
Geographical Distribution and Genetic Diversity of Bank Vole Hepaciviruses in Europe

 , , ,
, , ,  , , , , , add
Show full author list
, , , , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Small Mammals and Species Identification
2.2. Nucleic Acid Extraction and RT-PCR Analyses
2.3. BvHV Sequence, Phylogenetic, and Isolation-by-Distance Analyses
2.4. Statistical Analysis
3. Results
3.1. Detection of BvHV RNA in Small Mammals from European Countries
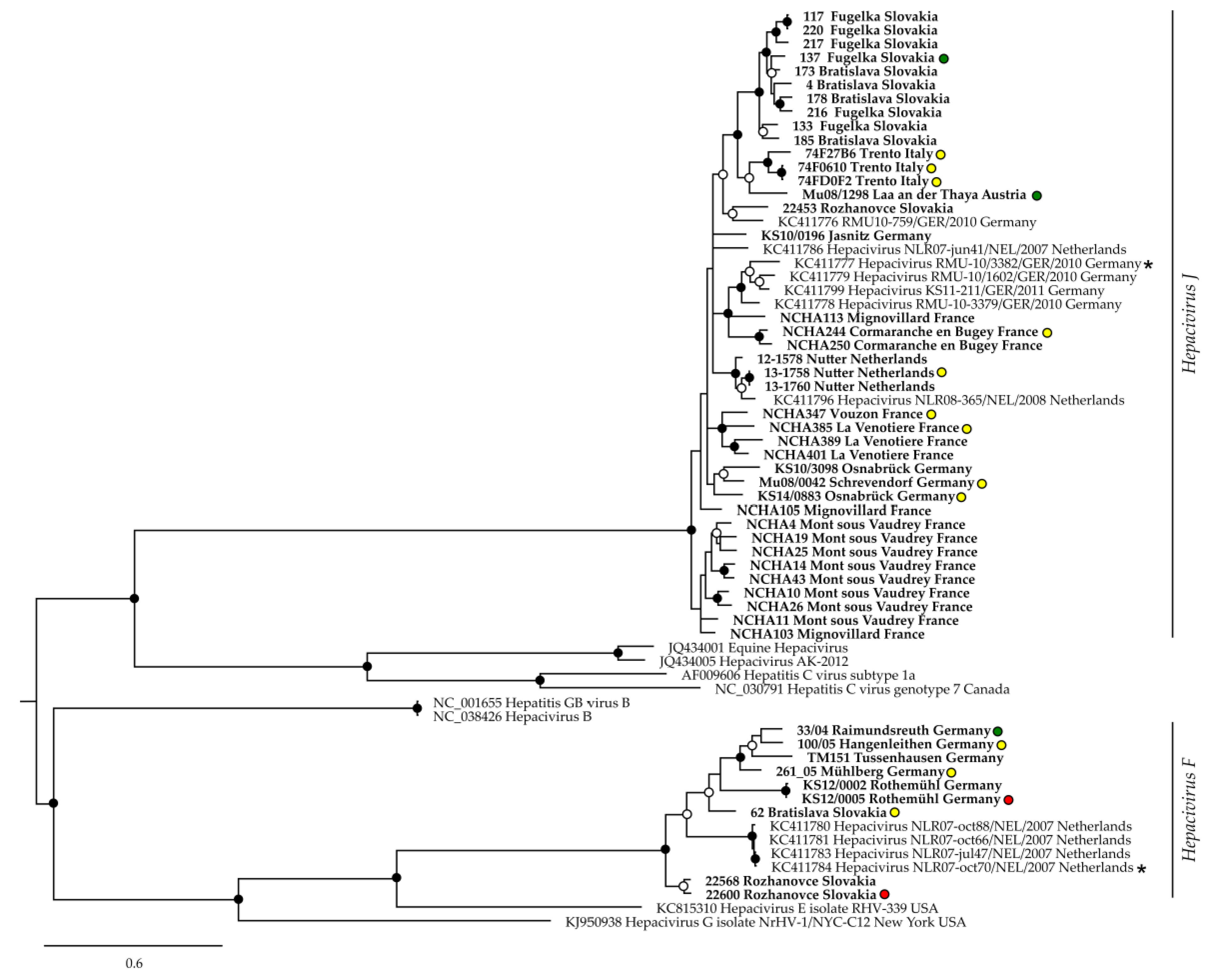
3.2. Sequence Divergence of BvHV
3.3. BvHV Association with Bank Vole Evolutionary Lineages
3.4. BvHV–Host Dynamics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Ethical Permits
Acknowledgments
Conflicts of Interest
References
- ICTV. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/w/flaviviridae/362/genus-hepacivirus (accessed on 1 December 2020).
