
Phylogenetic Analysis of the 2020 West Nile Virus (WNV) Outbreak in Andalusia (Spain)

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples, Molecular Diagnosis, and Culture Isolation
2.2. Viral Sequencing
2.3. Viral Genomic Data Processing
2.4. Phylogenetic Analysis
3. Results
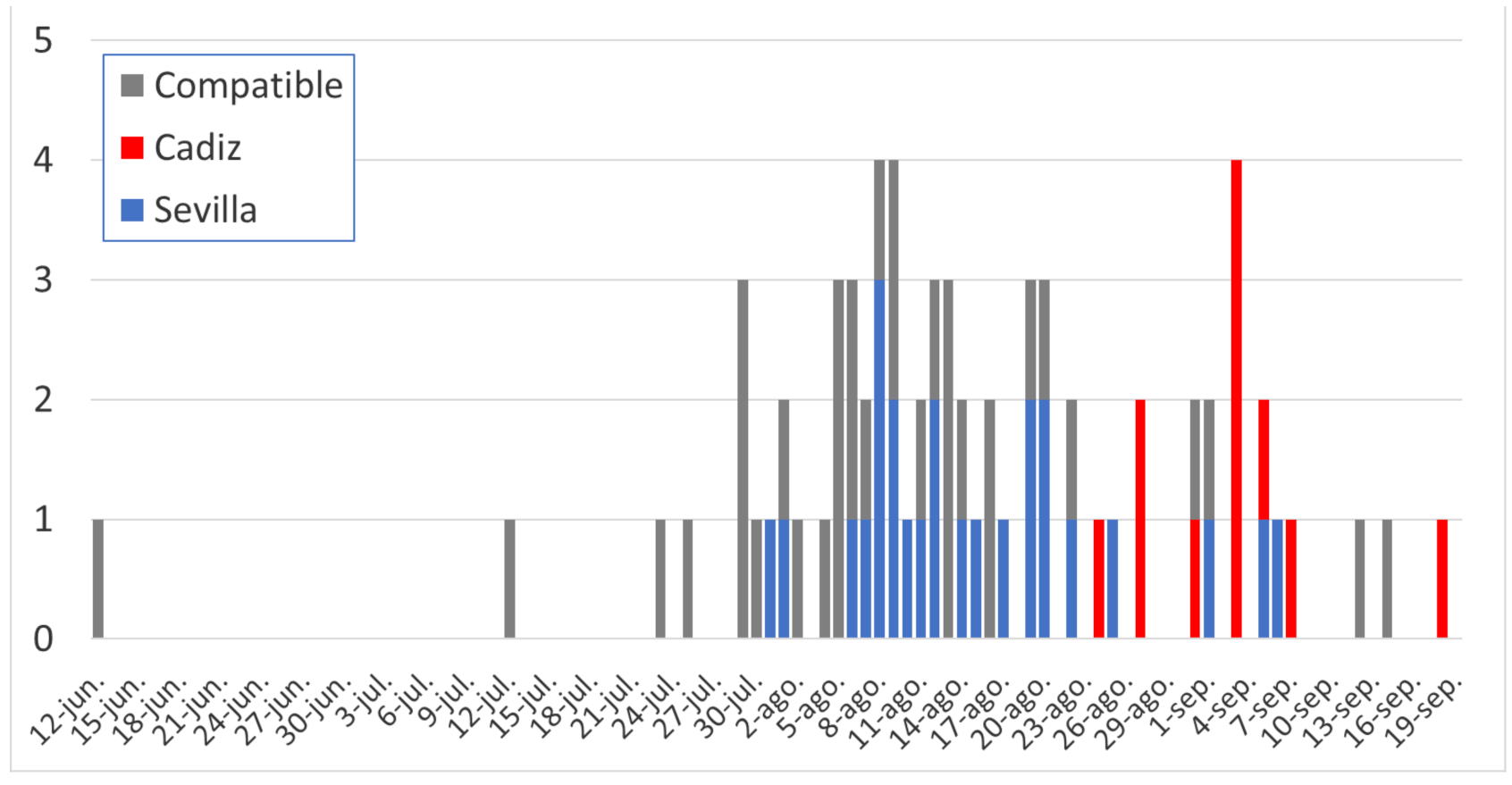
3.1. Sample Sequencing and Sequencing Data Collection
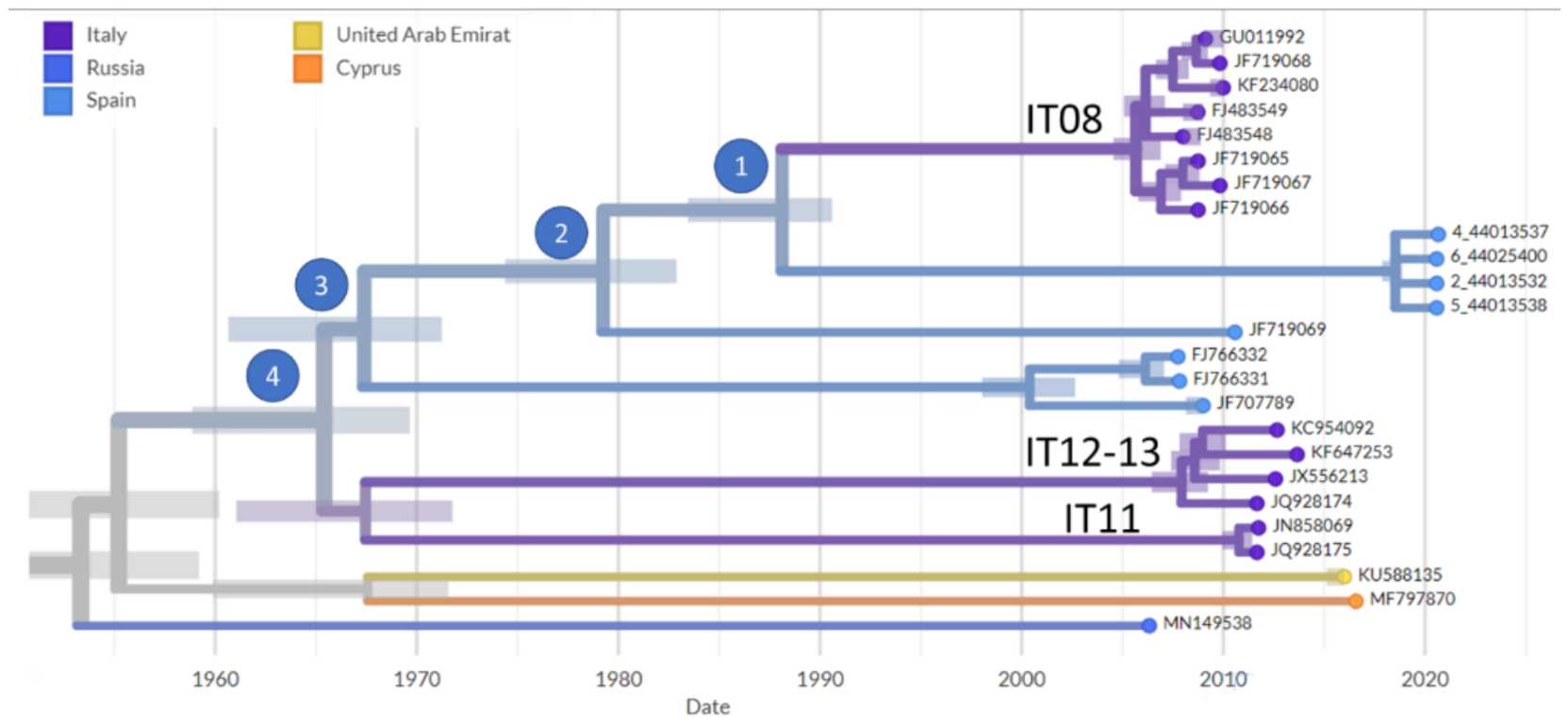
3.2. Phylogenetic Analysis
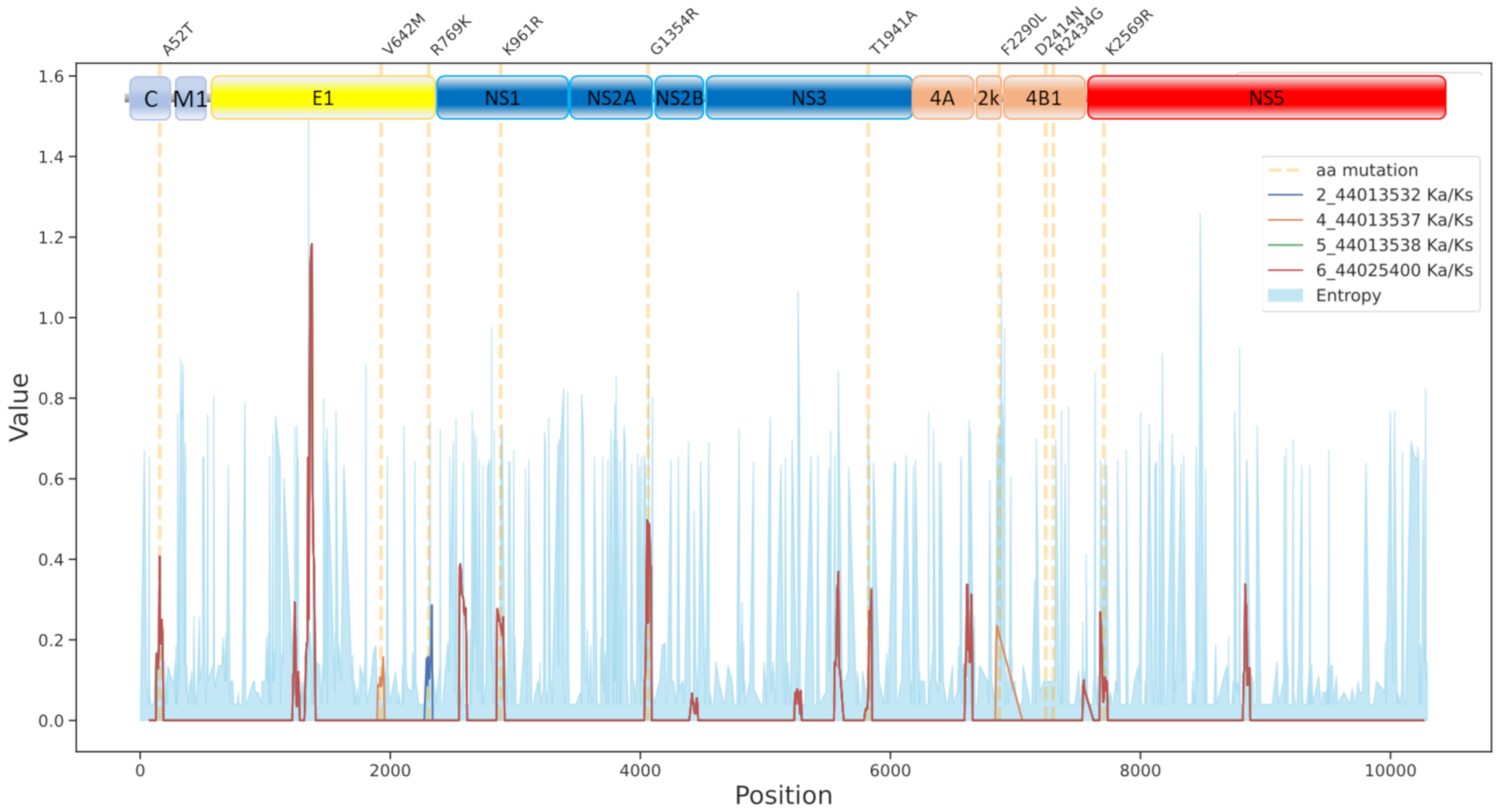
3.3. Mutational Patterns in the Spanish Outbreak
3.4. The SIEGA Nextstrain Server
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mackenzie, J.S.; Gubler, D.J.; Petersen, L.R. Emerging flaviviruses: The spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 2004, 10, S98–S109. [Google Scholar] [CrossRef] [PubMed]
- Kramer, L.D.; Styer, L.M.; Ebel, G.D. A global perspective on the epidemiology of West Nile virus. Annu. Rev. Entomol. 2008, 53, 61–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubálek, Z.; Halouzka, J. West Nile fever—A reemerging mosquito-borne viral disease in Europe. Emerg. Infect. Dis. 1999, 5, 643. [Google Scholar] [CrossRef]
- Fall, G.; Di Paola, N.; Faye, M.; Dia, M.; de Melo Freire, C.C.; Loucoubar, C.; de Andrade Zanotto, P.M.; Faye, O. Biological and phylogenetic characteristics of West African lineages of West Nile virus. PLoS Negl. Trop. Dis. 2017, 11, e0006078. [Google Scholar] [CrossRef]
