
The Structure of the Porcine Deltacoronavirus Main Protease Reveals a Conserved Target for the Design of Antivirals

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression and Protein Purification
2.2. Crystallization, Data Collection, Structure Determination and Refinement
2.3. Enzyme Activity and Inhibition Assays
3. Results
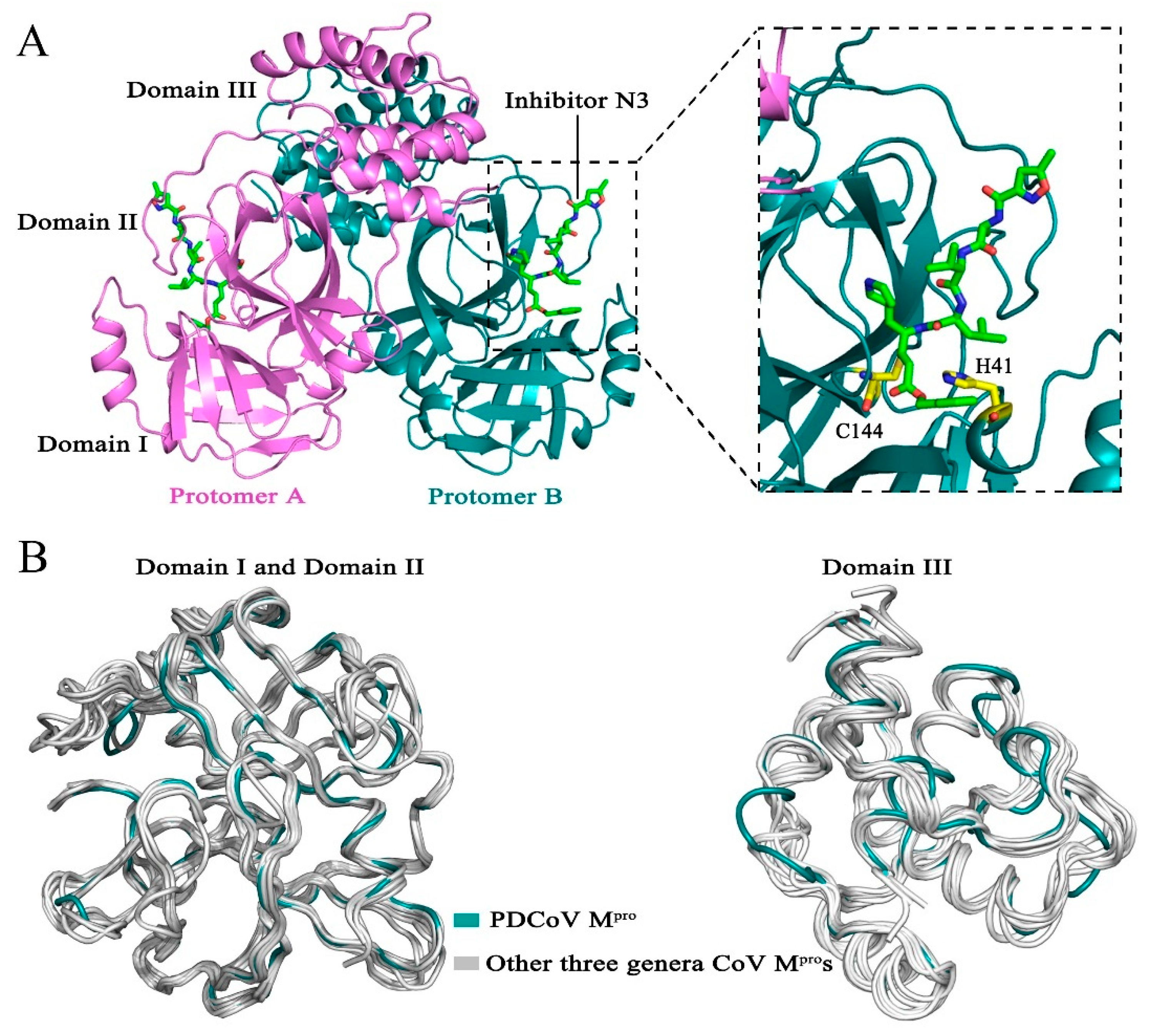