- Kim, C.W.; Chang, K.-M. Hepatitis C virus: Virology and life cycle. Clin. Mol. Hepatol. 2013, 19, 17–25. [Google Scholar] [CrossRef]
- Kaito, M.; Watanabe, S.; Tsukiyama-Kohara, K.; Yamaguchi, K.; Kobayashi, Y.; Konishi, M.; Yokoi, M.; Ishida, S.; Suzuki, S.; Kohara, M. Hepatitis C virus particle detected by immunoelectron microscopic study. J. Gen. Virol. 1994, 75 Pt 7, 1755–1760. [Google Scholar] [CrossRef]
- Shimizu, Y.K.; Feinstone, S.M.; Kohara, M.; Purcell, R.H.; Yoshikura, H. Hepatitis C virus: Detection of intracellular virus particles by electron microscopy. Hepatology 1996, 23, 205–209. [Google Scholar] [CrossRef]
- Penin, F. Structural biology of hepatitis C virus. Clin. Liver Dis. 2003, 7, 1–21. [Google Scholar] [CrossRef]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef]
- Bukh, J. Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology 2012, 142, 1279–1287.e3. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Lemon, S.M. Model systems for hepatitis C research: The cup half empty? Gastroenterology 2011, 141, 806–809. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Lukashev, A.; Gmyl, A.; Coutard, B.; Adam, A.; Ritz, D.; Leijten, L.M.; Van Riel, D.; et al. Evidence for novel hepaciviruses in rodents. PLoS Pathog. 2013, 9, e1003438. [Google Scholar] [CrossRef] [Green Version]
- Firth, C.; Bhat, M.; Firth, M.A.; Williams, S.H.; Frye, M.J.; Simmonds, P.; Conte, J.M.; Ng, J.; Garcia, J.; Bhuva, N.P.; et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. mBio 2014, 5, e01933-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsay, J.D.; Evanoff, R.; Mealey, R.H. Hepacivirus A Infection in Horses Defines Distinct Envelope Hypervariable Regions and Elucidates Potential Roles of Viral Strain and Adaptive Immune Status in Determining Envelope Diversity and Infection Outcome. J. Virol. 2018, 92, e00314-18. [Google Scholar] [CrossRef] [Green Version]
- Baechlein, C.; Fischer, N.; Grundhoff, A.; Alawi, M.; Indenbirken, D.; Postel, A.; Baron, A.L.; Offinger, J.; Becker, K.; Beineke, A.; et al. Identification of a Novel Hepacivirus in Domestic Cattle from Germany. J. Virol. 2015, 89, 7007–7015. [Google Scholar] [CrossRef] [Green Version]
- El-Attar, L.; Mitchell, J.A.; Brownlie, H.B.; Priestnall, S.L.; Brownlie, J. Detection of non-primate hepaciviruses in UK dogs. Virology 2015, 484, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, P.-L.; Firth, C.; Conte, J.M.; Williams, S.H.; Zambrana-Torrelio, C.M.; Anthony, S.J.; Ellison, J.A.; Gilbert, A.T.; Kuzmin, I.V.; Niezgoda, M.; et al. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc. Natl. Acad. Sci. USA 2013, 110, 8194–8199. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Soto, A.; Arroyo-Murillo, F.; Sander, A.-L.; Rasche, A.; Corman, V.; Tegtmeyer, B.; Steinmann, E.; Corrales-Aguilar, E.; Wieseke, N.; Avey-Arroyo, J.; et al. Cross-order host switches of hepatitis C-related viruses illustrated by a novel hepacivirus from sloths. Virus Evol. 2020, 6, veaa033. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.E.; Reeder, D.M. Mammal Species of The World: A Taxonomic and Geographic Reference, 3rd ed.; The Johns Hopkins University Press: Baltimore, MD, USA, 2005. [Google Scholar]
