Lactococcus Ceduovirus Phages Isolated from Industrial Dairy Plants—From Physiological to Genomic Analyses


Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawinskiego 5a, 02-106 Warsaw, Poland
*
Author to whom correspondence should be addressed.
Viruses 2020, 12(3), 280; https://doi.org/10.3390/v12030280
Submission received: 17 January 2020
/
Revised: 28 February 2020
/
Accepted: 28 February 2020
/
Published: 3 March 2020
(This article belongs to the Section Bacterial Viruses)
Abstract
:LactococcusCeduovirus (formerly c2virus) bacteriophages are among the three most prevalent phage types reported in dairy environments. Phages from this group conduct a strictly lytic lifestyle and cause substantial losses during milk fermentation processes, by infecting lactococcal host starter strains. Despite their deleterious activity, there are limited research data concerning Ceduovirus phages. To advance our knowledge on this specific phage group, we sequenced and performed a comparative analysis of 10 new Lactococcus lactis Ceduovirus phages isolated from distinct dairy environments. Host range studies allowed us to distinguish the differential patterns of infection of L. lactis cells for each phage, and revealed a broad host spectrum for most of them. We showed that 40% of the studied Ceduovirus phages can infect both cremoris and lactis strains. A preference to lyse strains with the C-type cell wall polysaccharide genotype was observed. Phage whole-genome sequencing revealed an average nucleotide identity above 80%, with distinct regions of divergence mapped to several locations. The comparative approach for analyzing genomic data and the phage lytic spectrum suggested that the amino acid sequence of the orf8-encoded putative tape measure protein correlates with host range. Phylogenetic studies revealed separation of the sequenced phages into two subgroups. Finally, we identified three types of phage origin of replication regions, and showed they are able to support plasmid replication without additional phage proteins.
1. Introduction
Ceduovirus phages infecting Lactococcus lactis starter strains are one of the three most prominent phage species in the dairy environment worldwide [1,2]. Phages classified to this type belong to the Siphoviridae family of the Caudovirales order, and are characterized by prolate heads and long non-contractile tails [3]. Members of the Ceduovirus (or prolate) group have a double-stranded (ds) DNA genome and conduct an obligatory lytic life cycle. Despite the acknowledged ubiquity of Ceduovirus phages in milk plants, there is a significant deficiency in the amount of experimental data concerning this group of phages. This stands in strong contrast to the well-characterized and abundantly sequenced Lactococcus Skunavirus (formerly 936-type or sk1virus) type [4,5,6]. Until now, less than 40 complete genome sequences of Ceduovirus phages were available in databases [7,8,9,10,11,12,13]. Based on previous studies, involving genome sequencing, Ceduoviruses were determined to represent a genetically similar phage group. Their genomes are organized into two divergently transcribed blocks comprising early and late genes, respectively, separated by a non-coding region containing the origin of phage replication [7,8,9,14]. In addition to the overall high genome conservation among prolate phages, certain regions of divergence can be observed, including (i) the early transcribed region (due to presence/lack of individual open reading frames—orfs), (ii) in the late region, particularly within structural genes implicated in host range determination, and (iii) in the non-coding intergenic region containing the origin of phage replication [8,9,14]. Determining the correlations between genomic data and host range patterns of Lactococcus phages has been the main objective of numerous studies, and is an important aspect in understanding the mechanisms of phage prevalence in the dairy environment [9,15,16,17]. Studies conducted on c2 and bIL67, the two model Ceduovirus phages, have appointed the respective gene regions l14-l15-l16 (locus_tag: c2p36-c2p37-c2p38) and orf34-orf35-orf36 (locus_tag: gp04-gp03-gp02) as host range determinants [9,15]. These genes are suggested to encode structural proteins, possibly tail fibers, but so far this has not been experimentally confirmed. Analysis of ORFll5c2/ORF35bIL67 and ORFl16c2/ORF36bIL67 using the HHPRED tool predicted structural similarity to proteins with carbohydrate-binding domains (5E7T_B for ORFll5c2/ORF35bIL67 and 3P6B_B for ORF36bIL67 and 1GU3_A for ORFl16c2), which assumes their engagement in adhesion to host cells. In turn, both, phage bIL67 orf34 and phage c2 l14 are annotated as encoding hypothetical proteins. By analogy, genes of other Ceduovirus phages localized in the same genetic context as l14-l15-l16 and orf34-orf35-orf36 are also expected to be crucial in defining their lytic spectrum. Additionally, genes l10 (locus_tag: c2p32) of phage c2 and orf31 (locus_tag: bIL67_gp07) of phage bIL67 have been reported to be implicated in host cell infection [8,15,18].
Efficient infection of L. lactis strains by Ceduovirus phages also relies on bacterial factors. Initiation of Ceduovirus phage infection is described as a two-step process relying on (i) reversible adsorption to the bacterial cell wall polysaccharide (CWPS) followed by (ii) irreversible binding to a proteinaceous receptor [19,20,21]. A study by Oliveira and co-workers [10] on 21 Ceduovirus phages determined their strong preference for bacterial hosts with the type C CWPS genotype. This suggests that although the interaction with the host CWPS is reversible, it may be crucial in the initial step of infection, ensuring a high rate of phage adsorption and effective propagation. Among the features differentiating the otherwise conserved Ceduovirus phage group is the ori region, which is localized between the early and late transcribed gene clusters [22]. Multiple sequence alignments of Ceduovirus phage genomes have allowed identification of three distinct ori groups: bIL67-type, c2-type, and 923-type [14]. A typical feature of all ori types is a non-coding sequence flanked by PE1 and PE2—the first two promoters from the early transcribed region. The nucleotide sequence between these two promoters is the major varying feature of Ceduovirus phage ori types. Based on studies of phage c2, the PE1 promoter was found to be involved in production of a non-coding transcript (PE1-T) which may have a crucial role in initiating phage replication in a transcription-mediated fashion [23,24]. Both the length and the sequence and/or structure of PE1-T were suggested to be crucial factors of each ori type—chimeric origins were non-functional [25].
We sequenced and characterized 10 Lactococcus Ceduovirus phages isolated from distinct dairy whey samples derived from disturbed production processes in Poland. By analyzing the phage infection patterns, a correlation with the subspecies of the L. lactis hosts and their cell wall polysaccharide (CWPS) profiles was observed. Comparative analysis of the sequenced phage genomes allowed us to establish the relatedness and determine several distinguishing features with respect to other Lactococcus Ceduovirus phages. Phylogeny studies revealed clustering of the analyzed phages with other Ceduovirus phages into two major subgroups, irrelevantly of their geographical settings. Moreover, by using comparative analysis we identified the putative origins of replication of the studied phages and classified them into one of the previously identified Ceduovirus phage ori groups (c2-, bIL67-, or 923-like). Functional studies confirm the ability of the phage oris to support plasmid replication in L. lactis strains.
2. Materials and Methods
2.1. Bacterial Strains, Bacteriophages, and Culture Conditions
Bacterial strains and bacteriophages used in study are listed in Table 1. Lactococcus lactis strains were grown at 30 °C on M17 broth (Oxoid) supplemented with 0.5% glucose (GM17) or on GM17 solid-agar (1.5%). When needed, GM17 plates were supplemented with chloramphenicol (Cm) at 10 µg mL−1. GM17 medium for phage propagation was supplemented with 10 mM CaCl2 (GM17-CaCl2). Escherichia coli strain was grown at 37 °C in Luria-Bertani (LB) broth with shaking or on LB solid-agar (1.5%). When needed, LB plates were supplemented with tetracycline (Tet) at 8 µg mL−1 and ampicillin (Amp) at 100 µg mL−1. Bacteriophages derived from whey samples collected from dairy plants over a period of 4 years from eight locations across Poland.
2.2. Bacteriophage Propagation
L. lactis host strains were cultured in 200 mL of GM17-CaCl2 broth at 30 °C until OD600 0.15–0.2. After reaching the desired OD600 the culture was infected by adding 0.2 mL of high titer phage lysate (≥109 plaque forming units, PFU, per mL) and further incubated at 30 °C until complete lysis (up to 2 h). The lysate was filtered using 0.45 μm filters (Stericup Millipore) and kept at 4 °C until further use.
2.3. Electron Microscopy
Phage lysates were concentrated for electron microscopy by centrifugation (14,000 rpm, 4 °C for 4 h), the pellet was washed with 100 mM ammonium acetate, then centrifuged (14,000 rpm, 4 °C for 1 h) three times and stained using phosphoric potassium tungstate (2% w/v) on Formvar-coated copper grid. Micrographs were performed using the JEM 1400 (JEOL Co., Japan) electron microscope equipped with energy-dispersive full range X-ray microanalysis system (EDS INCA Energy TEM, Oxford Instruments, UK), tomographic holder and 11 Megapixel TEM Camera MORADA G2 (EMSIS GmbH, Germany) in the Laboratory of Electron Microscopy at the Nencki Institute of Experimental Biology (Warsaw, Poland).
2.4. Host Range Studies
Phage infection specificity was tested against a collection of 57 L. lactis strains, comprising industrial (50), environmental (4), and laboratory (3) isolates by double-layer plate method, as described elsewhere [30]. Phage titer (PFU per mL) was determined by mixing the diluted phage lysates (10−2–10−8; 100 µL) with overnight-grown bacteria (200 µL) and plating in GM17 soft-agar (0.75%) on GM17-CaCl2 plates. After overnight (o/n) incubation at 30 °C, the number of plaques for each dilution was counted and the phage titer calculated. The same approach was applied for host range studies. Phage-sensitive strains were detected after o/n incubation at 30 °C by screening plates for the presence of lytic zones.
2.5. One-Step Growth Assay
One-step growth studies were done according to a previously established protocol [31]. Phage titer (PFU per mL) was calculated by counting the number of visible lysis zones after o/n incubation at 30 °C. Burst size was determined by dividing the final average count of liberated phage particles (at plateau) by the initial average count of infected bacterial cells. Time prior to the release of infection particles was established as the latent period. Burst time was inferred directly from the one-step growth curve as the time needed for the phage to complete one cycle. The assay was repeated three-fold.
2.6. PCR-Based Typing
Primers used in this study are listed in Table 2. Phages were assigned to the Ceduovirus phages based on PCR reaction using Ceduovirus-specific primers (c-2for/c-2rev) and phage lysate (1 μL) as template. Phage c2 and phage sk1 lysates served in the assay as the positive and negative control, respectively. PCR products were run on a 0.8% (w/v) agarose gel in 1xTAE (Tris acetate-EDTA) buffer stained with ethidium bromide and analyzed under UV light using the G:BOX instrument (Syngene).