- Pérez-Ramírez, E.; Llorente, F.; Del Amo, J.; Fall, G.; Lubisi, A.; Lecollinet, S.; Vázquez, A.; Jiménez-Clavero, M.Á. Pathogenicity evaluation of twelve West Nile virus strains belonging to four lineages from five continents in a mouse model: Discrimination between three pathogenicity categories. J. Gen. Virol. 2017, 98, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Bardos, V.; Adamcová, J.; Dedei, S.; Gjini, N.; Rosický, B.; Simkova, A. Neutralizing antibodies against some neurotropic viruses determined in human sera in Albania. J. Hyg. Epidemiol. Microbiol. Immunol. 1959, 3, 277–282. [Google Scholar]
- Joubert, L.; Oudar, J.; Hannoun, C.; Beytout, D.; Corniou, B.; Guillon, J.C.; Panthier, R. Epidemiology of the West Nile virus: Study of a focus in Camargue. IV. Meningo-encephalomyelitis of the horse. Ann. Inst Pasteur 1970, 118, 239–247. [Google Scholar]
- Zeller, H.; Schuffenecker, I. West Nile virus: An overview of its spread in Europe and the Mediterranean basin in contrast to its spread in the Americas. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Bakonyi, T.; Ivanics, É.; Erdélyi, K.; Ursu, K.; Ferenczi, E.; Weissenböck, H.; Nowotny, N. Lineage 1 and 2 strains of encephalitic West Nile virus, central Europe. Emerg. Infect. Dis. 2006, 12, 618. [Google Scholar] [CrossRef]
- Platonov, A.E.; Karan, L.S.; Shopenskaia, T.A.; Fedorova, M.V.; Koliasnikova, N.M.; Rusakova, N.M.; Shishkina, L.V.; Arshba, T.E.; Zhuravlev, V.I.; Govorukhina, M.V.; et al. Genotyping of West Nile fever virus strains circulating in southern Russia as an epidemiological investigation method: Principles and results. Zhurnal Mikrobiol. Epidemiol. Immunobiol. 2011, 2, 29–37. [Google Scholar]