- Burgin, C.J.; Wilson, D.E.; Mittermeier, R.A.; Rylands, A.B.; Lacher, T.E.; Sechrest, W. Illustrated Checklist of the Mammals of the World; Lynx Edicions: Barcelona, Spain, 2020. [Google Scholar]
- Voutilainen, L.; Kallio, E.R.; Niemimaa, J.; Vapalahti, O.; Henttonen, H. Temporal dynamics of Puumala hantavirus infection in cyclic populations of bank voles. Sci. Rep. 2016, 6, 21323. [Google Scholar] [CrossRef] [Green Version]
- Krebs, C.J.; Myers, J.H. Population cycles in small mammals. Adv. Ecol. Res. 1974, 8, 267–399. [Google Scholar]
- Ledevin, R.; Michaux, J.R.; Deffontaine, V.; Henttonen, H.; Renaud, S. Evolutionary history of the bank vole Myodes glareolus: A morphometric perspective. Biol. J. Linn. Soc. 2010, 100, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Wójcik, J.M.; Kawałko, S.; Marková, S.; Searle, J.B.; Kotlík, P. Phylogeographic signatures of northward post-glacial colonization from high-latitude refugia: A case study of bank voles using museum specimens. J. Zool. 2010, 281, 249–262. [Google Scholar] [CrossRef]
- Marková, S.; Horníková, M.; Lanier, H.C.; Henttonen, H.; Searle, J.B.; Weider, L.J.; Kotlík, P. High genomic diversity in the bank vole at the northern apex of a range expansion: The role of multiple colonizations and end-glacial refugia. Mol. Ecol. 2020, 29, 1730–1744. [Google Scholar] [CrossRef]
- Drewes, S.; Ali, H.S.; Saxenhofer, M.; Rosenfeld, U.M.; Binder, F.; Cuypers, F.; Schlegel, M.; Röhrs, S.; Heckel, G.; Ulrich, R.G. Host-Associated Absence of Human Puumala Virus Infections in Northern and Eastern Germany. Emerg. Infect. Dis. 2017, 23, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Röhrs, S.; Begeman, L.; Straub, B.K.; Boadella, M.; Hanke, D.; Drewes, S.; Hoffmann, B.; Keller, M.; Drexler, J.F.; Drosten, C.; et al. The bank vole (Clethrionomys glareolus)—Small animal model for hepacivirus infection. 2021. submitted. [Google Scholar]
- Fevola, C.; Rossi, C.; Rosso, F.; Girardi, M.; Rosà, R.; Manica, M.; Delucchi, L.; Rocchini, D.; Garzon-Lopez, C.X.; Arnoldi, D.; et al. Geographical Distribution of Ljungan Virus in Small Mammals in Europe. Vector-Borne Zoonotic Dis. 2020, 20, 692–702. [Google Scholar] [CrossRef]
- Fischer, S.; Mayer-Scholl, A.; Imholt, C.; Spierling, N.G.; Heuser, E.; Schmidt, S.; Reil, D.; Rosenfeld, U.M.; Jacob, J.; Nöckler, K.; et al. Leptospira Genomospecies and Sequence Type Prevalence in Small Mammal Populations in Germany. Vector-Borne Zoonotic Dis. 2018, 18, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Essbauer, S.S.; Mayer-Scholl, A.; Poppert, S.; Schmidt-Chanasit, J.; Klempa, B.; Henning, K.; Schares, G.; Groschup, M.H.; Spitzenberger, F.; et al. Multiple infections of rodents with zoonotic pathogens in Austria. Vector-Borne Zoonotic Dis. 2014, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Ali, H.S.; Drewes, S.; Sadowska, E.T.; Mikowska, M.; Groschup, M.H.; Heckel, G.; Koteja, P.; Ulrich, R.G. First molecular evidence for Puumala hantavirus in Poland. Viruses 2014, 6, 340–353. [Google Scholar] [CrossRef] [Green Version]