2.7. Phage Genome Sequencing
Phage genomic DNA was isolated from 200 mL of filtered phage lysates as described previously [33] and stored at 4 °C until further use. Sequencing of phage genomes was performed in the Laboratory of DNA Sequencing and Oligonucleotide Synthesis (IBB PAS) using Illumina Sequencing Technology. DNA was sheared mechanically using Covaris instrument (Covaris, Inc., Woburn, MA, USA) and TruSeq-like libraries were constructed using KAPA Library preparation kit (KAPA/Roche, Basel, Switzerland) according to the manufacturer’s instructions. Phage genomes were sequenced in paired-end mode (v3, 600 cycle chemistry kit) using the MiSeq instrument (Illumina, San Diego, CA, USA). Obtained sequence reads were filtered by quality using the fastp toolkit [34]. The Newbler version 3.0 software program (454 Life Sciences, Branford, CT, USA) was used for sequence assembly. Sequencing coverage depth was 100–120X for each assembled sample. Ambiguous sequence regions were amplified by PCR with specific primers and re-sequenced using the BigDye Terminator Mix v. 3.1 chemistry (Applied Biosystems, Foster City, CA, USA) and the ABI3730xl Genetic Analyzer (Life Technologies, Carlsbad, CA, USA). Terminal repeat regions were re-sequenced by Sanger sequencing with phage DNA as a template and primers complementary to the non-redundant parts at the end of the analyzed region. All sequence errors and mis-assemblies were corrected using the Seqman software (DNAStar, Madison, WI, USA). Assembled genomes were annotated automatically using the Rapid Annotation Subsystem Technology (RAST) server [35].
2.8. Bioinformatic Analyses
Further functional annotations of predicted amino acid sequences were performed manually by employing the BLASTp algorithm (cut-off E-value 0.0001) [36] and on-line programs accessible through the SIB ExPASy Bioinformatics Resource Portal (https://www.expasy.org) (UniProtKB, PROSITE, SwissModel) and MPI Bioinformatics Tool Kit (HHPRED with pdb, pfam, prk, tigr, and cogs databases) (https://toolkit.tuebingen.mpg.de/) [37,38]. Structural homologs were additionally searched using Phyre2 [39]. The ClustalOmega tool at default settings implemented by the EMBL-EBI website was used for comparative alignment of amino acid sequences predicted for each locus [40]. All-against-all comparison of phage nucleotide sequences was done by multiple pairwise alignment using BLAST (bl2seq) [36]. Schematic representation of the genomes was obtained using the Easyfig 2.1 software [41]. Multiple sequence alignments for proteins annotated as putative DNApol (with RecA-like NTPase superfamily domain cl28885), tail fiber (GP2 and GP4), major capsid (GP14), holin, lysin, and ERF-like (essential recombination function) recombinase were calculated with Mafft [42] and trimmed with trimAl [43] to remove poorly aligned regions. For each protein, a maximum likelihood tree was computed using PhyML [44], based on the model of evolution estimated individually for each multiple sequence alignment with ProtTest [45]. All trees were further used for building the supertree with URec [46]. Phylogenetic trees were visualized with iTol [47].
2.9. Construction of Phage Ori+ Plasmids
Phage-derived DNA fragments containing putative ori sequences representing c2-, bIL67-, and 923-like ori groups were amplified by PCR technique using suitable primers (Table 2) and respective phage (94p4, 27, p6/4 and c2) lysates as templates. The 5′ extremities of the primers were modified by adding sequences recognized by either EcoRI or EcoRV (forward) and HindIII (reverse) restriction enzymes. Amplified DNA fragments were cut and ligated with the pUN121 vector digested by the same respective enzymes and introduced into E. coli 2566 cells by means of electroporation. Transformants were selected on LB plates containing Tet (8 µg mL−1) and Amp (100 µg mL−1). The resultant recombinant plasmids (pUN121:ori27, pUN121:ori94p4, pUN121:orip6/4, pUN121:oric2) were confirmed by colony PCR using pUN121_F/ pUN121_R primers. The vector carries a tetracycline (Tet) and ampicillin (Amp) resistance markers and is able to replicate in E. coli cells but lacks a functional ori and an appropriate selection marker for L. lactis cells. To select for the ori-carrying plasmids in L. lactis cells, a chloramphenicol resistance marker (catR) was introduced. For this, the catR gene functional in L. lactis cells was amplified using the pG+host3 vector as a template and suitable primers carrying SalI and BamHI sites at the 5′ extremities (Table 2). The SalI/BamHI-digested catR product was ligated with the pUN121:ori27, pUN121:ori94p4, pUN121:orip6/4 and pUN121:oric2 plasmids cut with the same enzymes and introduced by electroporation into E. coli 2566 cells. Transformants were analyzed by colony PCR using primers pUN121_catR_F/pUN121_catR_R and the cloned fragments were verified by DNA sequencing.
2.10. Functional Analysis of Putative Phage Ori Regions
To confirm the ability of the cloned putative oris to carry out plasmid replication, L. lactis IL1403 and MG1363 cells were electroporated with 1000 ng of pUN121:catR:ori27, pUN121:catR:ori94p4, and pUN121:catR:orip6/4 plasmid DNA. Transformants were selected on GM17 plates supplemented with Cm at 10 µg mL−1. Relative transformation efficiency was calculated in respect to transformants carrying the pG+host3 plasmid which was used as a control for competence check. The pUN121:catR:oric2 vector carrying the previously identified phage c2 ori served for the method control.
3. Results
3.1. Phage Classification and Morphological Analysis
The phages under examination were derived from whey samples collected over a period of 4 years from disturbed fermentation processes in eight dairy plants across Poland (Figure 1A) [33,48]. For the study, we selected all phages previously classified as Ceduoviruses. DNA samples from these phages were subjected to restriction fragments length polymorphism (RFLP) analysis (Figure 1B). Based on the DNA restriction patterns, we distinguished and analyzed 10 distinct Ceduovirus phages. By PCR analysis using specific primers, we re-confirmed that all the analyzed phages were Ceduoviruses (Figure 1C).
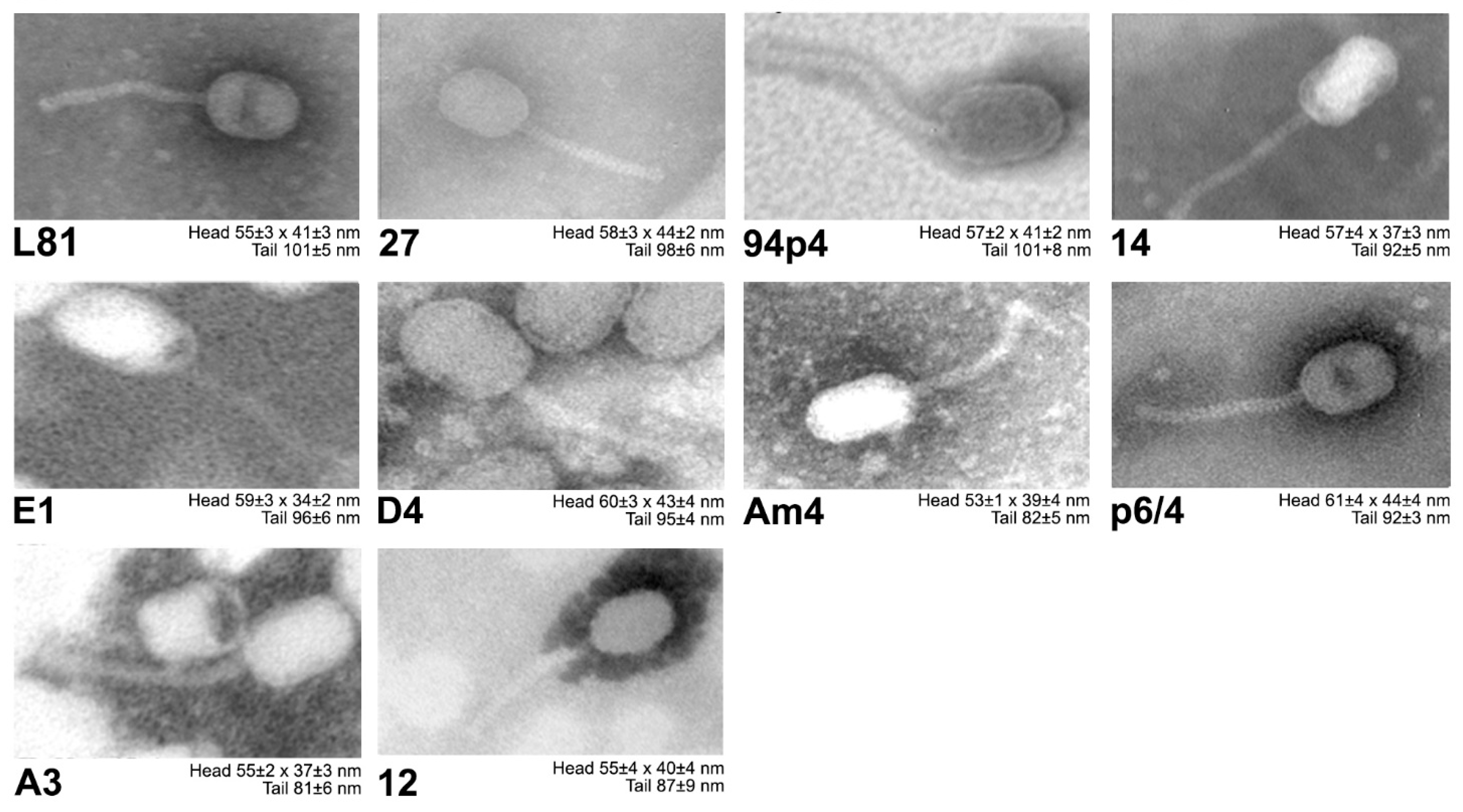
Electron microscopy imaging revealed that all of the studied phage isolates possess prolate heads (morphotype B2) and long non-contractile tails, both of which are distinctive for Siphoviridae Ceduovirus phages (Figure 2).