- Rizzo, C.; Napoli, C.; Venturi, G.; Pupella, S.; Lombardini, L.; Calistri, P.; Monaco, F.; Cagarelli, R.; Angelini, P.; Bellini, R. West Nile virus transmission: Results from the integrated surveillance system in Italy, 2008 to 2015. Eurosurveillance 2016, 21, 30340. [Google Scholar] [CrossRef] [Green Version]
- Barzon, L.; Pacenti, M.; Franchin, E.; Lavezzo, E.; Masi, G.; Squarzon, L.; Pagni, S.; Toppo, S.; Russo, F.; Cattai, M.; et al. Whole genome sequencing and phylogenetic analysis of West Nile virus lineage 1 and lineage 2 from human cases of infection, Italy, August 2013. Eurosurveillance 2013, 18, 20591. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.; Tryfonos, C.; Tourvas, A.; Floridou, D.; Paphitou, N.I.; Christodoulou, C. Complete Genome sequence of West Nile virus (WNV) from the first human case of neuroinvasive WNV infection in Cyprus. Genome Announc. 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Sotelo, E.; Fernandez-Pinero, J.; Llorente, F.; Vazquez, A.; Moreno, A.; Agüero, M.; Cordioli, P.; Tenorio, A.; Jimenez-Clavero, M.A. Phylogenetic relationships of Western Mediterranean West Nile virus strains (1996–2010) using full-length genome sequences: Single or multiple introductions? J. Gen. Virol. 2011, 92, 2512–2522. [Google Scholar] [CrossRef]
- López-Ruiz, N.; del Carmen Montaño-Remacha, M.; Durán-Pla, E.; Pérez-Ruiz, M.; Navarro-Marí, J.M.; Salamanca-Rivera, C.; Miranda, B.; Oyonarte-Gómez, S.; Ruiz-Fernández, J. West Nile virus outbreak in humans and epidemiological surveillance, west Andalusia, Spain, 2016. Eurosurveillance 2018, 23, 17-00261. [Google Scholar] [CrossRef] [Green Version]
