
Characterization of Prion Disease Associated with a Two-Octapeptide Repeat Insertion

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval, Data Collection
2.2. Genetic Testing
2.3. Reagents and Antibodies
2.4. Brain Tissue
2.5. Histology and PrP Immunohistochemistry
2.6. Preparation of Brain Homogenates and Detergent-Insoluble Fractions
2.7. Proteinase K Digestion and Western Blot (WB) Analysis
2.8. Penetrance Estimation
3. Results
3.1. Clinical Features and Genetics

{kind=link}
{kind=link}
| Case | Origin | Diagnosis | Codon 129 Genotype | resPrPD Type a | Age at Onset (Years) | Disease Duration (Months) | Gender | 14-3-3 Protein | Tau (pg/mL) b | RT-QuIC | PSWC on EEG | MRI c/w CJD | Family History | Clinical Phenotype |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | US | Definite | MM | 1 | 67 | 2 | Male | Pos. | 17,727 | NA | NA | Yes | None | Slurred speech, then a fulminant course including cognitive and cerebellar symptoms and myoclonus |
| 2 | US | Probable | MM | 1 | NA | NA | Male | Pos. | 8848 | NA | NA | Yes | None | NA |
| 3 | US | Definite | MM | 1 | 68 | 7 | Female | Pos. | 7990 | NA | Yes | Yes | Mother with several year slowly progressive dementia in her 60s, thought to be AD | Absence-like episodes, followed 5 months later by cognitive symptoms, personality change, and myoclonus |
| 4 | US | Probable | MV | 1–2 | 84 | 3 | Female | Unk. | Unk. | NA | NA | Yes | None | Myoclonus, unilateral weakness/spasticity, late cognitive symptoms |
| 5 | US | Definite | MM | 1–2 | NA | NA | Female | Pos. | 15,046 | Pos. | NA | Yes | None | NA |
| 6 | UK | Probable | MV | NA | 76 | 10 | Male | Neg. | Unk. | Pos. | No | Yes | Sister with 2-year history of Alzheimer’s dementia starting at age 89; sister with mild dementia in her 80s. | Gait ataxia, followed by cognitive symptoms, visual hallucinations, and myoclonus |
| 7 | UK | Definite | MM | 1 | 58 | 7 | Male | Unk. | Unk. | NA | No | NA | Unknown | Right hand paresthesia followed by unilateral weakness/spasticity |
| 8 | AU | Probable | MV | NA | 74 | 21 | Female | Pos. | Unk. | Pos. | No | Yes | Brother with 5-year history of dementia in his 60s. | Visual disturbances, followed by gait disturbance, Parkinsonian features, apraxia, and visuospatial difficulties |