3.2. Phage-Host Interactions
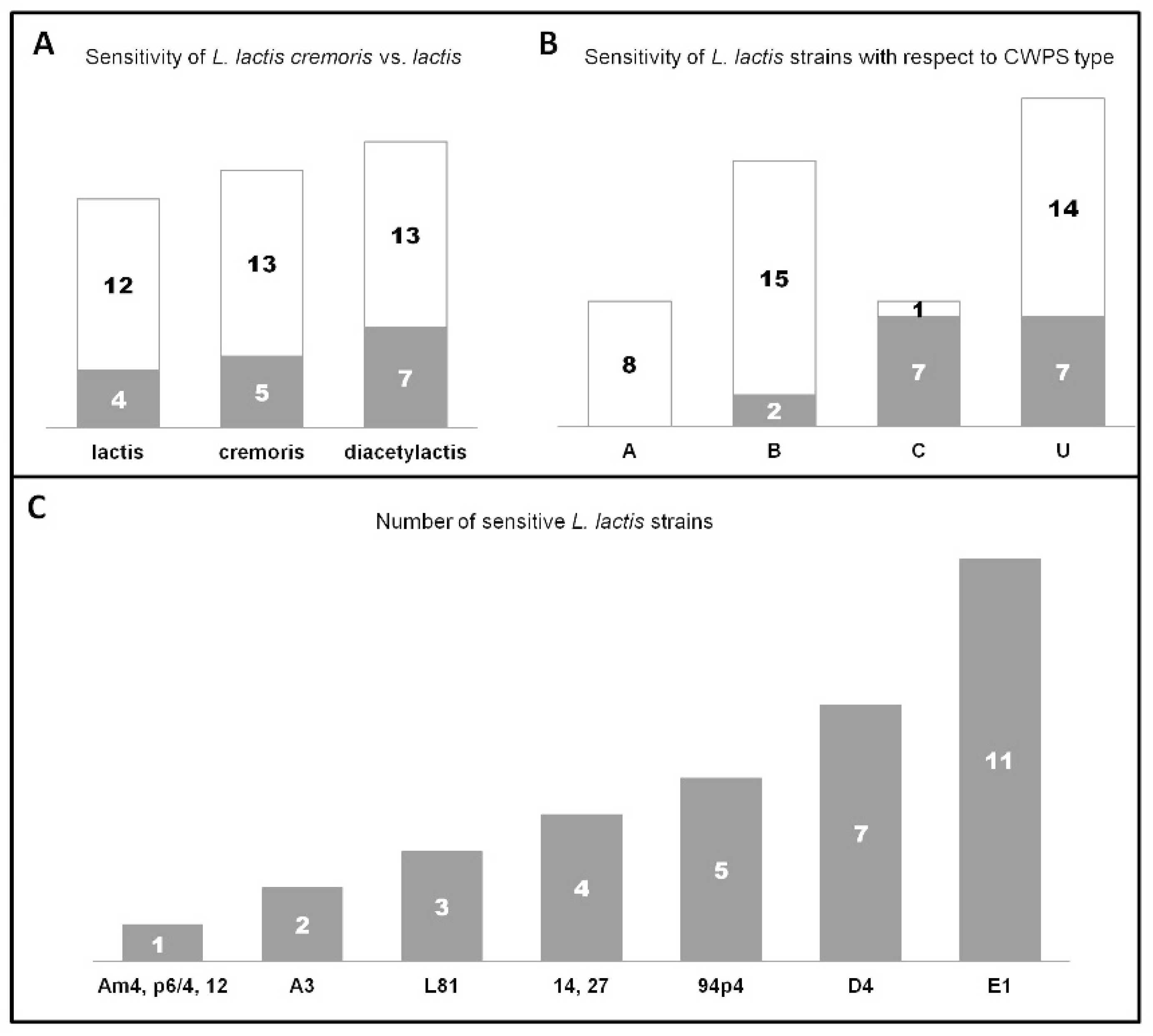
The capacity to lyse L. lactis cells was tested against a total of 54 isolates deriving from different environments (industrial and natural), and represented by a similar number of subsp. lactis (29.7%), subsp. lactis biovar. diacetylactis (37%), and subsp. cremoris (33.3%) strains (Supplementary Materials Table S1A). As a result of this assay, host infection ranges could be determined. The examined phages were found to possess distinct infection patterns, by lysing different strains. The sole exceptions were phages 14 and 27, for which identical lytic profiles were determined. Almost 30% of all L. lactis strains tested were susceptible to phage infection. Among these phage-sensitive strains, 75% of were lysed by two or more phages. Notably, two reference phages, c2 and bIL67, did not form plaques on any of the industrial or environmental strains tested. Moreover, results of this assay revealed that taking together all L. lactis subsp. lactis strains, they were more frequently lysed by the studied Ceduovirus phages (biovar. diacetylactis: 35%; subsp. lactis: 25%) than the cremoris strains (27.7%) (Figure 3A).
Five out of ten phages were determined to present a relatively broad host range lysing four or more strains (Figure 3C). In this aspect, phage E1 was the most exceptional, infecting a total of 11 strains. Only three phages (Am4, p6/4, and 12) exhibited a restricted host profile limited to one L. lactis strain. The phage infection pattern was additionally tested on three laboratory L. lactis strains: IL1403, MG1363 and NZ9000 (Supplementary Materials Table S1B). In this assay, a clear subspecies preference was observed. Phages 14, 27 and Am4, similarly to the reference phage bIL67, exhibited lytic activity only on the IL1403 (biovar. diacetylactis) strain. In contrast, phages A3, L81, D4, and 12 formed clear plaques on the cremoris strains MG1363 and NZ9000, like the reference phage c2. The three remaining phages, 94p4, E1, and p6/4, were most particular, as they poorly lysed the IL1403 strain (decreased titer compared to their reference strains) (Supplementary Materials Table S1B). Additionally, E1 and p6/4 could not develop on either MG1363 or NZ9000. Such subspecies correlation with the lytic spectrum was less evident for industrial and environmental L. lactis strains. Only phages 14 and 27 exhibited an explicit preference for biovar. diacetylactis, lysing four out of 20 strains (20%) from this subgroup, whereas the three narrow host range phages propagated on single strains: cremoris (phages 12), lactis (phage Am4), and biovar. diacetylactis (phage p6/4). In turn, phages with the widest host profile (94p4, D4, and E1) infected L. lactis strains from both subspecies.
Among phages with the widest host range (lysis of > 4 L. lactis strains), phages E1 and D4 were the only ones that infected bacteria deriving from natural environmental samples. These two phages presented also a similar lytic spectrum against cremoris and lactis strains, excluding biovar. diacetylactis (Supplementary Materials Table S1A). Moreover, phage E1 was the only isolate exhibiting an overlapping host range pattern with phages A3, D4, and L81 (group 1) and 14, 27, and 94p4 (group 2).
In previous works, cell wall polysaccharides (CWPS) of bacterial hosts were determined to be a crucial factor in the initial phase of phage adsorption [49,50]. Currently three CWPS types are recognized: A, B, and C, and group U of so-far unknown type(s) [19,32]. In order to elucidate the influence of this factor on phage adsorption, we analyzed the infection patterns with regards to the CWPS type of L. lactis cells. Among all industrial and environmental strains tested 31.5% possessed the B type CWPS, while C and A types were equally present in 14.8% of the cells, and 38.9% had an uncharacterized (U) CWPS genotype.
Our host infection study showed that among susceptible industrial strains (16 out of 51) an equal percentage (43.75%) had either the C or U type CWPS, while only 12.5% of phage-sensitive strains had a confirmed B-type genotype. None of the phages lysed A-type CWPS L. lactis strains (Figure 3B). When taking into account the whole set of L. lactis strains tested, four out of the ten studied phages infected only L. lactis strains of a specific type CWPS—phages A3 and 12 lysed only cells with type C CWPS, and phages Am4 and p6/4 with type B CWPS. The remaining six phages formed plaques on L. lactis strains with C and U, B and U, or B, C and U CWPS genotypes (Supplementary Materials Table S3).
3.3. Infection Dynamics in One-Step Growth Tests
The results of one-step growth assays determined for phages examined in this study a latent period of 19 ± 2 to 28 ± 0 min, and burst time of 27 ± 2 to 33 ± 0 min (Table 3). Despite the rather uniform latent periods and burst times, the burst size of the phages varied and ranged from 32 ± 3 to 197 ± 17 particles per cell. Phage lytic activity was also studied by examining the morphology and plaque size, which ranged between 2 and 5.3 mm. Additionally, for some phages, turbid halos around the plaques were detected. Such plaque morphology may be indicative of phage-encoded depolymerase activity on bacterial cell wall exopolysaccharides.
3.4. Phage Genome Sequencing and Comparative Analysis
Whole-genome sequencing of 10 Ceduovirus phages isolated from industrial whey samples revealed their genetic content and allowed comparison to other phages from this group. Genome sizes of the characterized phages ranged between 21,458–22,703 base pairs, with an average G-C content of 36% (Table 4). Phage genomes were determined to be organized into two oppositely-oriented blocks, corresponding to the early and late transcribed genes. These two regions were established to contain 18–21 early and 18 late open reading frames (orfs).
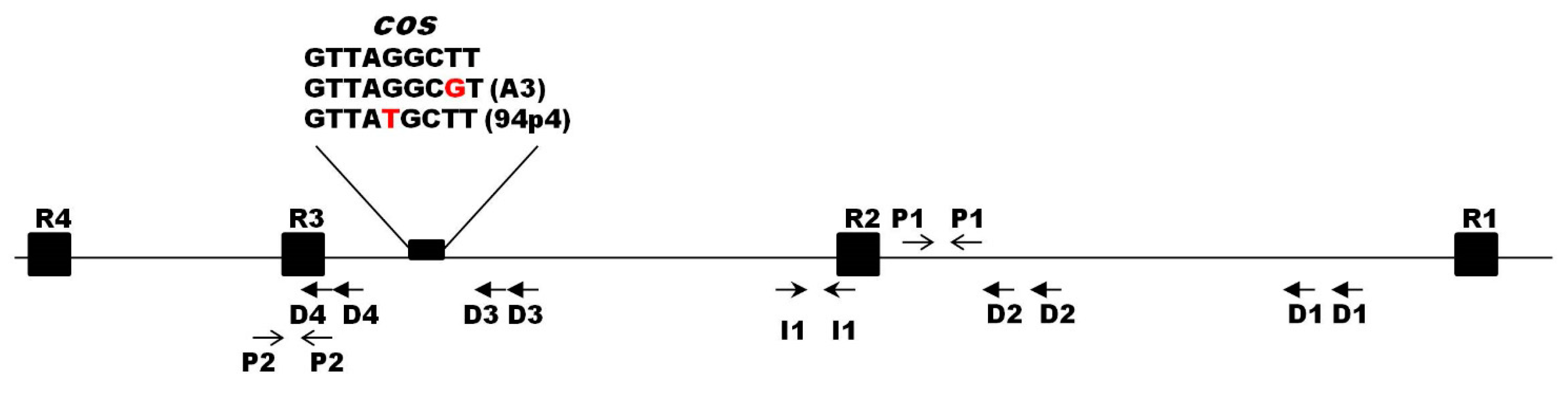
At the extreme regions of the analyzed phage genomes, 9-nt cohesive ends (cos) were recognized. For eight out of ten phages, the sequence of the cos sites was identical (5′-GTTAGCTT-3′) and matched that of phage c2. The cos sites of phages A3 and 94p4 differed by a single nucleotide each (Supplementary Materials Figure S1). Other features typical for the region surrounding the Ceduovirus cos sites were also identified and included four direct (D1–D4) and one inverted (I1) repeats, as well as two palindromic sequences (P1–P2) (Figure 4). Moreover, four 19-bp putative terminase binding sites (R1–R4) were recognized based on analogous sites previously reported for phage c2 [51].