- Busquets, N.; Laranjo-González, M.; Soler, M.; Nicolás, O.; Rivas, R.; Talavera, S.; Villalba, R.; San Miguel, E.; Torner, N.; Aranda, C. Detection of West Nile virus lineage 2 in north-eastern Spain (Catalonia). Transbound. Emerg. Dis. 2019, 66, 617–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Centre for Disease Prevention and Control. Epidemiological Update: West Nile Virus Transmission Season in Europe. Available online: https://www.ecdc.europa.eu/en/news-events/epidemiological-update-west-nile-virus-transmission-season-europe-2019 (accessed on 20 April 2021).
- Meningoencefalitis por Virus del Nilo Occidental en España (2a Actualización). Evaluación Rápida de Riesgo. Available online: https://www.mscbs.gob.es/profesionales/saludPublica/ccayes/alertasActual/docs/20201203_ERR_Nilo_Occidental.pdf (accessed on 20 April 2021).
- Protocolo de Vigilancia y Alerta de Fiebre del Nilo Occidental. Protocol for West Nile Fever Surveillance and Alert. Available online: https://juntadeandalucia.es/export/drupaljda/salud_5af95879cc803_fiebre_nilo250817.pdf (accessed on 20 April 2021).
- Martínez Yoldi, M.J.; Ruiz, M.; Sánchez-Seco Fariñas, M.P.; Vázquez González, A. Diagnóstico microbiológico de las principales arbovirosis importadas y autóctonas. In Procedimientos en Microbiología Clínica; Mansilla Cercenado, E., Moreno Cantón, R., Eds.; Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC): Madrid, Spain, 2020. [Google Scholar]