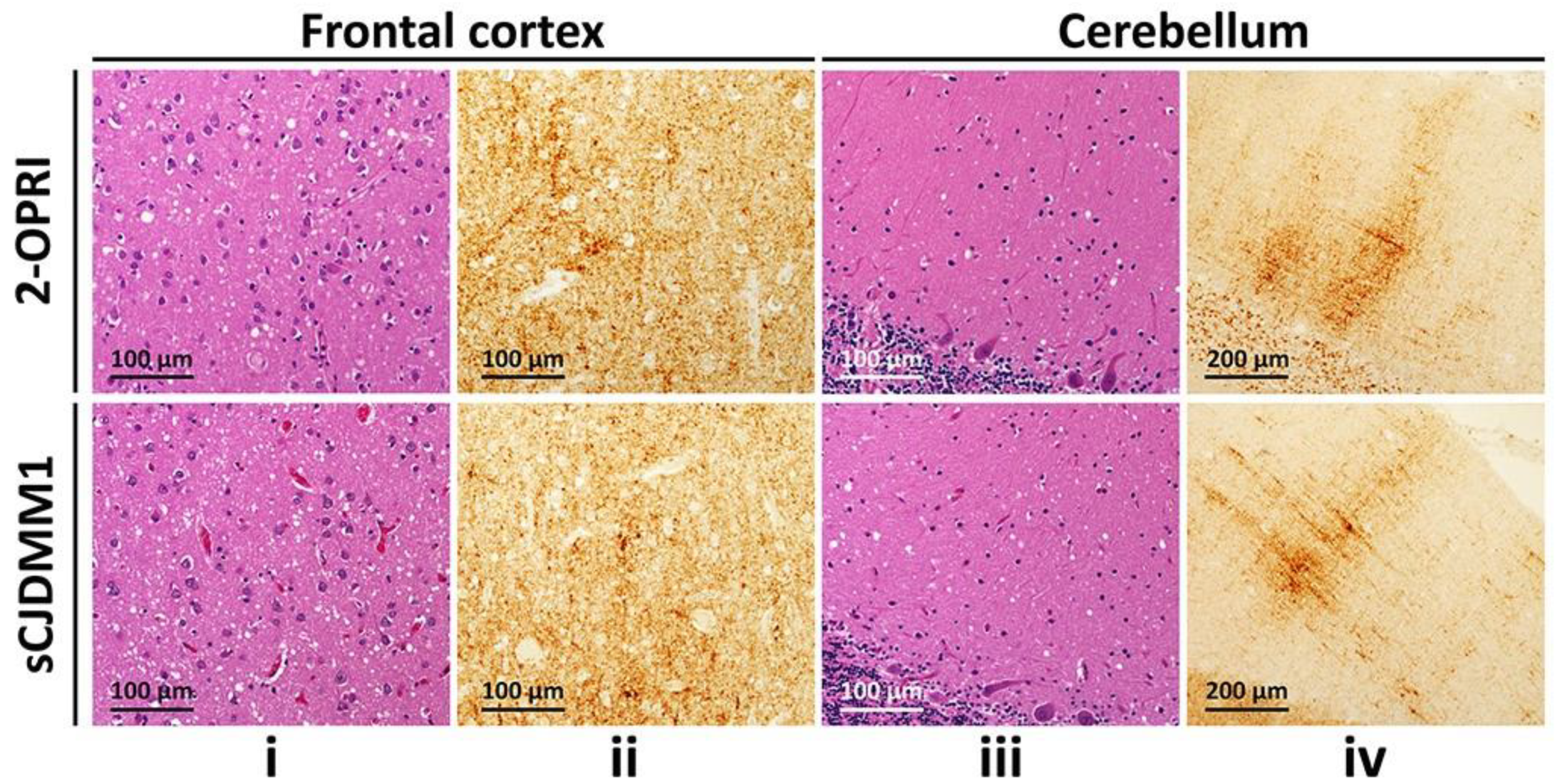
3.2. Histopathology and Immunohistochemistry
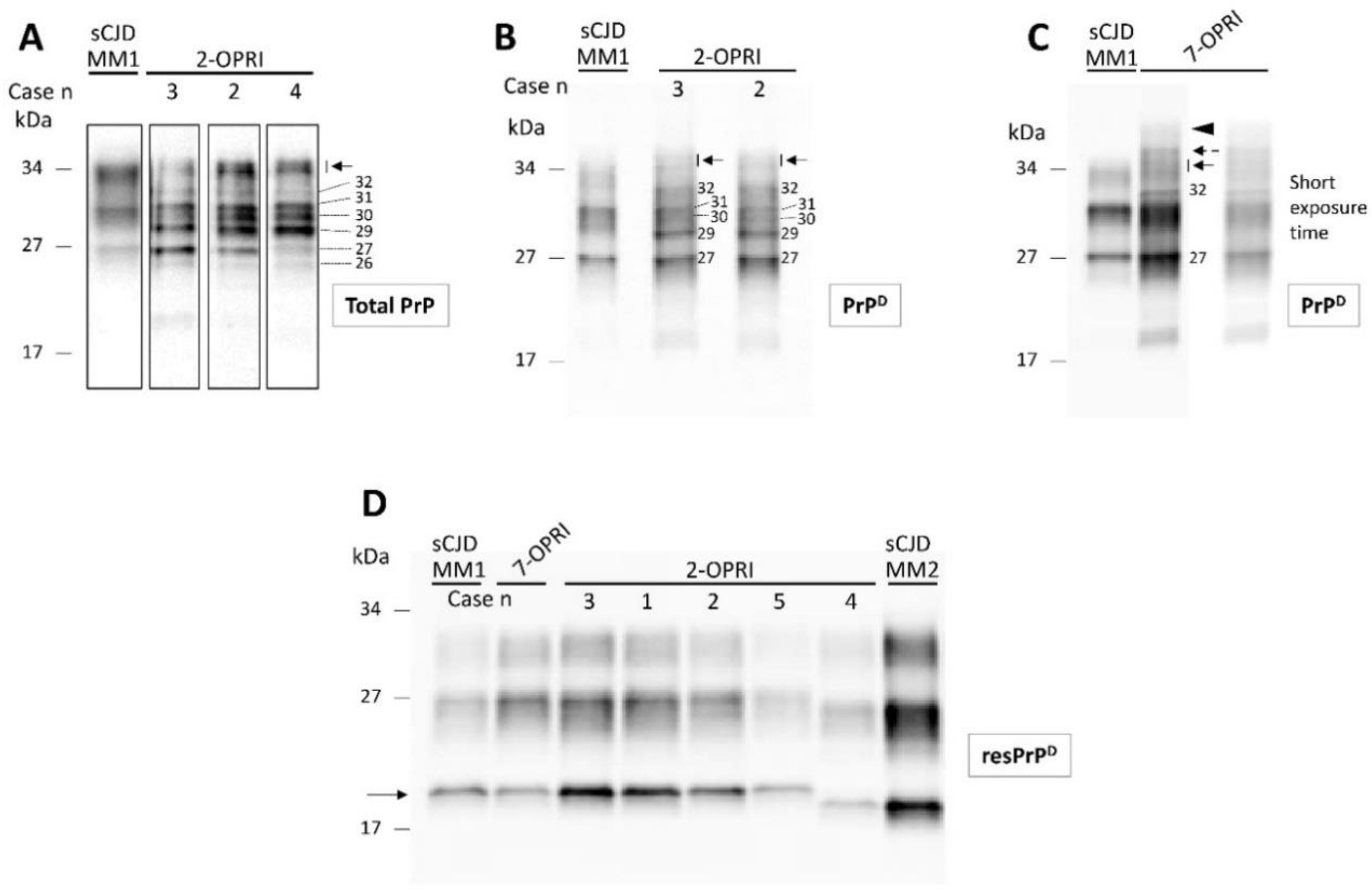
3.3. Characterization of Detergent-Insoluble and PK-Resistant PrPD (resPrPD)
3.4. Estimation of 2-OPRI Penetrance
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Takada, L.T.; Kim, M.-O.; Metcalf, S.; Gala, I.I.; Geschwind, M.D. Prion Disease. Handb. Clin. Neurol. 2018, 148, 441–464. [Google Scholar] [CrossRef]
- Kim, M.-O.; Takada, L.T.; Wong, K.; Forner, S.A.; Geschwind, M.D. Genetic PrP Prion Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033134. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.A.; Herzog, C.; Errett, J.; Kocisko, D.A.; Arnold, K.M.; Hayes, S.F.; Priola, S.A. Octapeptide Repeat Insertions Increase the Rate of Protease-Resistant Prion Protein Formation. Protein Sci. 2006, 15, 609–619. [Google Scholar] [CrossRef]
- Croes, E.; Theuns, J.; Houwing-Duisterma, J.; Dermaut, B.; Sleegers, K.; Roks, G.; Van den Broeck, M.; van Harten, B.; van Swieten, J.C.; Cruts, M.; et al. Octapeptide Repeat Insertions in the Prion Protein Gene and Early Onset Dementia. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, L.G.; Brown, P.; Little, B.W.; Cervenáková, L.; Kenney, K.; Gibbs, C.J.; Gajdusek, D.C. A New (Two-Repeat) Octapeptide Coding Insert Mutation in Creutzfeldt-Jakob Disease. Neurology 1993, 43, 2392–2394. [Google Scholar] [CrossRef]
- Van Harten, B.; Van Gool, W.A.; Van Langen, I.M.; Deekman, J.M.; Meijerink, P.H.; Weinstein, H.C. A New Mutation in the Prion Protein Gene: A Patient with Dementia and White Matter Changes. Neurology 2000, 55, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.; Boyd, A.; Fletcher, A.; Gonzales, M.; McLean, C.A.; Byron, K.; Masters, C.L. Creutzfeldt-Jakob Disease: Diagnostic Utility of 14-3-3 Protein Immunodetection in Cerebrospinal Fluid. J. Clin. Neurosci. 2000, 7, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, C.; Puoti, G.; Berri, S.; Sting, E.; Harris, C.; Cohen, M.; Spear, C.; Bizzi, A.; Debanne, S.M.; Rowland, D.Y. A Comparison of Tau and 14-3-3 Protein in the Diagnosis of Creutzfeldt-Jakob Disease. Neurology 2012, 79, 547–552. [Google Scholar] [CrossRef]