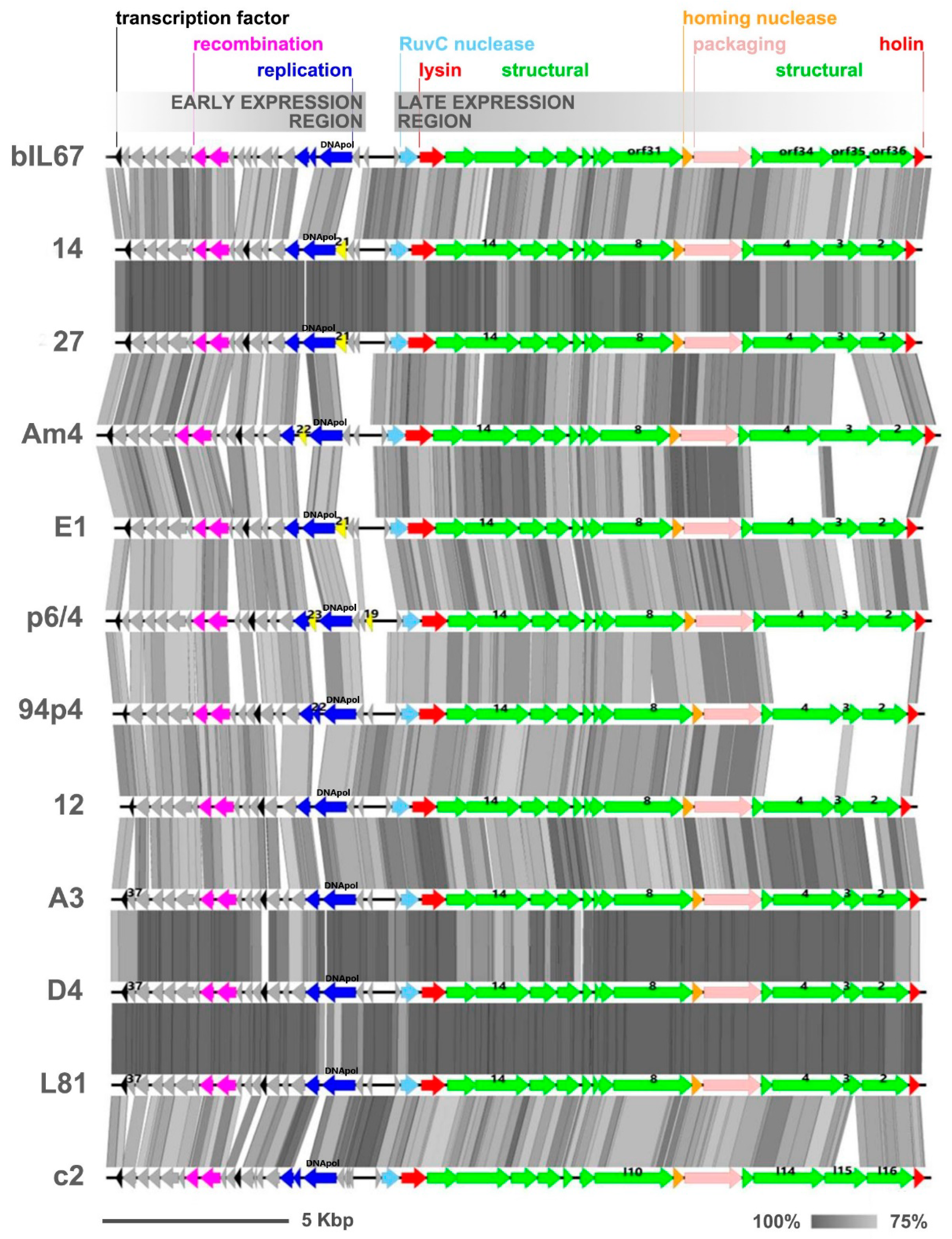
The genomes of the sequenced phages exhibited highly similar gene synteny, resembling that reported for two reference Ceduovirus phages, namely bIL67 and c2 [7,8] (Figure 5).
BLAST pairwise alignments with a set of whole phage genomes revealed an overall 86%–99% of nucleotide identity level between the individual phages (coverage 75%–100%). Average nucleotide identity (ANI) calculations confirmed over 80% of identity among the Polish phage isolates (Supplementary Materials Figure S2). Phages 27 and 14 (group A) and D4, A3, and L81 (group B) each shared more than 95% ANI, suggesting the highest similarity among these phages. Interestingly, while phages D4 and A3 from group B derived from the same dairy plant, L81 was isolated from a distant location. Also, group A phages were isolated from distinct geographical sites. An ANI level above 80% was also determined between the analyzed phages and other Ceduoviruses present in the GenBank database. In this case as well the nucleotide identity was often higher for phages from distant locations than for indigenous isolates; for instance phage 12 shares >95% ANI with phage CHPC1161. These observations were confirmed by constructing a supertree (Figure 6) based on separate multiple alignments of selected gene products (DNApol, Essential Recombination Function (ERF)-like recombinase, holin, lysin, GP2, GP4, and GP14) and homologous proteins of other Ceduovirus phages accessible in the GenBank database (ML trees for each multiple sequence alignments are shown in Supplementary Materials Figure S3A–G). The supertree shows separation of the phages into two major subgroups. The newly sequenced phages are present in both subgroups and show close clustering with other Ceduovirus phages from distinct geographical locations (Supplementary Materials Figure S3A–G).
All-against-all pairwise alignments of nucleotide sequences of the early transcribed gene regions revealed 85%–99% of identity (with 76%–100% coverage) among the studied phages. Within this region, two modules encoding putative recombination (ERF-like: pfam 04404 and SSB: pfam 16773) and two to three replication (DNApol) protein products (one with the RecA-like_NTPase superfamily domain cl28885) were annotated by RAST coupled with BLASTp and HHPRED analyses. In spite of the overall similarity, the sequence divergence between phage genomes was observed to result from variations in the length and in the number of identified orfs directly upstream of the recombination gene module and the sequence surrounding the putative replication genes (Figure 5). The product of one of these orfs, gp37, found in phages D4, A3, and L81, was predicted to contain a sterile alpha motif (SAM)-like fold (40.7% confidence, 66% coverage) characteristic for the RuvA domain 2-like superfamily. A truncated version of gp37, lacking the last nine nucleotides, was found in phage p6/4. Interestingly, GP37-like proteins with unknown function were detected in only four other Ceduovirus phages: bIL67, D4410, D4412, and M6654. Another distinct orf was detected in phages 94p4 (gp22), p6/4 (gp23), and Am4 (gp22) between two orfs constituting the putative replication module. The gp22-encoded product of phage 94p4 exhibited similarity to proteins of unknown function encoded by other Ceduovirus phages in the same genomic position. In contrast, the products encoded by the two remaining phages appeared to be largely unique: the p6/4 gp23 product exhibited no significant hits to any known proteins in the databases, whereas homologs of the Am4 gp22 product were identified only in phages LW81 and 949, belonging to another Lactococcus phage group (949-like). This finding indicates events of horizontal gene transfer (HGT) between different types of lytic lactococcal phages. Another distinct orf, gp21, was mapped in the early gene region in phages 14, 27, and E1. Its putative product was determined to be a homolog of a hypothetical protein from a Lactococcus P335-like phage 98204 (72%–73% identity, 97% coverage) as well as products encoded by various L. lactis strains, implying a possible HGT between prophage sequences and lytic phage genomes. Notably, the gp21 product was found to possess an EF-Ts (elongation factor Ts) domain-like fold. Alignment studies using ClustalOmega showed sequence conservation to the C’ terminal part of Escherichia coli EF-Ts (GI: 67462330) involved in dimerization and protein–protein interactions (data not shown). Gene products carrying the same fold have been previously detected in distant phages [52]. As EF-Ts catalyze the discharge of GDP from elongation factor Tu (EF-Tu) and facilitates EF-Tu binding to GTP, it was hypothesized that the phage-encoded EF-Ts-like protein may increase the rate of translation, facilitating phage propagation. Finally, a unique orf, designated as gp19, was detected at the end of the early gene region of phage p6/4 with no hits in BLASTp searches. Other sequence divergences in the early gene region of the studied phages were linked with the presence/lack of orfs, which putative products exhibit high sequence similarity to proteins encoded by other Ceduovirus phages, but which function remains unknown.
The late transcribed gene region of the studied phages encodes a conserved number of orfs. The nucleotide identity inferred from all-against-all comparison by pairwise alignment of this region was 86%–99% (with 69%–100% coverage). Comparative analysis of the late gene cluster revealed lower sequence conservation within the gp2-gp3-gp4 region, exhibiting analogy to orf36-orf35-orf34 of bIL67 (locus_tag: bIL67_gp02-gp03-gp04) and l16-l15-l14 of c2 (locus_tag: c2p38-c2p37-c2p36), suggested to encode structural proteins, possibly adhesion fibers, involved in host range determination [8,15]. Further divergences were noted within orfs for which potential products were annotated as putative tape measure protein (TMP; GP8) and major capsid protein (MCP; GP14). The deduced amino acid sequences of all products were subjected to careful examination in order to correlate them with the host spectrum.
The gp2-gp3-gp4 region showed the largest sequence divergence within gp3 (Figure 5), which resulted from variations both of the length (from 169 AA to 542 AA) and sequence of its putative product. Multiple sequence alignment of the gp3 product of the phages sequenced in this study, and respective proteins of other Ceduovirus phages, revealed high similarity at the N’ terminus and divergent middle and C’ parts (Supplementary Materials Figure S4). Notably, the N’ end of the gp3 product of phages 27, 14, 94p4, Am4, and E1 exhibited significant sequence conservation (>95% identity) to the first 100 AA of orf35 product (ORF35bIL67) and l15 product (ORFl15c2). For the remaining phages, the N’ terminus of the gp3 product demonstrated lower sequence similarity to ORF35bIL67 and ORFl15c2 (75%–77% identity), and resembled more the hypothetical protein (YP_009278611.1) of phage D4412 (>95% identity). Divergence was also noted in the C’ terminal part of GP3 and could be divided into products homologous to the last 65 AA of the C’ terminus of ORFl15c2 (>80% identity; phages E1, p6/4, A3, L81, D4, and 12) or 55 AA of ORF35bIL67 (>85% identity; phages 27, 14, 94p4, and Am4). In turn, BLASTp analyses of gp3 products longer than 300 AA showed that they are most divergent in the central part, but overall exhibit the highest similarity to ORF35bIL67 (59%–62% identity; 98%–100% coverage). The putative products of gp2 and gp4 showed minor variations in length and AA sequence, which included short deletions and/or substitutions. Based on the calculated size of GP4, phages in this study could be divided into those encoding a 628- (phages 94p4, Am4, 14, and 27) or 638-AA (phages E1, p6/4, 12, A3, D4, and L81) protein. BLASTp all-to-all comparison showed high similarity (>90% identity) of gp4-encoded products within each of these two groups, and a generally low intergroup similarity (39%–42% identity). The exceptions were phage 94p4 and p6/4, which both exhibited lower similarity (80% and 88% identity, respectively) to other gp4 products within their groups. In turn, comparative analysis of the deduced AA sequence of GP2 showed two conserved groups with low intergroup similarity (≤40% identity). In general, each of the two groups possessed a highly conserved N’ (first 60 AA) and C’ (last 200 AA) end, and a more variable central part. The first group comprised gp2-encoded products from phages Am4, 14, and 27 (≥90% sequence identity; 397 AA), and from phage 94p4 (72%–74% identity; 401 AA). The second group of GP2 products was more diverse in length (413–418AA) and in sequence (78%–99% identity). Within this group, a highly conserved (99%–100% identity) subclass of GP2 products could be distinguished for phages A3, L81, and D4.