- Antón, A.I.N.; de Ory Manchón, F.; Fariñas, M.P.S.-S.; Narváez, L.F.; Cámara, M.I.G.; Mari, J.M.N.; Matanzo, A.T. Diagnóstico microbiológico de arbovirosis y robovirosis emergentes. Enfermedades Infecciosas y Microbiologia Clinica 2015, 33, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, A.; Herrero, L.; Negredo, A.; Hernández, L.; Sánchez-Seco, M.P.; Tenorio, A. Real time PCR assay for detection of all known lineages of West Nile virus. J. Virol. Methods 2016, 236, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P. The Arboviruses: Epidemiology and Ecology; CRC Press: Boca Raton, FL, USA, 2020; Volume 1. [Google Scholar]
- Barzon, L.; Pacenti, M.; Franchin, E.; Squarzon, L.; Sinigaglia, A.; Ulbert, S.; Cusinato, R.; Palù, G. Isolation of West Nile virus from urine samples of patients with acute infection. J. Clin. Microbiol. 2014, 52, 3411–3413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepp, C.M.; Cocking, J.H.; Valentine, M.; Young, S.J.; Damian, D.; Samuels-Crow, K.E.; Sheridan, K.; Fofanov, V.Y.; Furstenau, T.N.; Busch, J.D. Phylogenetic analysis of West Nile Virus in Maricopa County, Arizona: Evidence for dynamic behavior of strains in two major lineages in the American Southwest. PLoS ONE 2018, 13, e0205801. [Google Scholar] [CrossRef] [Green Version]
- Nf-Core/Viralrecon: Nf-Core/Viralrecon v1.1.—Steel Pangolin (Version 1.1.0). Available online: https://zenodo.org/record/3905178#.YBumlOhKi71 (accessed on 20 April 2021).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Picard. A Set of Command Line Tools (in Java) for Manipulating High-Throughput Sequencing (HTS) Data and Formats Such as SAM/BAM/CRAM and VCF. Available online: http://broadinstitute.github.io/picard/ (accessed on 20 April 2021).