3.1. Overall Structure
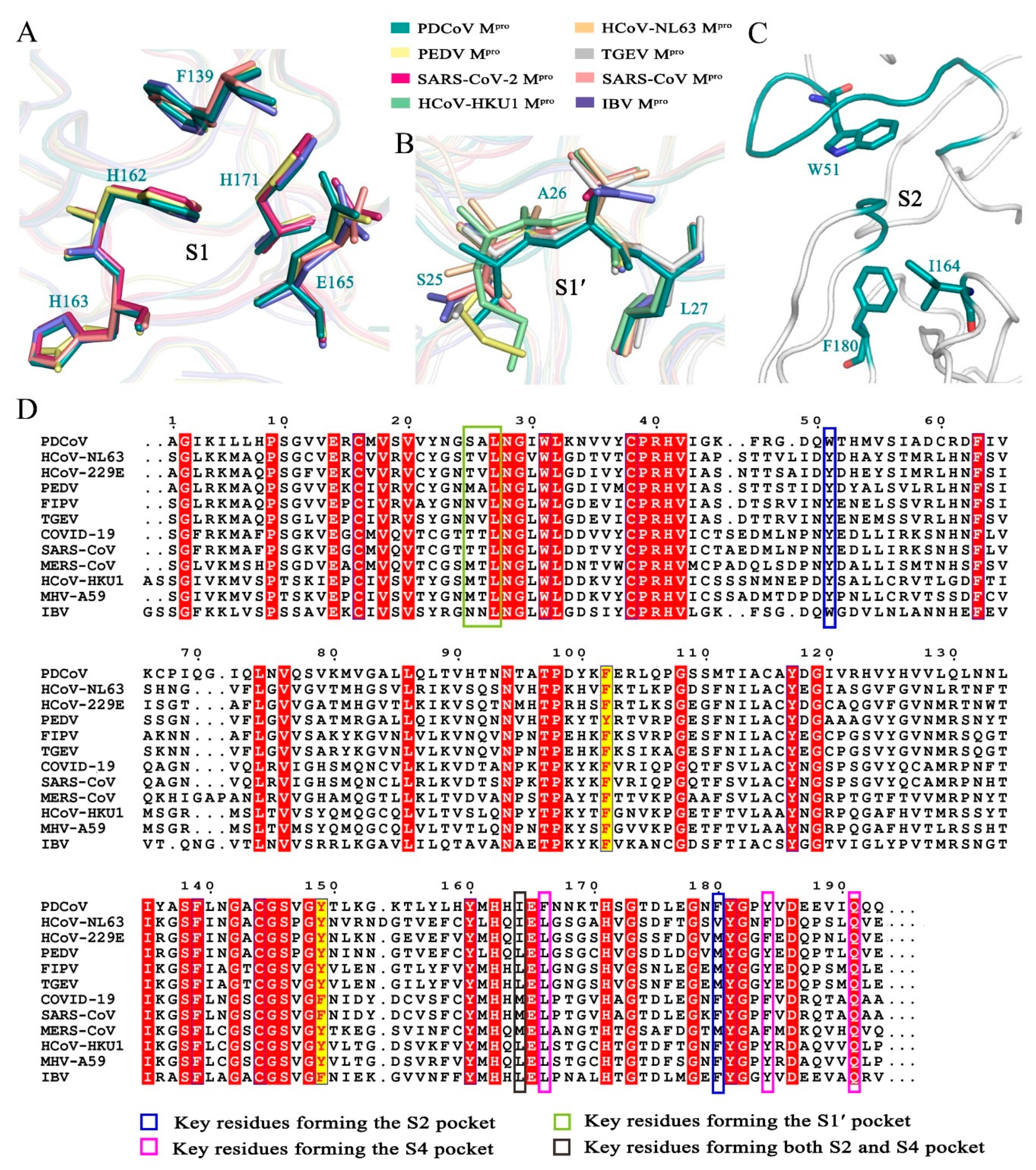
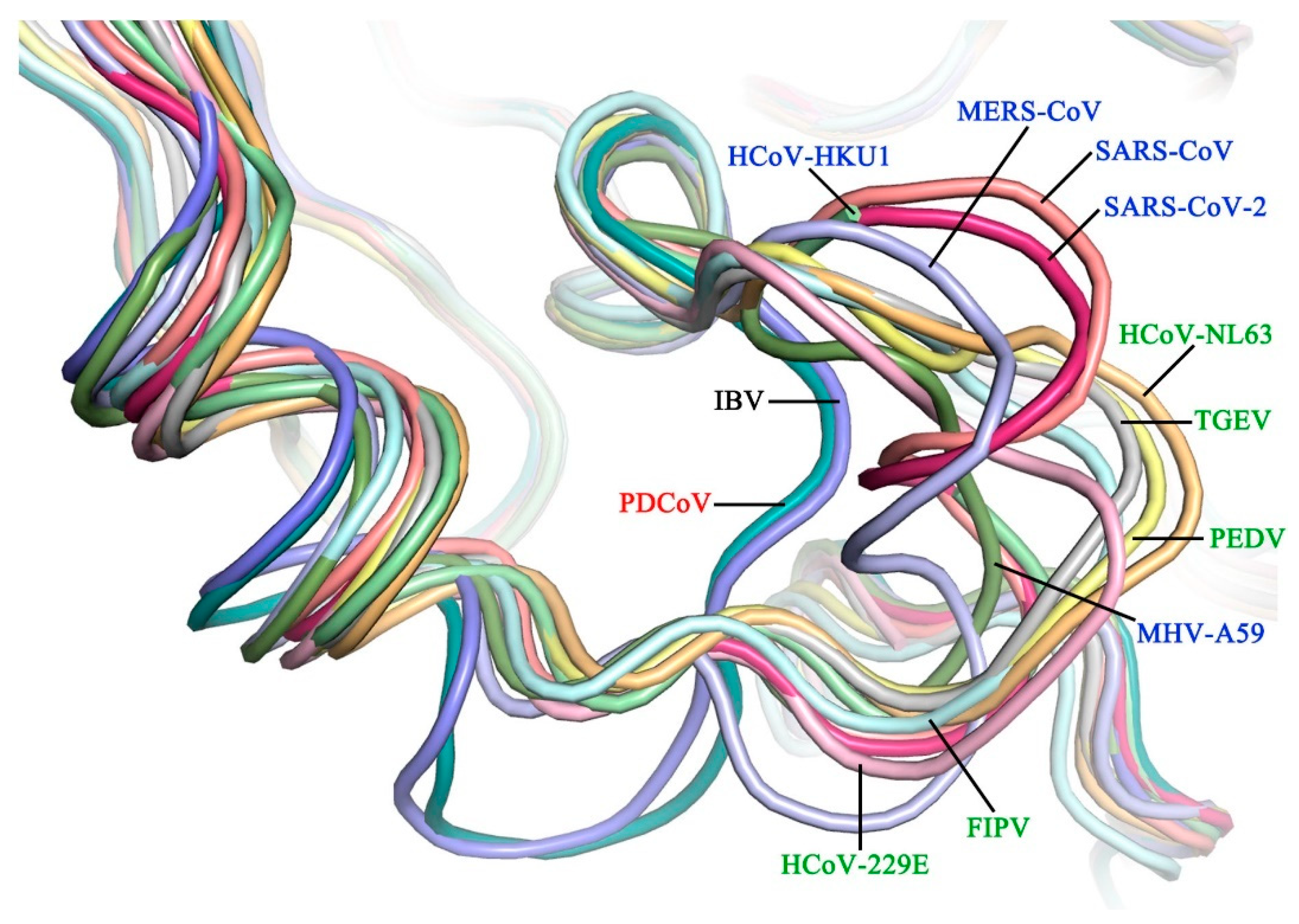
3.2. The Substrate-Binding Pocket PDCoV Mpro Is Structurally Conserved