As we believe the gp2-gp3-gp4 encode for tail fibers involved in adhesion, an attempt was made to correlate the sequencing data of their putative products with the host infection pattern of the studied phages (Supplementary Materials Table S3). Yet, we did not observe any direct link between the AA sequences of these products with the subspecies or the CWPS type of phage-susceptible strains. Notably, phages 14 and 27 infected the same hosts with CWPS of U type, but did not encode identical gp2-gp3-gp4 products, and, conversely, phages L81 and D4, for which the AA sequences of gp3 and gp4 products were 100% identical, propagated on different hosts. However, when results of host range studies were limited to reference laboratory strains, it was observed that phages with the smallest (169 AA) gp3 product produced clear plaques only on L. lactis subsp. cremoris, while phages encoding the longer protein version also infected L. lactis subsp. lactis. Further studies are needed to better understand this observation. Lower sequence conservation was also found within gp8, of which the product was annotated as a putative tape measure (TMP) due to the presence of a specific domain (TIGR02675) typical for phage proteins known or predicted to act as the tail tape measure. Dissimilarities were due to both the length and the deduced AA sequence (72%–96% identity) of the gp8 product. In phages bIL67 and c2, the corresponding products encoded by orf31 and l10 were suggested to play (along with orf34-orf35-orf36 and l14-l15-l16, respectively) a role in host range determination [8,15]. Comparative analyses accompanied by BLASTp searches allowed us to distinguish four groups of gp8-encoded products based on their high identity (>90%) with proteins of previously sequenced Ceduovirus phages: hypothetical protein of phage CHPC1161 (protein id: QGT52480.1)—L81, D4, A3; tail assembly protein of phage CHPC134 (protein id: QGT52852.1)—12, 94p4; phage CHL92 ORF5 (protein id: AAO15644.1)—p6/4; and phage bIL67 ORF31 (protein id: NP_042314.1)—Am4, E1, 27, and 14. The bIL67- and CHL92-like gp8 products are of the same length (620 AA) and differ by single AA substitutions. The CHPC1161- and CHPC134-like products are longer (706 AA) and carry an 86-AA insertion in the central part of the protein and exhibit high similarity to ORFl10 of phage c2. Otherwise, the AA sequence of the N’ and C’ terminal part for both groups are largely conserved. For discrimination purposes, in our analyses, we called these two groups: bIL67-like and c2-like. Relatedness of the gp8-encoded products was confirmed by phylogenetic analyses using the maximum-likelihood algorithm (Supplementary Materials Figure S5). The ML tree shows a clear separation of gp8 products into two branches, correlating with their size. Analysis of the deduced AA sequence allowed us to distinguish one full and one to two incomplete motifs (ER-hand calcium-binding domain) known to be implicated in binding Ca2+ and required for efficient adsorption of Ceduovirus phages to bacterial cells [53]. For phages 14, p6/4, 94p4 and Am4, a YqbO (lysozyme-like) domain was detected in the N terminal part of GP8, which we believe may play role in peptidoglycan-degradation of host cell walls. Remarkably, phages carrying this domain infected only the lactis subspecies, except 94p4 which also infected cremoris strains. For phages 27 and E1, the YqbO domain was not detected by computational tools, but the sequences were highly identical across the 1–420 AA region spanning the domain (only one AA mismatch). All of these results corroborate the role of GP8 in host recognition/adsorption and entry of phage DNA. Interestingly, a connection between the deduced AA length of GP8 and infection pattern could be observed (Supplementary Materials Table S3). Phages encoding 620-AA GP8 infected only L. lactis subsp. lactis strains, whereas phages encoding the longer product could propagate also on L. lactis subsp. cremoris hosts. The sole exception was phage E1, which encoded the shorter version of the putative GP8, but was able to lyse L. lactis from both the cremoris and lactis groups.
Lower sequence conservation was also detected within gp14, for which the potential product was annotated as a putative major capsid protein (MCP). Detailed multiple alignment studies allowed us to establish that these differences were rather minor—the overall AA sequence identity of GP14 was >90% (98%–100% coverage). The lowest similarity was observed for GP14 of phages 14 and 27, for which the deduced sequence contained an additional four AA in the central part compared to the other phages tested. Yet, this difference did not correlate with capsid size (Figure 2) or host range of the phages (Supplementary Materials Tables S1 and S3).
3.5. Identification and Functional Analysis of the Origin of Phage Replication
The origin of replication has been previously identified in Ceduovirus phages in the intergenic region between the two divergently-oriented modules comprising, respectively, the early and late genes [8]. Multiple alignments of the intergenic region from phages studied in this work and the previously identified replication oris of Ceduovirus phages allowed us to classify them into specific ori groups: c2-, bIL67-, or 923-like (Supplementary Materials Figure S6).
Overall, the replication origins within each group were highly conserved (>90% sequence identity) and presented no significant intergroup similarity. Among the three recognized ori types, the c2-like ori was most frequently represented and was found in six out of ten sequenced phages, the bIL67 ori type was determined in three, and 923-type in only one phage. For all phages, the analyzed region was found to be flanked by two putative promoter sequences, which are the first two promoters of the early transcribed gene region (PE1, PE2). The region within these two promoters did not contain any orfs ≥ 100 bp. No ribosome-binding sites downstream of the putative PE1 were detected. Both of these features imply that the examined DNA fragments do not encode any protein products. To confirm that these regions contain the putative phage origin of replication, we examined their capacity to carry out plasmid replication in a non-replicative vector in Lactococcus cells. For this, representatives of each ori type were cloned in the E. coli pUN121 vector, lacking a functional origin in L. lactis cells. A set of ori+ vectors were obtained: pUN121:ori27 (bIL67-ori type), pUN121:orip6/4 (923-ori type), pUN121:ori94p4 (c2-ori type), and pUN121:oric2 (method control). The ability of phage ori regions to support replication was tested by introducing the ori+ vectors into L. lactis cells and screening for transformants resistant to chloramphenicol. The results showed that all ori-test plasmids were successfully introduced into L. lactis IL1403 wt (subsp. lactis) and MG1363 (subsp. cremoris) (Supplementary Materials Table S2). This observation holds with the notion that the tested putative phage ori fragments are able to support plasmid DNA replication without additional phage functions in both L. lactis strains.
4. Discussion
Phages belonging to the Ceduovirus group are the second most persistent group of lytic Lactococcus phages in dairy plants worldwide. Biodiversity studies have shown the dominance of lytic Ceduovirus phages in whey samples only in several countries (e.g., Canada, Germany, Russia, Belarus, and the Czech Republic) [10,54,55]. This observation seems intriguing given the fact that Ceduovirus phages have been repeatedly described to have a much wider host range compared to Skunavirus phages, and it would be expected that they would encounter a permissive strain more frequently and spread in the environment more easily [10,33,56,57].
In order to gain more knowledge concerning Ceduovirus phages in the Polish dairy environment, we investigated the physiological features and performed whole-genome sequencing coupled with comparative analysis of 10 lactococcal phage isolates. The phages derived from whey samples collected over a four-year period in eight different geographical locations. The lytic nature of Ceduovirus phages was confirmed by observing a rapid lysis of Lactococcus lactis cells. The relatively short latent period of the studied phages is comparable to that of other lactococcal phages detected in dairy settings. In general, lactococcal phages exhibit a latent period of 20 to 60 min, and a burst size in the range of 42 to 400 plaques per cell [58]. The determined burst sizes (32 ± 3 to 197 ± 17 plaques per cell) and high titers (>1 × 109 PFU mL−1) on reference starter strains indicate effective phage propagation and an overall good adaptation to the dairy environment. This observation is further supported by the fact that two reference lactococcal phages, bIL67 and c2, were unable to lyse any of the industrial strains tested. It may also imply the ability of industrial phages to bypass potential bacterial phage-resistance mechanisms; however, this notion would need further investigation. A majority of the tested phages were found to infect from 4–11 Lactococcus strains of different origin (industrial, environmental, laboratory), which corresponds to a relatively wide host range. Among them, phages D4 and E1 presented the widest lytic spectrum. They were the only ones that could lyse L. lactis cells from natural sources, indicating a particular adaptation of these phages to both dairy and natural environments. The presence of a turbid halo around the plaques of D4 and several other phages may signify production of carbohydrate-degrading enzymes. This property is regarded as a competitive/adaptive advantage that increases the fitness of the phage to certain niches [59].
The preference of the studied phages to lyse either cremoris or lactis (including biovar. diacetylactis) strains was observed, and was especially clear when analyzing laboratory strains but less apparent when considering the whole subset of L. lactis strains. This discrepancy may possibly be due to phage-resistance mechanisms (e.g., plasmid- or prophage-encoded) carried by the starter bacteria. Overall, the highest percentage of L. lactis phage-sensitive strains belonged to biovar. diacetylactis. A similar preference for biovar. diacetylactis strains has been reported previously also for Polish Skunaviruses [31]. Such results may indicate the dominant use of biovar. diacetylactis starter strains in Polish dairy factories and, subsequently, adaptation of phages to this particular host type. Remarkably, not all phages were efficient in lysing the model, laboratory bacteria (IL1403, MG1363, and NZ9000). Phages E1, and p6/4 exhibited poor lysis (turbid, small plaques, low titer compared to their reference strains) of the IL1403 strain, while the cremoris type strains (MG1363 and NZ9000) were fully resistant to these phages. Recently, it has been shown that resident prophages may influence host susceptibility and the phage plaquing efficiency [60]. This effect may be due to a yet undermined prophage-encoded anti-Ceduovirus defense mechanism, similar to the described super-infection exclusion (Sie) phenomenon against Skunavirus phages. Thus, the potential factors influencing the lytic pattern of the analyzed phages, including the prophage effect, need further exploration.
The sequenced phages show a preference for strains with type C CWPS, which is in line with previous reports on Ceduoviruses [10]. Based on this analysis, they were grouped into two types (i) phages infecting hosts with a specific CWPS type (B—for phages Am4 and p6/4, and C—for phages A3 and 12) and (ii) those which lyse hosts with different saccharidic receptors (six remaining phages). We may speculate that the first group of phages recognizes and adsorbs to unique components of specific CWPS types, while phages from the second group interact with a common CWPS determinant. Additionally, all strains with type A CWPS tested in this study were phage-resistant. Given these results, it seems that the bacterial cell wall components may be a crucial parameter that should be taken into account when selecting for phage-resistant industrial starter strains.
All versus all genome comparisons revealed more than 80% average nucleotide identity between the studied phages and other Ceduoviruses found in GenBank. Several phages shared >95% ANI, a value which is generally regarded as a cut-off for species discrimination. Remarkably, no general correlation with their geographical origin could be found; often phages isolated from different settings exhibited high identity, including as examples phages 12 and CHPC1161 or 14 and 27. Phylogenetic studies showed separation of Ceduoviruses into two major subgroups and confirmed the relatedness of phages from distant locations. In the case of phages 14 and 27, their high similarity was also reflected in the results of our host range and one-step growth studies. It could be that these two phages originated from the same parental phage, which was carried over by humans or as a contamination in starter media to different dairy plants where it diverged. Assessment of the link between phage phylogeny and the site of their isolation may be also impaired by the global distribution of common industrial starters. Only further analysis could provide informative data concerning the occurrence and evolution of related phages in geographically separate regions.