- Huddleston, J.; Hadfield, J.; Sibley, T.R.; Lee, J.; Fay, K.; Ilcisin, M.; Harkins, E.; Bedford, T.; Neher, R.A.; Hodcroft, E.B. Augur: A bioinformatics toolkit for phylogenetic analyses of human pathogens. J. Open Source Softw. 2021, 6, 2906. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- To, T.-H.; Jung, M.; Lycett, S.; Gascuel, O. Fast dating using least-squares criteria and algorithms. Syst. Biol. 2016, 65, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Kames, J.; Holcomb, D.D.; Kimchi, O.; DiCuccio, M.; Hamasaki-Katagiri, N.; Wang, T.; Komar, A.A.; Alexaki, A.; Kimchi-Sarfaty, C. Sequence analysis of SARS-CoV-2 genome reveals features important for vaccine design. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- SIEGA. West Nile Virus Epidemiological Local Server—SIEGA. Available online: http://nextstrain.clinbioinfosspa.es/wnv (accessed on 20 April 2021).
- Barzon, L.; Pacenti, M.; Franchin, E.; Squarzon, L.; Lavezzo, E.; Toppo, S.; Martello, T.; Cattai, M.; Cusinato, R.; Palù, G. Novel West Nile virus lineage 1a full genome sequences from human cases of infection in north-eastern Italy, 2011. Clin. Microbiol. Infect. 2012, 18, E541–E544. [Google Scholar] [CrossRef] [Green Version]
- Barzon, L.; Squarzon, L.; Cattai, M.; Franchin, E.; Pagni, S.; Cusinato, R.; Palu, G. West Nile virus infection in Veneto region, Italy, 2008–2009. EuroSurveillance 2009, 14. [Google Scholar] [CrossRef] [Green Version]
- Barzon, L.; Pacenti, M.; Franchin, E.; Pagni, S.; Lavezzo, E.; Squarzon, L.; Martello, T.; Russo, F.; Nicoletti, L.; Rezza, G. Large human outbreak of West Nile virus infection in north-eastern Italy in 2012. Viruses 2013, 5, 2825–2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Ng, M. Infectious entry of West Nile virus occurs through a clathrin-mediated endocytic pathway. J. Virol. 2004, 78, 10543–10555. [Google Scholar] [CrossRef] [Green Version]
- Shiryaev, S.A.; Kozlov, I.A.; Ratnikov, B.I.; Smith, J.W.; Lebl, M.; Strongin, A.Y. Cleavage preference distinguishes the two-component NS2B-NS3 serine proteinases of Dengue and West Nile viruses. Biochem. J. 2007, 401, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.; Bhattacharya, T.; Foley, B. Tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Surveillance Program for the West Nile Virus 2021. Programa de Vigilancia de Fiebre del Nilo Occidental 2021. Available online: https://www.mapa.gob.es/es/ganaderia/temas/sanidad-animal-higiene-ganadera/programafiebredelnilooccidental2021_tcm30-437515.pdf (accessed on 20 April 2021).