- Foutz, A.; Appleby, B.S.; Hamlin, C.; Liu, X.; Yang, S.; Cohen, Y.; Chen, W.; Blevins, J.; Fausett, C.; Wang, H.; et al. Diagnostic and Prognostic Value of Human Prion Detection in Cerebrospinal Fluid. Ann. Neurol. 2017, 81, 79–92. [Google Scholar] [CrossRef]
- Green, A.J.E. RT-QuIC: A New Test for Sporadic CJD. Pract. Neurol. 2019, 19, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Zerr, I.; Kallenberg, K.; Summers, D.M.; Romero, C.; Taratuto, A.; Heinemann, U.; Breithaupt, M.; Varges, D.; Meissner, B.; Ladogana, A.; et al. Updated Clinical Diagnostic Criteria for Sporadic Creutzfeldt Jakob Disease. Brain 2009, 132, 2659–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadsworth, J.D.F.; Adamson, G.; Joiner, S.; Brock, L.; Powell, C.; Linehan, J.M.; Beck, J.A.; Brandner, S.; Mead, S.; Collinge, J. Methods for Molecular Diagnosis of Human Prion Disease. In Prions: Methods and Protocols; Lawson, V.A., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 311–346. ISBN 978-1-4939-7244-9. [Google Scholar]
- Piazza, M.; Prior, T.W.; Khalsa, P.S.; Appleby, B. A Case Report of Genetic Prion Disease with Two Different PRNP Variants. Mol. Genet. Genomic Med. 2020, e1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfarb, L.G.; Brown, P.; McCombie, W.R.; Goldgaber, D.; Swergold, G.D.; Wills, P.R.; Cervenakova, L.; Baron, H.; Gibbs, C.J.; Gajdusek, D.C. Transmissible Familial Creutzfeldt-Jakob Disease Associated with Five, Seven, and Eight Extra Octapeptide Coding Repeats in the PRNP Gene. Proc. Natl. Acad. Sci. USA 1991, 88, 10926–10930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kascsak, R.J.; Rubenstein, R.; Merz, P.A.; Tonna-DeMasi, M.; Fersko, R.; Carp, R.I.; Wisniewski, H.M.; Diringer, H. Mouse Polyclonal and Monoclonal Antibody to Scrapie-Associated Fibril Proteins. J. Virol. 1987, 61, 3688–3693. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.-Q.; Langeveld, J.; Xiao, X.; Chen, S.; McGeer, P.L.; Yuan, J.; Payne, M.C.; Kang, H.-E.; McGeehan, J.; Sy, M.-S.; et al. PrP Conformational Transitions Alter Species Preference of a PrP-Specific Antibody. J. Biol. Chem. 2010, 285, 13874–13884. [Google Scholar] [CrossRef] [Green Version]
