expressed in mol of cAMP produced per mol of enzyme per sec) at the indicated CaM concentrations were fitted to equation: A = (AMax × [CaM])/(KD + [CaM]). Maximal activities (AMax) of 2165 ± 30 and 1183 ± 22 s−1 were calculated for AC384 and EF, respectively, while the CaM concentration at half-maximal activation (≈KD) were 0.11 ± 0.01 and 1.04 ± 0.13 nM for AC384 and EF, respectively.

Figure 3.
Salt and urea concentration dependence of AC384 activity. NaCl (Panel (A)) and urea (Panel (B)) concentration dependence of B. pertussis AC activity. Reactions were carried out for 10 min with 0.32 nM AC384 in the presence of 2 mM ATP and 1 μM CaM (mean and STD of 4 independent experiments). Control experiments (not shown) showed that ATP and cAMP binding to the Al2O3 were not modified by the presence of NaCl or urea at the tested concentrations. Panel (C): B. pertussis AC384 activities were measured at the indicated CaM concentrations in standard conditions (i.e., in Tris buffer, blue square) or in the presence of 0.2 M NaCl (green circle) or 0.2 M urea (red diamond). Data were fitted as in Figure 2. Maximal activities (in mol of cAMP per mol of AC384 per sec) were 2225 ± 95, 1351 ± 50, and 1440 ± 32 s−1 for assays in Tris buffer, in 0.2 M NaCl, and 0.2 M urea, respectively, while the CaM concentrations at half-maximal activation (≈KD) were 0.20 ± 0.06, 0.32 ± 0.08, and 0.24 ± 0.04 nM, respectively.
Figure 3.
Salt and urea concentration dependence of AC384 activity. NaCl (Panel (A)) and urea (Panel (B)) concentration dependence of B. pertussis AC activity. Reactions were carried out for 10 min with 0.32 nM AC384 in the presence of 2 mM ATP and 1 μM CaM (mean and STD of 4 independent experiments). Control experiments (not shown) showed that ATP and cAMP binding to the Al2O3 were not modified by the presence of NaCl or urea at the tested concentrations. Panel (C): B. pertussis AC384 activities were measured at the indicated CaM concentrations in standard conditions (i.e., in Tris buffer, blue square) or in the presence of 0.2 M NaCl (green circle) or 0.2 M urea (red diamond). Data were fitted as in Figure 2. Maximal activities (in mol of cAMP per mol of AC384 per sec) were 2225 ± 95, 1351 ± 50, and 1440 ± 32 s−1 for assays in Tris buffer, in 0.2 M NaCl, and 0.2 M urea, respectively, while the CaM concentrations at half-maximal activation (≈KD) were 0.20 ± 0.06, 0.32 ± 0.08, and 0.24 ± 0.04 nM, respectively.

Figure 4.
Characterization of a detoxified CyaAE5-OVA vaccine. AC activity of the purified CyaAE5-OVA protein (stored at 5.8 μM in 6.6 M urea, 20 mM Hepes-Na) at the indicated concentrations was monitored at 30 °C in the presence of 2 mM ATP and 1 μM CaM for 10 min (bars 1 to 6). As a control, in bars 4–6, 0.32 nM AC384 were spiked in the reaction mixtures in addition to the CyaAE5-OVA protein, in order to check that the residual urea concentration in each assay (indicated on the bottom line) did not affect the enzymatic activity. Results are the mean and STD of four independent experiments.
Figure 4.
Characterization of a detoxified CyaAE5-OVA vaccine. AC activity of the purified CyaAE5-OVA protein (stored at 5.8 μM in 6.6 M urea, 20 mM Hepes-Na) at the indicated concentrations was monitored at 30 °C in the presence of 2 mM ATP and 1 μM CaM for 10 min (bars 1 to 6). As a control, in bars 4–6, 0.32 nM AC384 were spiked in the reaction mixtures in addition to the CyaAE5-OVA protein, in order to check that the residual urea concentration in each assay (indicated on the bottom line) did not affect the enzymatic activity. Results are the mean and STD of four independent experiments.