- Alexander, N.; Allepuz, A.; Alten, B.; Bødker, R.; Bonnet, S.; Carpenter, S.; Cetre-Sossah, C.; Chirouze, E.; Depaquit, J.; Dressel, K.; et al. The Impact of a Decade of Research on Vector Borne Disease; CIRAD: Montpellier, France, 2015. [Google Scholar]
- Schlegel, M.; Ali, H.S.; Stieger, N.; Groschup, M.H.; Wolf, R.; Ulrich, R.G. Molecular identification of small mammal species using novel cytochrome B gene-derived degenerated primers. Biochem. Genet. 2011, 50, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Braaker, S.; Heckel, G. Transalpine colonisation and partial phylogeographic erosion by dispersal in the common vole (Microtus arvalis). Mol. Ecol. 2009, 18, 2518–2531. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Hoehna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Filipi, K.; Marková, S.; Searle, J.; Kotlík, P. Mitogenomic phylogenetics of the bank vole Clethrionomys glareolus, a model system for studying end-glacial colonization of Europe. Mol. Phylogenet. Evol. 2015, 82 Pt A, 245–257. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxenhofer, M.; de Melo, V.W.; Ulrich, R.G.; Heckel, G. Revised time scales of RNA virus evolution based on spatial information. Proc. R. Soc. B Boil. Sci. 2017, 284, 20170857. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Dray, S.; Dufour, A.-B. The ade4 Package: Implementing the Duality Diagram for Ecologist. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Kesäniemi, J.; Lavrinienko, A.; Tukalenko, E.; Mappes, T.; Watts, P.C.; Jurvansuu, J. Infection Load and Prevalence of Novel Viruses Identified from the Bank Vole Do Not Associate with Exposure to Environmental Radioactivity. Viruses 2019, 12, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxenhofer, M.; Schmidt, S.; Ulrich, R.G.; Heckel, G. Secondary contact between diverged host lineages entails ecological speciation in a European hantavirus. PLoS Biol. 2019, 17, e3000142. [Google Scholar] [CrossRef] [Green Version]
- De Melo, V.W.; Ali, H.S.; Freise, J.; Kühnert, D.; Essbauer, S.; Mertens, M.; Wanka, K.M.; Drewes, S.; Ulrich, R.G.; Heckel, G. Spatiotemporal dynamics of Puumala hantavirus associated with its rodent host, Myodes glareolus. Evol. Appl. 2015, 8, 545–559. [Google Scholar] [CrossRef] [Green Version]
- Hiltbrunner, M.; Heckel, G. Assessing Genome-Wide Diversity in European Hantaviruses through Sequence Capture from Natural Host Samples. Viruses 2020, 12, 749. [Google Scholar] [CrossRef]
- Schmidt, S.; Saxenhofer, M.; Drewes, S.; Schlegel, M.; Wanka, K.M.; Frank, R.; Klimpel, S.; Von Blanckenhagen, F.; Maaz, D.; Herden, C.; et al. High genetic structuring of Tula hantavirus. Arch. Virol. 2016, 161, 1135–1149. [Google Scholar] [CrossRef]