- Mauro, C.; Giaccone, G.; Piscosquito, G.; Lavorgna, A.; Nigro, M.; Di Fede, G.; Leonardi, A.; Coppola, C.; Formisano, S.; Tagliavini, F.; et al. A Novel Insertional Mutation in the Prion Protein Gene: Clinical and Bio-Molecular Findings. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1395–1398. [Google Scholar] [CrossRef]
- Cali, I.; Cracco, L.; Saracino, D.; Occhipinti, R.; Coppola, C.; Appleby, B.S.; Puoti, G. Case Report: Histopathology and Prion Protein Molecular Properties in Inherited Prion Disease With a De Novo Seven-Octapeptide Repeat Insertion. Front. Cell Neurosci. 2020, 14, 150. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Joiner, S.; Wadsworth, J.D.; Sidle, K.C.; Bell, J.E.; Budka, H.; Ironside, J.W.; Collinge, J. Molecular Classification of Sporadic Creutzfeldt-Jakob Disease. Brain 2003, 126, 1333–1346. [Google Scholar] [CrossRef]
- Cali, I.; Miller, C.J.; Parisi, J.E.; Geschwind, M.D.; Gambetti, P.; Schonberger, L.B. Distinct Pathological Phenotypes of Creutzfeldt-Jakob Disease in Recipients of Prion-Contaminated Growth Hormone. Acta Neuropathol. Commun. 2015, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Cali, I.; Castellani, R.; Yuan, J.; Al-Shekhlee, A.; Cohen, M.L.; Xiao, X.; Moleres, F.J.; Parchi, P.; Zou, W.-Q.; Gambetti, P. Classification of Sporadic Creutzfeldt-Jakob Disease Revisited. Brain 2006, 129, 2266–2277. [Google Scholar] [CrossRef] [Green Version]
- Cali, I.; Castellani, R.; Alshekhlee, A.; Cohen, Y.; Blevins, J.; Yuan, J.; Langeveld, J.P.M.; Parchi, P.; Safar, J.G.; Zou, W.-Q.; et al. Co-Existence of Scrapie Prion Protein Types 1 and 2 in Sporadic Creutzfeldt-Jakob Disease: Its Effect on the Phenotype and Prion-Type Characteristics. Brain 2009, 132, 2643–2658. [Google Scholar] [CrossRef]
- Parchi, P.; Castellani, R.; Capellari, S.; Ghetti, B.; Young, K.; Chen, S.G.; Farlow, M.; Dickson, D.W.; Sima, A.A.; Trojanowski, J.Q.; et al. Molecular Basis of Phenotypic Variability in Sporadic Creutzfeldt-Jakob Disease. Ann. Neurol. 