Apart from host factors, results of our analyses suggest that the gp8 product of the studied phages may be correlated with their lytic spectrum. GP8 has been annotated as a putative tape measure based on the presence of TIGR02785 domain typical for phage TMPs. In other phages, tape measure proteins have been implicated in several crucial steps of phage infection, including tail length determination and entry of phage DNA into the host cell. The ORFl10 of c2 and ORF31 of bIL67, encoded by genes located in the same genetic context, were suggested to play a role in host range determination [8,15]. Based on the similarity of GP8 to these two products, two (c2-like and bIL67-like) groups varying in the length of AA sequence were detected. Remarkably, phages encoding longer GP8 (c2-like) were capable of lysing cremoris and, in some cases, lactis strains of C or U CWPS type. Shorter products (bIL67-like) were restricted to phages infecting solely subsp. lactis cells of B or U CWPS type. The exceptions were phages 94p4 and E1, which both formed plaques on lactis and cremoris strains (B, C, or U type), but encoded for GP8 of a different length. These observations allow us to assume that the recognition of particular lactococci hosts with a specific CWPS type is dependent on the AA composition/length of the GP8. Yet, the exact function of GP8 remains obscure. In general, phage TMPs are acknowledged to play role in DNA injection by forming a channel that reaches through the thick peptidoglycan layer, typical for Gram-positive lactococci, into the host cell. The presence of a Ca2+ binding motif and peptidoglycan-degrading (YqbO) domain (for shorter products) within the GP8 support the notion of its function as a tape measure protein. Further data will be needed to resolve this issue. Generating a series of hybrid gp8 products, differing in length and presence of specific motifs and domains, may provide more data on the crucial sequence elements that correlate with host range.
As suggested by previous studies on c2 and bIL67 model Ceduovirus phages, product(s) of the l16-l15-l14 and orf36-orf35-orf34 gene clusters, respectively, might encode for another host range determinant(s) [15]. Our comparative analysis of analogous phage products encoded by gp2-gp3-gp4 did not reveal any correlation of AA sequence divergence with host range. Within the gp2-gp3-gp4 region, the gp3 product exhibits the largest variation in length and might have the biggest impact on the phage host range. However, this assumption would need further examination, as phage Am4 encoding the longest (542 AA) putative gp3 product identified so far in Ceduovirus phages lysed only 1 out of 54 examined L. lactis strains. The widest spectrum of infection was noted for phage E1, of which the gp3 product was most similar in AA sequence to that of phage 27, able to effectively lyse only four strains. Thus, it seems that there are other proteins (or factors) engaged in host range determination. Notably, the lytic spectrum of phage E1 overlapped with that of phages infecting exclusively cremoris or lactis/diacetylactis strains. Thus, it may be that phage E1 is a hybrid between c2-like and bIL67-like phages. Yet, we cannot exclude that the broad host range of E1 is rather the effect of phage evolution due to starter strain rotation—a common routine applied in dairy plants to limit phage infections. Such an approach in the long term is suggested to favor the appearance of phages with an extended host range [18].
Sequence analysis of the putative ori region confirmed the presence of three distinct ori types, with c2 ori type the most frequently observed. This is in line with previous studies on a much larger set of phages from New Zealand where the c2 ori type also prevailed [14]. To date, no recombinant oris have been detected, which strongly implies a general sequence conservation of the three ori types, irrespective of geographical location. Our studies prove that the identified ori regions serve the same function and support plasmid replication without any additional phage proteins. Moreover, we show that the ori regions are functional in both lactis and cremoris strains, regardless of the lytic spectrum of the phage itself. This suggests that other host-phage factors than ori incompatibility influence the observed host range.
5. Conclusions
The role of Ceduovirus phages in shaping dairy environments should not be underestimated. With our study, we anticipated to deepen our knowledge on host-phage interactions and genetic diversity in relation to already sequenced Ceduovirus phages. The results of our study suggest a link between the putative tape measure protein and host range. Phylogenetic studies and average nucleotide identity calculations have shown that highly similar phages can occur in distant geographical locations. To our knowledge, this is the first reported research on Ceduovirus phages that have emerged from dairy plants in Poland. Data on Ceduovirus phage characteristics, including preference for specific L. lactis hosts and the presence of unique features, provides valuable information that may be used to develop efficient methods of eliminating them from dairy plants and, at the same time, creates a basis for future phage studies.
Supplementary Materials
The following are available online at https://www.mdpi.com/1999-4915/12/3/280/s1.
Author Contributions
M.C.-J. was engaged in all experimental works, including phage DNA isolation and genome annotations. She also drafted the figures and participated in writing of the manuscript. A.K.S. coordinated the experiments, performed physiological and bioinformatic studies, critically analyzed the data, prepared graphs and tables, and drafted the main body text of the manuscript. J.K.B. was involved in critical review of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the European Funds Portal Innovative Economy “Centre of medicinal product biotechnology. Package of innovative biopharmaceuticals for human and animal therapy and prophylactics” grant number POIG.01.01.02-00-007/08.
Acknowledgments
We would like to extend out thanks to Szymon Suski from the Laboratory of Electron Microscopy at the Nencki Institute of Experimental Biology (Warsaw, Poland) for help with electron micrographs as well as Janek Gawor and Karolina Żuchniewicz from the DNA Sequencing and Oligonucleotide Synthesis Laboratory, IBB Polish Academy of Sciences, for their technical assistance during genome sequencing and assembly. We would like to appreciate the help of Kamil Steczkiewicz in phylogenetic analyses and figure drawings.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Labrie, S.; Moineau, S. Multiplex PCR for detection and identification of lactococcal bacteriophages. Appl. Environ. Microbiol. 2000, 66, 987–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deveau, H.; Labrie, S.J.; Chopin, M.C.; Moineau, S. Biodiversity and classification of lactococcal phages. Appl. Environ. Microbiol. 2006, 72, 4338–4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, A.W.; Fitzgerald, G.F.; Mata, M.; Mercenier, A.; Neve, H.; Powell, I.B.; Ronda, C.; Saxelin, M.; Teuber, M. Species and type phages of lactococcal bacteriophages. Intervirology 1991, 32, 2–9. [Google Scholar] [CrossRef]
- McGrath, S.; Neve, H.; Seegers, J.F.; Eijlander, R.; Vegge, C.S. Anatomy of a lactococcal phage tail. J. Bacteriol. 2006, 188, 3972–3982. [Google Scholar] [CrossRef] [Green Version]
- Castro-Nallar, E.; Chen, H.; Gladman, S.; Moore, S.C.; Seemann, T.; Powell, I.B.; Hillier, A.; Crandall, K.A.; Chandry, P.S. Population genomics and phylogeography of an Australian dairy factory derived lytic bacteriophage. Genome Biol. Evol. 2012, 4, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.; Bottacini, F.; Mahony, J.; Kelleher, P.; Neve, H.; Zomer, A.; Nauta, A.; van Sinderen, D. Comparative genomics and functional analysis of the 936 group of lactococcal Siphoviridae phages. Sci. Rep. 2016, 6, 21345. [Google Scholar] [CrossRef] [Green Version]
- Schouler, C.; Ehrlich, S.D.; Chopin, M.C. Sequence and organization of the lactococcal prolate-headed bIL67 phage genome. Microbiology 1994, 140, 3061–3069. [Google Scholar] [CrossRef] [Green Version]
- Lubbers, M.W.; Waterfield, N.R.; Beresford, T.P.; Le Page, R.W.; Jarvis, A.W. Sequencing and analysis of the prolate-headed lactococcal bacteriophage c2 genome and identification of the structural genes. Appl. Environ. Microbiol. 1995, 61, 4348–4356. [Google Scholar] [CrossRef] [Green Version]
- Millen, A.M.; Romero, D.A. Genetic determinants of lactococcal c2viruses for host infection and their role in phage evolution. J. Gen. Virol. 2016, 97, 1998–2007. [Google Scholar] [CrossRef]
- Oliveira, J.; Mahony, J.; Hanemaaijer, L.; Kouwen, T.R.H.M.; van Sinderen, D. Biodiversity of bacteriophages infecting Lactococcus lactis starter cultures. J. Dairy Sci. 2018, 101, 96–105. [Google Scholar] [CrossRef]
- Hayes Whey Sample Dairy Phages. Available online: https://www.ncbi.nlm.nih.gov/nuccore/?term=Hayes%20Whey%20sample%20dairy%20phages&utm_source=gquery&utm_medium=search (accessed on 2 March 2020).
- Marcelli, B.; de Jong, A.; Kuipers, O.P. Genome Sequences of 31 Lactococcus lactis Bacteriophages Isolated from Foods. Available online: https://www.ncbi.nlm.nih.gov/nuccore/?term=Genome+sequences+of+31+Lactococcus+lactis+bacteriophages+isolated+from+foods (accessed on 2 March 2020).
- Damnjanovic, D.M. Lactococcus Phage vB_LacS_15, Complete Genome. Available online: https://www.ncbi.nlm.nih.gov/nuccore/1740136454? report=genbank (accessed on 3 January 2019).