- Binder, F.; Ryll, R.; Drewes, S.; Jagdmann, S.; Reil, D.; Hiltbrunner, M.; Rosenfeld, U.M.; Imholt, C.; Jacob, J.; Heckel, G.; et al. Spatial and Temporal Evolutionary Patterns in Puumala Orthohantavirus (PUUV) S Segment. Pathogens 2020, 9, 548. [Google Scholar] [CrossRef]
- Tersago, K.; Schreurs, A.; Linard, C.; Verhagen, R.; Van Dongen, S.; Leirs, H. Population, environmental, and community effects on local bank vole (Myodes glareolus) Puumala virus infection in an area with low human incidence. Vector-Borne Zoonotic Dis. 2008, 8, 235–244. [Google Scholar] [CrossRef]
- Reil, D.; Rosenfeld, U.M.; Imholt, C.; Schmidt, S.; Ulrich, R.G.; Eccard, J.A.; Jacob, J. Puumala hantavirus infections in bank vole populations: Host and virus dynamics in Central Europe. BMC Ecol. 2017, 17, 9. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.; Ecke, F.; Evander, M.; Bucht, G.; Hörnfeldt, B. Population Dynamics of Bank Voles Predicts Human Puumala Hantavirus Risk. EcoHealth 2019, 16, 545–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gather, T.; Walter, S.; Todt, D.; Pfaender, S.; Brown, R.J.P.; Postel, A.; Becher, P.; Moritz, A.; Hansmann, F.; Baumgaertner, W.; et al. Vertical transmission of hepatitis C virus-like non-primate hepacivirus in horses. J. Gen. Virol. 2016, 97, 2540–2551. [Google Scholar] [CrossRef]
- Preciado, M.V.; Valva, P.; Escobar-Gutierrez, A.; Rahal, P.; Ruiz-Tovar, K.; Yamasaki, L.; Chacon, C.A.V.; Martinez-Guarneros, A.; Carpio-Pedroza, J.C.; Fonseca-Coronado, S.; et al. Hepatitis C virus molecular evolution: Transmission, disease progression and antiviral therapy. World J. Gastroenterol. 2014, 20, 15992. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Family | Species | No. of Hepacivirus-Positive/Tested Animals | % RT-qPCR Positive | Country (Total Number of Animals) |
|---|---|---|---|---|---|
| Rodentia | Cricetidae | Lemmus lemmus | 0/21 | 0 | FIN (21) |
| Cricetidae | Microtus agrestis | 0/8 | 0 | GER (8) | |
| Cricetidae | Microtus arvalis | 0/129 | 0 | GER (129) | |
| Cricetidae | Microtus oeconomus (syn. Alexandromys oeconomus) | 0/2 | 0 | FIN (2) | |
| Cricetidae | Myodes glareolus (syn. Clethrionomys glareolus) | 442/1838 | 24.0 | GBR (61), GER (1297), FRA (99), ITA (25), NED (20), AUT (33), POL (15), SWE (223), SVK (65) | |
| Cricetidae | Myodes rutilus (syn. Clethrionomys rutilus) | 1/7 | 14.3 | FIN (7) | |
| Cricetidae | Myopus schisticolor | 0/9 | 0 | FIN (9) | |
| Muridae | Apodemus spp. | 0/31 | 0 | CRO (30), ITA (1) | |
| Muridae | Apodemus agrarius | 0/5 | 0 | CRO (2), GER (3) | |
| Muridae | Apodemus flavicollis | 0/209 | 0 | GER (206), ITA (3) | |
| Muridae | Apodemus sylvaticus | 0/39 | 0 | GER (39) | |
| Muridae | Micromys minutus | 0/1 | 0 | GER (1) | |
| Muridae | Mus musculus | 0/22 | 0 | GER (1), EST (8), FIN (3), LAT (8), LTU (2) | |
| Muridae | Mus spp. | 0/2 | 0 | CRO (2) | |
| Muridae | Rattus spp. | 0/4 | 0 | CRO (4) | |
| Carnivora | Mustelidae | Mustela nivalis | 0/1 | 0 | ITA (1) |
| Eulipotyphla | Erinaceidae | Erinaceus europaeus | 0/1 | 0 | GER (1) |