1996, 39, 767–778. [Google Scholar] [CrossRef]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of Sporadic Creutzfeldt-Jakob Disease Based on Molecular and Phenotypic Analysis of 300 Subjects. Ann. Neurol 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.C.; Gambetti, P.; et al. Quantifying Prion Disease Penetrance Using Large Population Control Cohorts. Sci. Transl. Med. 2016, 8, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.H.; Koriath, C.; Jaunmuktane, Z.; Campbell, T.; Joiner, S.; Wadsworth, J.D.F.; Hosszu, L.L.P.; Brandner, S.; Parvez, A.; Truelsen, T.C.; et al. Evaluating the Causality of Novel Sequence Variants in the Prion Protein Gene by Example. Neurobiol. Aging 2018, 71, 265.e1–265.e7. [Google Scholar] [CrossRef] [PubMed]
- Vital, C.; Gray, F.; Vital, A.; Parchi, P.; Capellari, S.; Petersen, R.B.; Ferrer, X.; Jarnier, D.; Julien, J.; Gambetti, P. Prion Encephalopathy with Insertion of Octapeptide Repeats: The Number of Repeats Determines the Type of Cerebellar Deposits. Neuropathol. Appl. Neurobiol. 1998, 24, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Reiniger, L.; Mirabile, I.; Lukic, A.; Wadsworth, J.D.; Linehan, J.M.; Groves, M.; Lowe, J.; Druyeh, R.; Rudge, P.; Collinge, J.; et al. Filamentous White Matter Prion Protein Deposition Is a Distinctive Feature of Multiple Inherited Prion Diseases. Acta Neuropathol. Commun. 2013, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Hill, A.F.; Joiner, S.; Beck, J.A.; Campbell, T.A.; Dickinson, A.; Poulter, M.; Wadsworth, J.D.F.; Collinge, J. Distinct Glycoform Ratios of Protease Resistant Prion Protein Associated with PRNP Point Mutations. Brain 2006, 129, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Mead, S.; Poulter, M.; Beck, J.; Webb, T.E.F.; Campbell, T.A.; Linehan, J.M.; Desbruslais, M.; Joiner, S.; Wadsworth, J.D.F.; King, A.; et al. Inherited Prion Disease with Six Octapeptide Repeat Insertional Mutation—Molecular Analysis of Phenotypic Heterogeneity. Brain 2006, 129, 2297–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. BioRxiv 2020, 531210. [Google Scholar] [CrossRef] [Green Version]
- Minikel, E.V.; Zhao, H.T.; Le, J.; O’Moore, J.; Pitstick, R.; Graffam, S.; Carlson, G.A.; Kavanaugh, M.P.; Kriz, J.; Kim, J.B.; et al. Prion Protein Lowering Is a Disease-Modifying Therapy across Prion Disease Stages, Strains and Endpoints. Nucleic Acids Res. 2020, 48, 10615–10631. [Google Scholar] [CrossRef]