- Bhuvanakantham, R.; Ng, M.L. West Nile virus and dengue virus capsid protein negates the antiviral activity of human Sec3 protein through the proteasome pathway. Cell Microbiol. 2013, 15, 1688–1706. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. Methods Mol. Biol. 2016, 1374, 23–54. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.; Cerutti, L.; de Castro, E.; Langendijk-Genevaux, P.S.; Bulliard, V.; Bairoch, A.; Hulo, N. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 2010, 38, D161–D166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuerdo de 9 de Marzo de 2021, del Consejo de Gobierno, por el que se Toma Conocimiento del Programa de Vigilancia y Control Integral de Vectores de la Fiebre del Nilo Occidental. Available online: https://www.juntadeandalucia.es/boja/2021/48/4 (accessed on 20 April 2021).
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, 19, 9. [Google Scholar] [CrossRef]
- Integrated System for Genomic Epidemiology of Andalusia. Sistema Integrado de Epidemiologia Genomica de Andalucia—SIEGA. Available online: http://clinbioinfosspa.es/projects/siega/ (accessed on 20 April 2021).




{kind=link}
{kind=link}
{kind=link}
| Date | Code | Hospital | Sample | PCR | Culture | SAMPLE_ID | Depth | HCV | Coverage |
|---|---|---|---|---|---|---|---|---|---|
| 13 August 2020 | 44013531 | HUVR | Urine | Positive | No | 1_44013531 | 835x | 351 | 57.00% |
| 13 August 2020 | 44013532 | HUVR | Urine | Positive | No | 2_44013532 | 1190x | 461 | 96.10% |
| 2013 Au-gust 2020 | 44013536 | HUVR | Urine | Positive | No | 3_44013536 | 465x | 88 | 19.10% |
| 2013 Au-gust 2020 | 44013537 | HUVR | Urine | Positive | No | 4_44013537 | 1043x | 420 | 79.70% |
| 2013 Au-gust 2020 | 44013538 | HUVR | Urine | Positive | Negative | 5_44013538 | 1205x | 453 | 94.50% |
| 9 September 2020 | 44025400 | HPR | CSF | Positive | Negative | - | |||
| 9 September 2020 | 44025400 | HPR | Urine | Positive | Positive | 6_44025400 | 1145x | 451 | 94.70% |
| 2_44013532 | 4_44013537 | 5_44013538 | 6_44025400 | FJ766331 | FJ766332 | JF707789 | JF719069 | KU588135 | MF797870 | MN149538 | IT081 | IT12-132 | IT113 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2_44013532 | 0.0 | 8.0 | 5.0 | 5.0 | 226.0 | 224.0 | 240.0 | 182.0 | 330.0 | 293.0 | 252.0 | 136.0 | 238.5 | 248.0 |
| 4_44013537 | 8.0 | 0.0 | 8.0 | 7.0 | 209.0 | 207.0 | 222.0 | 165.0 | 302.0 | 265.0 | 234.0 | 127.0 | 219.5 | 224.0 |
| 5_44013538 | 5.0 | 8.0 | 0.0 | 4.0 | 221.0 | 219.0 | 235.0 | 178.0 | 320.0 | 289.0 | 250.0 | 133.0 | 232.5 | 244.0 |
| 6_44025400 | 5.0 | 7.0 | 4.0 | 0.0 | 221.0 | 219.0 | 235.0 | 178.0 | 322.0 | 289.0 | 250.0 | 133.0 | 233.5 | 242.0 |
| FJ766331 | 226.0 | 209.0 | 221.0 | 221.0 | 0.0 | 6.0 | 37.0 | 182.0 | 268.0 | 243.0 | 185.0 | 179.0 | 174.0 | 192.0 |
| FJ766332 | 224.0 | 207.0 | 219.0 | 219.0 | 6.0 | 0.0 | 35.0 | 180.0 | 266.0 | 241.0 | 185.0 | 177.0 | 172.0 | 190.0 |