| Soricidae | Crocidura leucodon | 0/1 | 0 | ITA (1) | |
| Soricidae | Crocidura russula | 0/1 | 0 | GER (1) | |
| Soricidae | Neomys fodiens | 0/1 | 0 | ITA (1) | |
| Soricidae | Sorex araneus | 0/37 | 0 | GER (37) | |
| Soricidae | Sorex alpinus | 0/1 | 0 | ITA (1) | |
| Soricidae | Sorex antinorii | 0/7 | 0 | ITA (7) | |
| Soricidae | Sorex coronatus | 0/26 | 0 | GER (26) | |
| Soricidae | Sorex minutus | 0/25 | 0 | GER (25) | |
| Total | 443/2428 | 18.2% |
| Country | Trapping Site | rodHCVeur Assays (Hepacivirus J) | RHV-NS3-Line4 Assay (Hepacivirus F) | Both Assays (Hepacivirus F and Hepacivirus J) | Positive/Tested Per Trapping Site | Positive/Tested Per Country |
|---|---|---|---|---|---|---|
| Austria | Laa an der Thaya | 3 | 10 | 1 | 12/33 | 12/33 |
| Finland | Pallasjärvi | 0 * | 1 * | 0 * | 1/7 * | 1/7 * |
| France | Cormaranche-en-Bugey | 12 | 5 | 3 | 14/20 | 61/99 |
| La Venotiere | 10 | 7 | 4 | 13/20 | ||
| Mignovillard | 9 | 5 | 3 | 11/20 | ||
| Mont-sous-Vaudrey | 9 | 2 | 2 | 9/20 | ||
| Vouzon | 8 | 12 | 6 | 14/19 | ||
| Germany | Ahlhorn | 1 | 0 | 0 | 1/2 | 286/1297 |
| Bad Waldsee | 0 | 0 | 0 | 0/5 | ||
| Bierhütte | 0 | 0 | 0 | 0/2 | ||
| Billerbeck | 67 | 8 | 0 | 75/285 | ||
| Bogen | 0 | 0 | 0 | 0/1 | ||
| Bremerhaven | 3 | 0 | 0 | 3/19 | ||
| Falkenstein | 0 | 2 | 0 | 2/5 | ||
| Freyung | 0 | 0 | 0 | 0/5 | ||
| Geversdorf | 0 | 0 | 0 | 0/4 | ||
| Glashütte | 0 | 0 | 0 | 0/1 | ||
| Gotha | 56 | 61 | 51 | 66/319 | ||
| Hangenleithen | 0 | 3 | 0 | 3/6 | ||
| Jasnitz | 5 | 0 | 0 | 5/20 | ||
| Jeeser | 41 | 3 | 2 | 42/155 | ||
| Langenfurth | 0 | 0 | 0 | 0/1 | ||
| Lucka (bei Groitzsch) | 3 | 1 | 0 | 4/17 | ||
| Mühlberg, Spiegelau | 0 | 1 | 0 | 1/1 | ||
| Mutzenwinkel | 0 | 1 | 0 | 1/2 | ||
| Oberndorf (Hemmoor) | 0 | 2 | 0 | 2/8 | ||
| Osnabrück | 11 | 8 | 4 | 15/23 | ||
| Raimundsreuth | 0 | 3 | 0 | 3/14 | ||
| Reinberg | 0 | 0 | 0 | 0/1 | ||
| Rothemühl | 0 | 2 | 0 | 2/4 | ||
| Schrevendorf | 9 | 2 | 0 | 11/20 | ||
| Steinheim am Albuch | 0 | 0 | 0 | 0/20 | ||
| Treben (Altenburg) | 0 | 0 | 0 | 0/3 | ||
| Tussenhausen | 0 | 4 | 0 | 4/9 | ||
| Weissach | 41 | 4 | 1 | 44/332 | ||
| Wolbrechtshausen | 1 | 0 | 0 | 1/2 | ||
| Wolfertschlag | 0 | 1 | 0 | 1/5 | ||
| Zußdorf | 0 | 0 | 0 | 0/5 | ||
| Zwiesel | 0 | 0 | 0 | 0/1 | ||
| Great Britain | Cumbria | 0 | 2 | 0 | 2/51 | 5/61 |
| Pentland Hills | 0 | 3 | 0 | 5/10 | ||
| Italy | Brescia | 0 | 0 | 0 | 0/5 | 5/25 |
| Trento | 3 | 2 | 0 | 5/20 | ||
| The Netherlands | Nutter | 11 | 4 | 2 | 13/20 | 13/20 |
| Poland | Mikołajki | 0 | 0 | 0 | 0/5 | 1/15 |
| Dobskie island | 0 | 1 | 0 | 1/5 | ||
| Dejguny island | 0 | 0 | 0 | 0/5 | ||
| Slovakia | Bratislava | 8 | 9 | 1 | 16/22 | 42/65 |
| Fugelka | 10 | 7 | 3 | 14/23 | ||
| Rozhanovce | 6 | 10 | 4 | 12/20 | ||
| Sweden | Ammarnäs | 0 | 0 | 0 | 0/20 | 17/223 |
| Grimsö | 0 | 4 | 0 | 4/20 | ||
| Haparanda | 0 | 2 | 0 | 2/20 | ||
| Harads | 0 | 3 | 0 | 3/20 | ||
| Öster Malma | 0 | 1 | 0 | 1/20 | ||
| Umeå | 0 | 2 | 0 | 2/42 | ||