- Parchi, P.; Strammiello, R.; Notari, S.; Giese, A.; Langeveld, J.P.M.; Ladogana, A.; Zerr, I.; Roncaroli, F.; Cras, P.; Ghetti, B.; et al. Incidence and Spectrum of Sporadic Creutzfeldt-Jakob Disease Variants with Mixed Phenotype and Co-Occurrence of PrPSc Types: An Updated Classification. Acta Neuropathol. 2009, 118, 659–671. [Google Scholar] [CrossRef] [Green Version]
- Maddox, R.A.; Person, M.K.; Blevins, J.E.; Abrams, J.Y.; Appleby, B.S.; Schonberger, L.B.; Belay, E.D. Prion disease incidence in the United States: 2003–2015. Neurology 2020, 94, e153–e157. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Prion Diseases of Humans and Animals: Their Causes and Molecular Basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, J.A.; Poulter, M.; Campbell, T.A.; Adamson, G.; Uphill, J.B.; Guerreiro, R.; Jackson, G.S.; Stevens, J.C.; Manji, H.; Collinge, J.; et al. PRNP Allelic Series from 19 Years of Prion Protein Gene Sequencing at the MRC Prion Unit. Hum. Mutat. 2010, 31, E1551–E1563. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Rülicke, T.; Raeber, A.; Sailer, A.; Moser, M.; Oesch, B.; Brandner, S.; Aguzzi, A.; Weissmann, C. Prion Protein (PrP) with Amino-Proximal Deletions Restoring Susceptibility of PrP Knockout Mice to Scrapie. EMBO J. 1996, 15, 1255–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B.; Groth, D.F.; Bolton, D.C.; Kent, S.B.; Hood, L.E. Purification and Structural Studies of a Major Scrapie Prion Protein. Cell 1984, 38, 127–134. [Google Scholar] [CrossRef]
- Wells, M.A.; Jackson, G.S.; Jones, S.; Hosszu, L.L.P.; Craven, C.J.; Clarke, A.R.; Collinge, J.; Waltho, J.P. A Reassessment of Copper(II) Binding in the Full-Length Prion Protein. Biochem. J. 2006, 399, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Kaski, D.N.; Pennington, C.; Beck, J.; Poulter, M.; Uphill, J.; Bishop, M.T.; Linehan, J.M.; O’Malley, C.; Wadsworth, J.D.F.; Joiner, S.; et al. Inherited Prion Disease with 4-Octapeptide Repeat Insertion: Disease Requires the Interaction of Multiple Genetic Risk Factors. Brain 2011, 134, 1829–1838. [Google Scholar] [CrossRef]