Figure 5.
Assay of CyaA binding to erythrocyte. Panels A and B: Erythrocytes (RBC) were incubated at 30 °C with 2 mM CaCl2, for 30 min without CyaA (Panel (A)) or with 11.2 nM CyaA (panel (B)), and then extensively washed as described in the Methods section. After lysis, 20 μL of RBC lysates were tested in standard AC assays for the indicated times and the absorption of the Al2O3 supernatants were recorded at 260, 340, 405, and 595 nm. The cyclic AMP content in the Al2O3 supernatants was determined for sample A after 30 min of incubation and for sample B after 10 min of incubation by a specific ELISA assay and compared to the cAMP quantification deduced from the absorption at 260 nm (Abs260). Panel (C): Erythrocytes were incubated without (no CyaA, black) or with 11.2 nM CyaA at 4 or 30 °C with 2 mM CaCl2 or 2 mM EDTA for 30 min (as indicated). After extensive washing and cell lysis, 20 μL of RBC lysates were tested in standard AC assays as above for the indicated times. The relative fractions of “bound” CyaA activity, as compared to the total CyaA added to each cell suspension, correspond to about 2.4% for the incubation at 30 °C in the presence of CaCl2 (blue), about 1.1% for the incubation at 4 °C in the presence of CaCl2 (green), and less than 0.15% for the incubation at 30 °C in the presence of EDTA (red).
Figure 5.
Assay of CyaA binding to erythrocyte. Panels A and B: Erythrocytes (RBC) were incubated at 30 °C with 2 mM CaCl2, for 30 min without CyaA (Panel (A)) or with 11.2 nM CyaA (panel (B)), and then extensively washed as described in the Methods section. After lysis, 20 μL of RBC lysates were tested in standard AC assays for the indicated times and the absorption of the Al2O3 supernatants were recorded at 260, 340, 405, and 595 nm. The cyclic AMP content in the Al2O3 supernatants was determined for sample A after 30 min of incubation and for sample B after 10 min of incubation by a specific ELISA assay and compared to the cAMP quantification deduced from the absorption at 260 nm (Abs260). Panel (C): Erythrocytes were incubated without (no CyaA, black) or with 11.2 nM CyaA at 4 or 30 °C with 2 mM CaCl2 or 2 mM EDTA for 30 min (as indicated). After extensive washing and cell lysis, 20 μL of RBC lysates were tested in standard AC assays as above for the indicated times. The relative fractions of “bound” CyaA activity, as compared to the total CyaA added to each cell suspension, correspond to about 2.4% for the incubation at 30 °C in the presence of CaCl2 (blue), about 1.1% for the incubation at 4 °C in the presence of CaCl2 (green), and less than 0.15% for the incubation at 30 °C in the presence of EDTA (red).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of AC assay techniques.