| Vålådalen | 0 | 0 | 0 | 0/20 | ||
| Västernorrland | 0 | 0 | 0 | 0/20 | ||
| Växjö | 0 | 4 | 0 | 4/21 | ||
| Vindeln | 0 | 1 | 0 | 1/20 | ||
| Total | 327 | 202 + 1 * | 87 |
| Year | Season | Number of BvHV RNA-Positive/Total Number of Bank Voles | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Jeeser | Billerbeck | Gotha | Weissach | ||||||
| 2010 | spring | 5/11 | 26/82 | 24/84 | 57/242 | 8/48 | 23/218 | 15/131 | 24/262 |
| summer | 8/28 | 15/84 | 6/65 | 5/77 | |||||
| autumn | 13/43 | 18/74 | 9/105 | 4/54 | |||||
| 2011 | spring | 0/1 | 9/59 | 1/1 | 7/32 | 3/6 | 22/89 | 0/0 | 5/31 |
| summer | 0/23 | 3/17 | 10/45 | 5/23 | |||||
| autumn | 9/35 | 3/14 | 9/38 | 0/8 | |||||
| 2012 | spring | 6/15 | 3/11 | 10/15 | 10/39 | ||||
| Total | 41/156 | 67/285 | 55/322 | 39/332 | |||||
| Model | Factors | Model Statistics | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Population | Individual | |||||||||||
| Abundance (Bank Vole) | Abundance (Yellow-Necked Field Mouse) | Season | Year | Reproductive Activity | Sex | Mass | df | logLik | AICc | ΔAICc | Model Weight | |
| 1 | −0.0339 | + | 5 | −468.021 | 946.1 | 0 | 0.06 | |||||
| 2 | −0.0327 | + | 0.0248 | 6 | −467.137 | 946.4 | 0.26 | 0.053 | ||||
| 3 | −0.0335 | + | + | 0.0270 | 7 | −466.347 | 946.8 | 0.7 | 0.042 | |||
| 4 | −0.0346 | + | + | 6 | −467.379 | 946.8 | 0.74 | 0.041 | ||||
| 5 | + | 4 | −469.513 | 947.1 | 0.96 | 0.037 | ||||||
| 6 | + | 0.0263 | 5 | −468.525 | 947.1 | 1.01 | 0.036 | |||||
| 7 | −0.0350 | + | + | 0.0349 | 7 | −466.604 | 947.3 | 1.22 | 0.033 | |||
| 8 | −0.0360 | + | + | + | 0.0378 | 8 | −465.743 | 947.6 | 1.53 | 0.028 | ||
| 9 | + | + | 0.0284 | 6 | −467.808 | 947.7 | 1.6 | 0.027 | ||||
| 10 | + | + | 5 | −468.927 | 947.9 | 1.81 | 0.024 | |||||
| 11 | + | + | 5 | −468.936 | 947.9 | 1.83 | 0.024 | |||||
| 12 | −0.0306 | + | + | 6 | −467.967 | 948 | 1.92 | 0.023 | ||||
| 13 | −0.0346 | + | + | 6 | −467.978 | 948 | 1.94 | 0.023 | ||||
| 14 | −0.0021 | −0.0320 | + | 6 | −468.004 | 948.1 | 1.99 | 0.022 | ||||
| 15 | −0.0101 | + | 5 | −469.044 | 948.1 | 2.05 | 0.022 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, J.; Hoffmann, B.; Fevola, C.; Schmidt, M.L.; Imholt, C.; Fischer, S.; Ecke, F.; Hörnfeldt, B.; Magnusson, M.; Olsson, G.E.; et al. Geographical Distribution and Genetic Diversity of Bank Vole Hepaciviruses in Europe. Viruses 2021, 13, 1258. https://doi.org/10.3390/v13071258
Schneider J, Hoffmann B, Fevola C, Schmidt ML, Imholt C, Fischer S, Ecke F, Hörnfeldt B, Magnusson M, Olsson GE, et al. Geographical Distribution and Genetic Diversity of Bank Vole Hepaciviruses in Europe. Viruses. 2021; 13(7):1258. https://doi.org/10.3390/v13071258
Chicago/Turabian StyleSchneider, Julia, Bernd Hoffmann, Cristina Fevola, Marie Luisa Schmidt, Christian Imholt, Stefan Fischer, Frauke Ecke, Birger Hörnfeldt, Magnus Magnusson, Gert E. Olsson, and et al. 2021. "Geographical Distribution and Genetic Diversity of Bank Vole Hepaciviruses in Europe" Viruses 13, no. 7: 1258. https://doi.org/10.3390/v13071258