- Mead, S.; Webb, T.E.F.; Campbell, T.A.; Beck, J.; Linehan, J.M.; Rutherfoord, S.; Joiner, S.; Wadsworth, J.D.F.; Heckmann, J.; Wroe, S.; et al. Inherited Prion Disease with 5-OPRI: Phenotype Modification by Repeat Length and Codon 129. Neurology 2007, 69, 730–738. [Google Scholar] [CrossRef]
- Jones, E.; Hummerich, H.; Viré, E.; Uphill, J.; Dimitriadis, A.; Speedy, H.; Campbell, T.; Norsworthy, P.; Quinn, L.; Whitfield, J.; et al. Identification of Novel Risk Loci and Causal Insights for Sporadic Creutzfeldt-Jakob Disease: A Genome-Wide Association Study. Lancet Neurol. 2020, 19, 840–848. [Google Scholar] [CrossRef]


| Case Number | Codon 129 Genotype | Repeat Sequence a | resPrPD Type b | sCJD Histotype b |
|---|---|---|---|---|
| 1 | MM | R1-R2-R2-R2-R2-R3-R4 | 1 | MM(MV)1 |
| 2 | MM | R1-R2-R2-R3-R2-R3-R4 | 1 | MM(MV)1 |
| 3 | MM | R1-R2-R2-R2-R2-R3-R4 | 1 | MM(MV)1 |
| 4 | MV | R1-R2-R2-R2-R2-R3-R4 | 1-2 | MV2C + 1 c |
| 5 | MM | R1-R2-R2-R3-R2a-R2a-R4 | 1 | MM1-2c |
| 6 (UK) | MV | R1-R2-R2-R2-R2-R3-R4 | NA | NA |
| 7 (UK) | MM | R1-R2-R2-R3-R2a-R2a-R4 | 1 | MM(MV)1. |
| 8 (AUS) | MV | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brennecke, N.; Cali, I.; Mok, T.H.; Speedy, H.; Genomics England Research Consortium; Hosszu, L.L.P.; Stehmann, C.; Cracco, L.; Puoti, G.; Prior, T.W.; et al. Characterization of Prion Disease Associated with a Two-Octapeptide Repeat Insertion. Viruses 2021, 13, 1794. https://doi.org/10.3390/v13091794
Brennecke N, Cali I, Mok TH, Speedy H, Genomics England Research Consortium, Hosszu LLP, Stehmann C, Cracco L, Puoti G, Prior TW, et al. Characterization of Prion Disease Associated with a Two-Octapeptide Repeat Insertion. Viruses. 2021; 13(9):1794. https://doi.org/10.3390/v13091794
Chicago/Turabian StyleBrennecke, Nicholas, Ignazio Cali, Tze How Mok, Helen Speedy, Genomics England Research Consortium, Laszlo L. P. Hosszu, Christiane Stehmann, Laura Cracco, Gianfranco Puoti, Thomas W. Prior, and et al. 2021. "Characterization of Prion Disease Associated with a Two-Octapeptide Repeat Insertion" Viruses 13, no. 9: 1794. https://doi.org/10.3390/v13091794