| Assay Principle | Characteristics | Drawbacks | References |
|---|---|---|---|
| Monitor production of cNMP product | |||
| Radioactive: conversion of radioactive NTP into radioactive cNMP separated by Al2O3 chromatography and quantified in a scintillation counter | highly sensitive (a) (0.1 to 10 fmol) highly specific straightforward | Legal and environmental constraints of using radioactivity Expensive | [4,6,14] |
| Spectrometric: conversion of NTP into cNMP separated by Al2O3 chromatography and quantified by absorption at 260 nm | Sensitive (1 to 100 fmol of AC) straightforward inexpensive, | Possible application in complex media but requires proper controls | Present study |
| ELISA: Conversion of NTP into cNMP which is quantified by immunodetection | Highly Sensitive (0.01 to 10 fmol) Highly specific | Immunodetection (ELISA) assays are cumbersome and expensive Not high throughput | [15] |
| LC/MS: Conversion of NTP into cNMP which is quantified by liquid chromatography and Mass spectrometry | Highly sensitive (0.01 to 10 fmol) Highly specific Application to all cNMP | Special equipment (LC/MS) required Not high throughput | [16,17] |
| Monitor production of PPi product(b) | |||
| Colorimetric: detection of PPi product after hydrolysis into 2 × Pi quantified by colorimetry | Sensitive (30 to 100 fmol) Mid to high throughput | Not applicable in complex media or cell extracts (b) Requirement of coupled enzyme (PPiase) | [18,19] |
| Fluorometric: detection of PPi product after hydrolysis into 2 × Pi and coupling to production of fluorescent resorufin | Sensitive (1 to 100 fmol) Mid to high throughput Kinetic mode | Not applicable in complex media Requirement of coupled enzymes (PPiase, PNP, HRP) | [20] |
| Conductimetry: detection of PPi product after hydrolysis into 2 × Pi quantified by conductimetry | Low sensitivity (0.1 to 10 pmol) Kinetic mode | Not applicable in complex media Requirement of coupled enzyme (PPiase) | [21] |
| Monitor disappearance of ATP substrate(c) | |||
| Fluorescent: monitoring of ATP decrease with fluorescent probe Tb(III)–norfloxacin | Sensitive (10 to 100 fmol) Kinetic mode Mid to high throughput inexpensive, straightforward | Applicability in complex media? Possible interference with ATP consumption or production by other enzymes. | [22] |
| Luminescence: monitoring of ATP decrease with luciferase (d) | Sensitive (10 to 100 fmol) Mid to high throughput | Applicability in complex media? Requirement of coupled enzyme (luciferase) | [23] |
| Reverse reaction: conversion of cAMP and PPi into ATP | |||
| Spectrophotometric: ATP produced from cAMP and PPi-coupled by hexokinase and glucose-6-phosphate dehydrogenase-to the formation of NADPH measured by absorption at 334 nm | Low sensitivity (0.1 to 1 pmol) Kinetic mode Mid to high throughput | Interference with ATP production or consumption by other enzymes. Applicability in complex media? Requirement of coupled enzymes (hexokinase; glucose-6-phosphate dehydrogenase) | [24,25] |
(a) Sensitivity: the values correspond to the lowest levels of CyaA (AC) or EF enzymes robustly detected in standard assays; (b) assays monitoring production of PPi product are usually not applicable in complex media or extracts due to presence of either Pi or Pi-generating reactions (any NTP hydrolyzing enzymes); (c) assays monitoring the consumption of ATP substrate will require that at least 20% of initial ATP is converted into cAMP in order to obtain reliable measurements. Moreover, these assays are usually not applicable in complex media or extracts due to the potential presence of ATP hydrolyzing or producing enzymes; and (d) in the luminescent assay described by Israeli et al. [23], there is an apparent discrepancy between the consumption of ATP (monitored by decrease of luminescence) and the appearance of cAMP which remains unclarified.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Davi, M.; Sadi, M.; Pitard, I.; Chenal, A.; Ladant, D. A Robust and Sensitive Spectrophotometric Assay for the Enzymatic Activity of Bacterial Adenylate Cyclase Toxins. Toxins 2022, 14, 691. https://doi.org/10.3390/toxins14100691
AMA Style
Davi M, Sadi M, Pitard I, Chenal A, Ladant D. A Robust and Sensitive Spectrophotometric Assay for the Enzymatic Activity of Bacterial Adenylate Cyclase Toxins. Toxins. 2022; 14(10):691. https://doi.org/10.3390/toxins14100691
Chicago/Turabian StyleDavi, Marilyne, Mirko Sadi, Irene Pitard, Alexandre Chenal, and Daniel Ladant. 2022. "A Robust and Sensitive Spectrophotometric Assay for the Enzymatic Activity of Bacterial Adenylate Cyclase Toxins" Toxins 14, no. 10: 691. https://doi.org/10.3390/toxins14100691
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.