- Rakonjac, J.; Ward, L.J.H.; Schiemann, A.H.; Gardner, P.P.; Lubbers, M.W.; O’Toole, P.W. Sequence diversity and functional conservation of the origin of replication in lactococcal prolate phages. Appl. Environ. Microbiol. 2003, 69, 5104–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuer-Lauridsen, B.; Janzen, T.; Schnabl, J.; Johansen, E. Identification of the host determinant of two prolate-headed phages infecting lactococcus lactis. Virology 2003, 309, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Mahony, J.; Oliveira, J.; Collins, B.; Hanemaaijer, L.; Lugli, G.A.; Neve, H.; Ventura, M.; Kouwen, T.R.; Cambillau, C.; van Sinderen, D. Genetic and functional characterisation of the lactococcal P335 phage-host interactions. Bmc Genom. 2017, 18, 146. [Google Scholar] [CrossRef] [Green Version]
- Hayes, S.; Duhoo, Y.; Neve, H.; Murphy, J.; Noben, J.P.; Franz, C.M.A.P.; Cambillau, C.; Mahony, J.; Nauta, A.; van Sinderen, D. Identification of dual receptor binding protein systems in lactococcal 936 group phages. Viruses 2018, 10, 668. [Google Scholar] [CrossRef] [Green Version]
- Rakonjac, J.; O’Toole, P.W.; Lubbers, M. Isolation of lactococcal prolate phage-phage recombinants by an enrichment strategy reveals two novel host range determinants. J. Bacteriol. 2005, 187, 3110–3121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ainsworth, S.; Sadovskaya, I.; Vinogradov, E.; Courtin, P.; Guerardel, Y.; Mahony, J.; Grard, T.; Cambillau, C.; Chapot-Chartier, M.F.; van Sinderen, D. Differences in lactococcal cell wall polysaccharide structure are major determining factors in bacteriophage sensitivity. MBio 2014, 5, e00880-e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapot-Chartier, M.P.; Kulakauskas, S. Cell wall structure and function in lactic acid bacteria. Microb. Cell Fact. 2014, 13 (Suppl. 1), S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCabe, O.; Spinelli, S.; Farenc, C.; Labbé, M.; Tremblay, D.; Blangy, S.; Oscarson, S.; Moineau, S.; Cambillau, C. The targeted recognition of Lactococcus lactis phages to their polysaccharide receptors. Mol. Microbiol. 2015, 96, 875–886. [Google Scholar] [CrossRef]
- Waterfield, N.R.; Lubbers, M.W.; Polzin, K.M.; Le Page, R.W.; Jarvis, A.W. An origin of DNA replication from Lactococcus lactis bacteriophage c2. Appl. Environ. Microbiol. 1996, 62, 1452–1453. [Google Scholar] [CrossRef] [Green Version]
- Lubbers, M.W.; Schofield, K.; Waterfield, N.R.; Polzin, K.M. Transcription analysis of the prolate-headed lactococcal bacteriophage c2. J. Bacteriol. 1998, 180, 4487–4496. [Google Scholar] [CrossRef] [Green Version]
- Del Solar, G.; Giraldo, R.; Ruiz-Echevarria, M.J.; Espinosa, M.; Diaz-Orejas, R. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 1998, 62, 434–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiemann, A.; Rakonjac, J.; Callanan, M.; Gordon, J.; Polzin, K.; Lubbers, M.W.; O’Toole, P.W. Essentiality of the early transcript in the replication origin of the lactococcal prolate phage c2. J. Bacteriol. 2004, 186, 8010–8017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopin, A.; Chopin, M.-C.; Moillo-Batt, A.; Langella, P. Two plasmid-determined restriction and modification systems in Streptococcus lactis. Plasmid 1984, 11, 260–263. [Google Scholar] [CrossRef]
- Gasson, M.J. Plasmid complements of Streptococcus lactis NCDO 712 and other lactic streptococci after protoplast-induced curing. J. Bacteriol. 1983, 154, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguin, E.; Duwat, P.; Hege, T.; Ehrlich, D.; Gruss, A. New thermosensitive plasmid for gram-positive bacteria. J. Bacteriol. 1992, 174, 5633–5638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, B.; Uhlén, M.; Josephson, S.; Gatenbeck, S.; Philipson, L. An improved positive selection plasmid vector constructed by oligonucleotide mediated mutagenesis. NAR 1983, 11, 8019–8030. [Google Scholar] [CrossRef] [Green Version]
- Dupont, K.; Vogensen, F.K.; Josephsen, J. Detection of lactococcal 936-species bacteriophages in whey by magnetic capture hybridization PCR targeting a variable region of receptor-binding protein genes. J. Appl. Microbiol. 2005, 98, 1001–1009. [Google Scholar] [CrossRef]
- Chmielewska-Jeznach, M.; Bardowski, J.K.; Szczepankowska, A.K. Molecular, physiological and phylogenetic traits of Lactococcus 936-type phages from distinct dairy environments. Sci. Rep. 2018, 22, 12540. [Google Scholar] [CrossRef]
- Mahony, J.; Kot, W.; Murphy, J.; Ainsworth, S.; Neve, H.; Hansen, L.H.; Heller, K.J.; Sørensen, S.J.; Hammer, K.; Cambillau, C.; et al. Investigation of the relationship between lactococcal host cell wall polysaccharide genotype and 936 phage receptor binding protein phylogeny. Appl. Environ. Microbiol. 2013, 79, 4385–4392. [Google Scholar] [CrossRef] [Green Version]
- Szczepanska, A.K.; Hejnowicz, M.S.; Kołakowski, P.; Bardowski, J. Biodiversity of Lactococcus lactis bacteriophages in Polish dairy environment. Acta Biochim. Pol. 2007, 54, 151–158. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M. The RAST Server: Rapid annotations using subsystems technology. Bmc Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI Bioinformatics Toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Kelley, L.A.; Sternberg, M.J.E. Protein structure prediction on the web: A case study using the Phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.G.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualiser. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Górecki, P.; Tiuryn, J. URec: A system for unrooted reconciliation. Bioinformatics 2007, 23, 511–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Krysa, W. Identification and Characterization of Virulent Lactococcal Bacteriophages. Master’s Thesis, University of Warsaw and the Institute of Biochemistry and Biophysics, Warsaw, Poland, 26 September 2002. (In Polish). [Google Scholar]
- Monteville, M.R.; Ardestani, B.; Geller, B.L. Lactococcal bacteriophages require host cell wall carbohydrate and a plasma membrane protein for adsorption and ejection of DNA. Appl. Environ. Microbiol. 1994, 60, 3204–3211. [Google Scholar] [CrossRef] [Green Version]
- Dupont, K.; Janzen, T.; Vogensen, F.K.; Josephsen, J.; Stuer-Lauridsen, B. Identification of Lactococcus lactis genes required for bacteriophage adsorption. Appl. Environ. Microbiol. 2004, 70, 5825–5832. [Google Scholar] [CrossRef] [Green Version]
- Perrin, R.; Billard, P.; Branlant, C. Comparative analysis of the genomic DNA terminal regions of the lactococcal bacteriophages from species c2. Res. Microbiol. 1997, 148, 573–583. [Google Scholar] [CrossRef]
- Gill, J.J.; Berry, J.D.; Russell, W.K.; Lessor, L.; Escobar-Garcia, D.A.; Hernandez, D.; Kane, A.; Keene, J.; Maddox, M.; Martin, R. The Caulobacter crescentus phage phiCbK: Genomics of a canonical phage. Bmc Genom. 2012, 13, 542. [Google Scholar] [CrossRef] [Green Version]
- Lubbers, M.W.; Wardm, L.J.; Beresford, T.P.; Jarvis, B.D.; Jarvis, A.W. Sequencing and analysis of the cos region of the lactococcal bacteriophage c2. Mol. Gen. Genet. 1994, 245, 160–166. [Google Scholar] [CrossRef]
- Boucher, I.; Moineau, S. Phages of Lactococcus lactis: An ecological and economical equilibrium. Recent Res. Dev. Virol. 2001, 3, 243–256. [Google Scholar]
- Raiski, A.; Belyasova, N. Biodiversity of Lactococcus lactis bacteriophages in the Republic of Belarus. Int. J. Food Microbiol. 2009, 130, 1–5. [Google Scholar] [CrossRef]
- Heap, H.A.; Jarvis, A.W. A comparison of prolate- and isometric- headed lactic streptococcal bacteriophages. N. Z. J. Dairy Sci. Technol. 1980, 15, 75–81. [Google Scholar]
- Moineau, S.; Fortier, J.; Ackermann, H.W.; Pandian, S. Characterization of lactococcal bacteriophages from Quebec cheese plants. Can. J. Microbiol. 1992, 38, 875–882. [Google Scholar] [CrossRef]
- Depuis, M.-E.; Moineau, S. Genome organization and characterization of the virulent lactococcal phage 1358 and its similarities to Listeria phage. Appl. Environ. Microbiol. 2010, 76, 1623–1632. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.P.; Oliveira, H.; Melo, L.D.R.; Sillankorva, S.; Azeredo, J. Bacteriophage-encoded depolymerases: Their diversityand biotechnological applications. Appl. Microbiol. Biotechnol. 2016, 100, 2141–2151. [Google Scholar] [CrossRef] [Green Version]
- Aucouturier, A.; Chain, F.; Langella, P.; Bidnenko, E. Characterization of a prophage-free derivative strain of Lactococcus lactis ssp. lactis IL1403 reveals the importance of prophages for phenotypic plasticity of the host. Front. Microbiol. 2018, 9, 2032. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Geographical distribution, restriction fragments length polymorphism (RFLP) and PCR analysis of Ceduovirus phages. (A) Map shows the geographical site of isolation of each individual phage. (B) Restriction fragments length polymorphism (RFLP) analysis of Ceduovirus phages using the EcoRV enzyme. DNA samples were digested o/n at 37 °C with EcoRV. To dissociate cos ends prior to electrophoresis 50% formamide was added. M: 1-kb DNA Ladder (Fermentas). Unique phage DNA restriction patterns are as follows: 1, L81; 2, 14; 3, 27; 4, 94p4; 5, p6/4; 6, 12; 7, A3; 8, Am4; 9, D4; 10, E1. (C) PCR-based typing of Lactococcus phages analyzed in this study. Unique fragments of phage genomes were amplified using primers complementary to the conserved genomic region of Ceduovirus phages (mcp gene). The arrow indicates the position of a 474-bp band specific to the Ceduovirus group. M: 1-kb DNA Ladder (Fermentas), lanes 1–10: DNA fragments generated from respective phage lysates which served as templates: 14, 27, 94p4, L81, p6/4, 12, E1, D4, Am4, and A3, lane 11: DNA fragment generated from c2 (Ceduovirus phage; positive control), lane 12: DNA fragment generated from sk1 (Skunavirus phage; negative control).
Figure 1.
Geographical distribution, restriction fragments length polymorphism (RFLP) and PCR analysis of Ceduovirus phages. (A) Map shows the geographical site of isolation of each individual phage. (B) Restriction fragments length polymorphism (RFLP) analysis of Ceduovirus phages using the EcoRV enzyme. DNA samples were digested o/n at 37 °C with EcoRV. To dissociate cos ends prior to electrophoresis 50% formamide was added. M: 1-kb DNA Ladder (Fermentas). Unique phage DNA restriction patterns are as follows: 1, L81; 2, 14; 3, 27; 4, 94p4; 5, p6/4; 6, 12; 7, A3; 8, Am4; 9, D4; 10, E1. (C) PCR-based typing of Lactococcus phages analyzed in this study. Unique fragments of phage genomes were amplified using primers complementary to the conserved genomic region of Ceduovirus phages (mcp gene). The arrow indicates the position of a 474-bp band specific to the Ceduovirus group. M: 1-kb DNA Ladder (Fermentas), lanes 1–10: DNA fragments generated from respective phage lysates which served as templates: 14, 27, 94p4, L81, p6/4, 12, E1, D4, Am4, and A3, lane 11: DNA fragment generated from c2 (Ceduovirus phage; positive control), lane 12: DNA fragment generated from sk1 (Skunavirus phage; negative control).

Figure 2.
Visualization of the Lactococcus Ceduovirus phages using electron microscopy. Head diameter and tail size ± SD given under each electron micrograph were calculated using measure IT 5.0 (Olympus Soft Imaging Solutions GmbH) software as a mean from n = 5–10 phages.
Figure 2.
Visualization of the Lactococcus Ceduovirus phages using electron microscopy. Head diameter and tail size ± SD given under each electron micrograph were calculated using measure IT 5.0 (Olympus Soft Imaging Solutions GmbH) software as a mean from n = 5–10 phages.

Figure 3.
Infectivity of Ceduovirus phages towards a set of industrial and environmental L. lactis strains. Sensitive strains are marked in gray, resistant strains in white. (A) Sensitivity of L. lactis cremoris vs. lactis subspecies to infection by the analyzed phages. (B) Sensitivity of L. lactis strains with respect to their cell wall polysaccharide (CWPS) type. Clear lytic zones obtained on double-layer plates containing o/n cultures of a specific L. lactis strain (200 µL) and phage lysates (100 µL) were indicative of bacterial sensitivity. (C) Number of L. lactis strains (from 54 tested) sensitive to infection by individual phages.