| JF707789 | 240.0 | 222.0 | 235.0 | 235.0 | 37.0 | 35.0 | 0.0 | 197.0 | 277.0 | 260.0 | 204.0 | 193.5 | 190.0 | 208.5 |
| JF719069 | 182.0 | 165.0 | 178.0 | 178.0 | 182.0 | 180.0 | 197.0 | 0.0 | 298.0 | 250.0 | 219.0 | 137.0 | 197.0 | 202.0 |
| KU588135 | 330.0 | 302.0 | 320.0 | 322.0 | 268.0 | 266.0 | 277.0 | 298.0 | 0.0 | 246.0 | 258.0 | 295.0 | 277.5 | 298.0 |
| MF797870 | 293.0 | 265.0 | 289.0 | 289.0 | 243.0 | 241.0 | 260.0 | 250.0 | 246.0 | 0.0 | 210.0 | 258.5 | 233.5 | 260.0 |
| MN149538 | 252.0 | 234.0 | 250.0 | 250.0 | 185.0 | 185.0 | 204.0 | 219.0 | 258.0 | 210.0 | 0.0 | 216.0 | 197.5 | 220.5 |
| IT08 1 | 136.0 | 127.0 | 133.0 | 133.0 | 179.0 | 177.0 | 193.5 | 137.0 | 295.0 | 258.5 | 216.0 | 10.5 | 198.0 | 206.3 |
| IT12-13 2 | 238.5 | 219.5 | 232.5 | 233.5 | 174.0 | 172.0 | 190.0 | 197.0 | 277.5 | 233.5 | 197.5 | 198.0 | 9.75 | 192.3 |
| IT11 3 | 248.0 | 224.0 | 244.0 | 242.0 | 192.0 | 190.0 | 208.5 | 202.0 | 298.0 | 260.0 | 220.5 | 206.3 | 192.25 | 2.0 |
| Mutation | Protein | Function |
|---|---|---|
| A52T | Capsid protein C | Form the nucleocapsid |
| V642M | Envelope E | Binding host cell surface and mediates membranes fusion |
| R769K | Envelope E | Binding host cell surface and mediates membranes fusion |
| K961R | NS1 | Role in early RNA replication |
| G1354R | NS2A | Component of the viral RNA replication complex |
| T1941A | NS3 | In association with NS2B, performs its autocleavage |
| F2290L | 2k | Signal peptide for NS4B |
| D2414N | NS4B | Induces the formation of ER-derived membrane vesicles |
| R2434G | NS4B | Induces the formation of ER-derived membrane vesicles |
| K2569R | NS5 | RNA-directed RNA polymerase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casimiro-Soriguer, C.S.; Perez-Florido, J.; Fernandez-Rueda, J.L.; Pedrosa-Corral, I.; Guillot-Sulay, V.; Lorusso, N.; Martinez-Gonzalez, L.J.; Navarro-Marí, J.M.; Dopazo, J.; Sanbonmatsu-Gámez, S. Phylogenetic Analysis of the 2020 West Nile Virus (WNV) Outbreak in Andalusia (Spain). Viruses 2021, 13, 836. https://doi.org/10.3390/v13050836
Casimiro-Soriguer CS, Perez-Florido J, Fernandez-Rueda JL, Pedrosa-Corral I, Guillot-Sulay V, Lorusso N, Martinez-Gonzalez LJ, Navarro-Marí JM, Dopazo J, Sanbonmatsu-Gámez S. Phylogenetic Analysis of the 2020 West Nile Virus (WNV) Outbreak in Andalusia (Spain). Viruses. 2021; 13(5):836. https://doi.org/10.3390/v13050836
Chicago/Turabian StyleCasimiro-Soriguer, Carlos S., Javier Perez-Florido, Jose L. Fernandez-Rueda, Irene Pedrosa-Corral, Vicente Guillot-Sulay, Nicola Lorusso, Luis Javier Martinez-Gonzalez, Jose M. Navarro-Marí, Joaquin Dopazo, and Sara Sanbonmatsu-Gámez. 2021. "Phylogenetic Analysis of the 2020 West Nile Virus (WNV) Outbreak in Andalusia (Spain)" Viruses 13, no. 5: 836. https://doi.org/10.3390/v13050836