Figure 3.
Infectivity of Ceduovirus phages towards a set of industrial and environmental L. lactis strains. Sensitive strains are marked in gray, resistant strains in white. (A) Sensitivity of L. lactis cremoris vs. lactis subspecies to infection by the analyzed phages. (B) Sensitivity of L. lactis strains with respect to their cell wall polysaccharide (CWPS) type. Clear lytic zones obtained on double-layer plates containing o/n cultures of a specific L. lactis strain (200 µL) and phage lysates (100 µL) were indicative of bacterial sensitivity. (C) Number of L. lactis strains (from 54 tested) sensitive to infection by individual phages.

Figure 4.
Schematic representation of the cos region of the sequenced Ceduovirus phages. A small black rectangle marks the position of the cos site, and its 9-nt sequence is given above (differences in nucleotides are marked in red); black squares (R) mark putative terminase binding sites; arrows depict palindromic sequences (P), direct (D), and indirect (I) repeats.
Figure 4.
Schematic representation of the cos region of the sequenced Ceduovirus phages. A small black rectangle marks the position of the cos site, and its 9-nt sequence is given above (differences in nucleotides are marked in red); black squares (R) mark putative terminase binding sites; arrows depict palindromic sequences (P), direct (D), and indirect (I) repeats.

Figure 5.
Synteny analysis of complete genomes of Lactococcus Ceduovirus phages sequenced in this study. The two transcriptional regions are oriented divergently to each other and are marked above the scheme. Arrows represent open reading frames (orfs); gray arrows indicate orfs of unknown function, yellow indicates unique orfs with unknown function, and the remaining colours indicate orfs with annotated functions. The genomes were aligned with respect to the position of the cos sites. Each genome is compared with the succeeding genome. The genomes of phages c2 and bIL67 are shown for reference. The gray shading between genomes indicates regions of similarity based on tblastx comparison. Numbers above individual orfs are given as reference to the genes discussed in the text.
Figure 5.
Synteny analysis of complete genomes of Lactococcus Ceduovirus phages sequenced in this study. The two transcriptional regions are oriented divergently to each other and are marked above the scheme. Arrows represent open reading frames (orfs); gray arrows indicate orfs of unknown function, yellow indicates unique orfs with unknown function, and the remaining colours indicate orfs with annotated functions. The genomes were aligned with respect to the position of the cos sites. Each genome is compared with the succeeding genome. The genomes of phages c2 and bIL67 are shown for reference. The gray shading between genomes indicates regions of similarity based on tblastx comparison. Numbers above individual orfs are given as reference to the genes discussed in the text.

Figure 6.
Cladogram of the Ceduovirus phages based on the reconciled trees for the following proteins: holin, lysin, ERF-like recombinase, DNApol, tail fiber (GP2 and GP4), major capsid (GP14).
Figure 6.
Cladogram of the Ceduovirus phages based on the reconciled trees for the following proteins: holin, lysin, ERF-like recombinase, DNApol, tail fiber (GP2 and GP4), major capsid (GP14).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Bacterial strains, plasmids, and bacteriophages used in study.
| Bacterial Strains | Relevant Features | Source/Reference |
| IL1403 | L. lactis subsp. lactis wild-type strain | [26] |
| MG1363 | L. lactis subsp. cremoris wild-type strain | [27] |
| NZ9000 | L. lactis subsp. cremoris MG1363 derivative pepN::nisRnisK strain | MoBiTec® |
| ER2655 | E. coli strain F- λ- fhuA2 [lon] ompT lacZ::T7p07 gal sulA11 Δ(mcrC-mrr)114::IS10 R(mcr-73::miniTn10-TetS)2 R(zgb-210::Tn10)(TetS) endA1 [dcm] | NEB® |
| IBB338, 339, 342, 695-700, 704, 732-739, 741-758, 760-764, 1219, 1267, 1280, 1283, 1298, 1784, 1787-1792, 1796 | L. lactis strains | IBB PAS collection |
| Plasmids | Relevant Features | Source/Reference |
| pG+host3 | E. coli vector, Ts derivative of pGK12, catR | [28] |
| pUN121 | E. coli vector, tetR, ampR non-replicative in L. lactis cells | [29] |
| pUN121:catR:ori27 | recombinant plasmid with putative ori from L. lactis phage 27, catR | this study |
| pUN121:catR:ori94p4 | recombinant plasmid with putative ori from L. lactis phage 94p4, catR | this study |
| pUN121:catR:orip6/4 | recombinant plasmid with putative ori from L. lactis phage p6/4, catR | this study |
| pUN121:catR:oric2 | recombinant plasmid with putative ori from L. lactis phage c2, catR | this study |
| Bacteriophages | GenBank Accession Number* | Source/Reference |
| bIL67 | NC_001629.1 | [7] |
| c2 | NC_001706.1 | [8] |
| bIBB_L81 (L81) | MH779526 * | this study |
| bIBB_12 (12) | MH779518 * | “ |
| bIBB_14 (14) | MH779519 * | “ |
| bIBB_94p4 (94p4) | MH779521 * | “ |
| bIBB_27 (27) | MH779520 * | “ |
| bIBB_p6/4 (p6/4) | MH779527 * | “ |
| bIBB_A3 (A3) | MH779522 * | “ |
| bIBB_D4 (D4) | MH779524 * | “ |
| bIBB_Am4 (Am4) | MH779523 * | “ |
| bIBB_E1 (E1) | MH779525 * | “ |
* Complete whole-genome sequences of phages analyzed in this study were deposited in GenBank.
Table 2.
Primers used in study.
| Name | 5′-3′ Sequence * |
|---|---|
| c-2for | CAGGTGTAAAAGTTCGAGAACT |
| c-2rev | CAGATAATGCACCTGAATCA |
| ori27_F | CCGGAATTCCAAGCTATCAAATATTTC |
| ori27_R | CCCAAGCTTCATACCAACAAAAG |
| orip6/4_F | CCCGATATCCAATGTGTTTTTGTG |
| orip6/4_R | CCCAAGCTTCCTACAAAAAATTTAG |
| ori94p4_F | CCGGAATTCTARTTACYTTGCTAAAGGG |
| ori94p4_R | CCCAAGCTTTCCCTCWTTGATTATG |
| oric2_F | CCGGAATTCTARTTACYTTGCTAAAGGG |
| oric2_R | CCCAAGCTTTCCCTCWTTGATTATG |
| catR_F | CGCGGATCCATGAAGAAAGCAGACAAGTAAG |
| catR_R | ACGCGTCGACGTAAAAAGTACAGTC |
| pUN121_F | GTCTGGCTATGCAGAAATCC |
| pUN121_R | GCTTATAACGCCGCATTGCT |
| pUN121_catR_F | TATCGACTACGCGATCATGG |
| pUN121_catR_R | GCGTGCAAGATTCCGAATAC |
| CWPS_Afor | GTGCCTATGCTCCGTTAGTC |
| CWPS_Arev | CGAGGGCCAATCTCTTTACC |
| CWPS_Bfor | GATTCAGTTGCACGGCCG |
| CWPS_Brev | AGTAAGGGGGCGGATTGTG |
| CWPS_Cfor | AAAGCTCATCTTTCCCCTGTTGT |
| CWPS_Crev | GCACCATAGTCTGGAATAAGACC |
| CTL_for (U-type) | GTACACTATGTTTATAACAATCATCCAG |
| CTL_rev (U-type) | GCAAACCAGATTCAAAGTCAGTATG |
* underlined are restriction sites added to the primer sequence.
Table 3.
Phage infection parameters calculated from one-step growth curves.
| Phage | Burst Size * [Plaques Per Infected Cell] | Latent Period * [min] | Burst Time * [min] | Plaque Diameter [mm] | Halo [mm] |
|---|---|---|---|---|---|
| 14 | 66 ± 3 | 20 ± 0 | 32 ± 1 | 2.5 | 0.5 |
| 27 | 67 ± 2 | 20 ± 0 | 29 ± 3 | 2 | 0.5 |
| 94p4 | 64 ± 3 | 28 ± 0 | 31 ± 0 | 2 | nd |
| L81 | 32 ± 3 | 20 ± 0 | 27 ± 2 | 2 | 1 |
| p6/4 | 34 ± 3 | 22 ± 3 | 29 ± 1 | 1.5 | nd |
| 12 | 197 ± 17 | 22 ± 3 | 29 ± 2 | 3 | nd |
| E1 | 109 ± 8 | 26 ± 2 | 33 ± 0 | 5.3 | nd |
| D4 | 94 ± 4 | 19 ± 2 | 28 ± 3 | 5 | 1 |
| Am4 | 33 ± 3 | 20 ± 0 | 29 ± 2 | 3.5 | 0.5 |
| A3 | 91 ± 3 | 23 ± 3 | 33 ± 0 | 4 | 0.5 |
* Results are a means of at least three experiments ± SD; nd—none determined.
Table 4.
Characteristics of phage genomes based on sequencing data.
| Phage (GenBank Accession No.) | Propagation Strain (Phage Titer; PFU mL−1) | Genome Length (bp) | GC Content (%) | Orf Count |
|---|---|---|---|---|
| 14 (MH779519) | 738 (2.4 × 1010) | 21,710 | 36 | 37 |
| 27 (MH779520) | 738 (8 × 108) | 21,713 | 36.1 | 37 |
| 94p4 (MH779521) | 753 (1.1 × 1010) | 21,834 | 36.1 | 38 |
| L81 (MH779526) | MG1363 (8.4 × 109) | 21,915 | 36.0 | 38 |
| p6/4 (MH779527) | 739 (1.3 × 109) | 22,220 | 36.0 | 39 |
| 12 (MH779418) | 1787 (6.1 × 109) | 21,458 | 36.2 | 36 |
| E1 (MH779525) | 1283 (1.3 × 1010) | 21,749 | 36.1 | 37 |
| D4 (MH779524) | 1280 (1.4 × 1010) | 21,907 | 36.0 | 38 |
| Am4 (MH779523) | 1219 (2.4 × 109) | 22,703 | 35.9 | 38 |
| A3 (MH779522) | 1267 (7.2 × 109) | 21,906 | 36.2 | 39 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chmielewska-Jeznach, M.; Bardowski, J.K.; Szczepankowska, A.K. Lactococcus Ceduovirus Phages Isolated from Industrial Dairy Plants—From Physiological to Genomic Analyses. Viruses 2020, 12, 280. https://doi.org/10.3390/v12030280
AMA Style
Chmielewska-Jeznach M, Bardowski JK, Szczepankowska AK. Lactococcus Ceduovirus Phages Isolated from Industrial Dairy Plants—From Physiological to Genomic Analyses. Viruses. 2020; 12(3):280. https://doi.org/10.3390/v12030280
Chicago/Turabian StyleChmielewska-Jeznach, Magdalena, Jacek K. Bardowski, and Agnieszka K. Szczepankowska. 2020. "Lactococcus Ceduovirus Phages Isolated from Industrial Dairy Plants—From Physiological to Genomic Analyses" Viruses 12, no. 3: 280. https://doi.org/10.3390/v12030280
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.