Relative to Mpros from the Other Three Genera
3.3. The Peptidomimetic Inhibitor N3 Efficiently Inhibits PDCoV Mpro
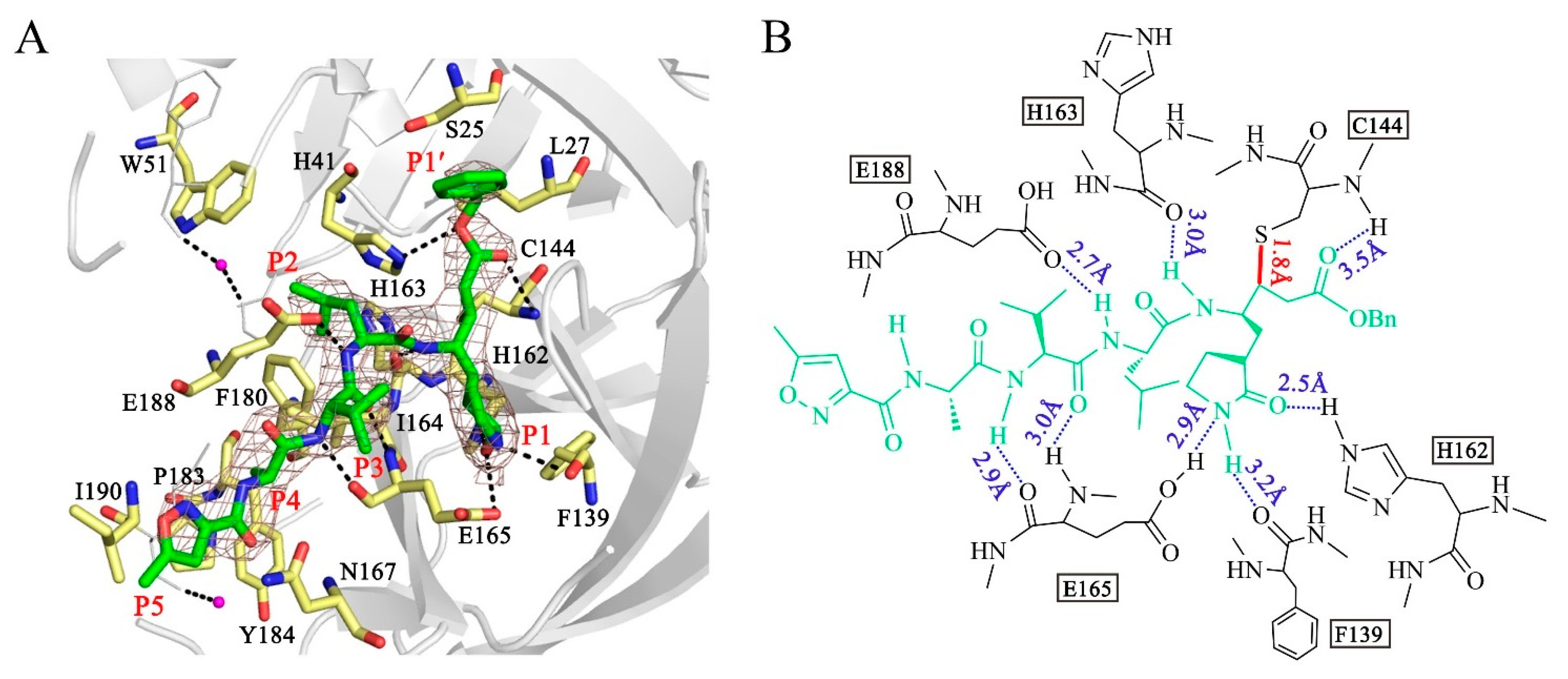
3.4. Binding of N3 to PDCoV Mpro
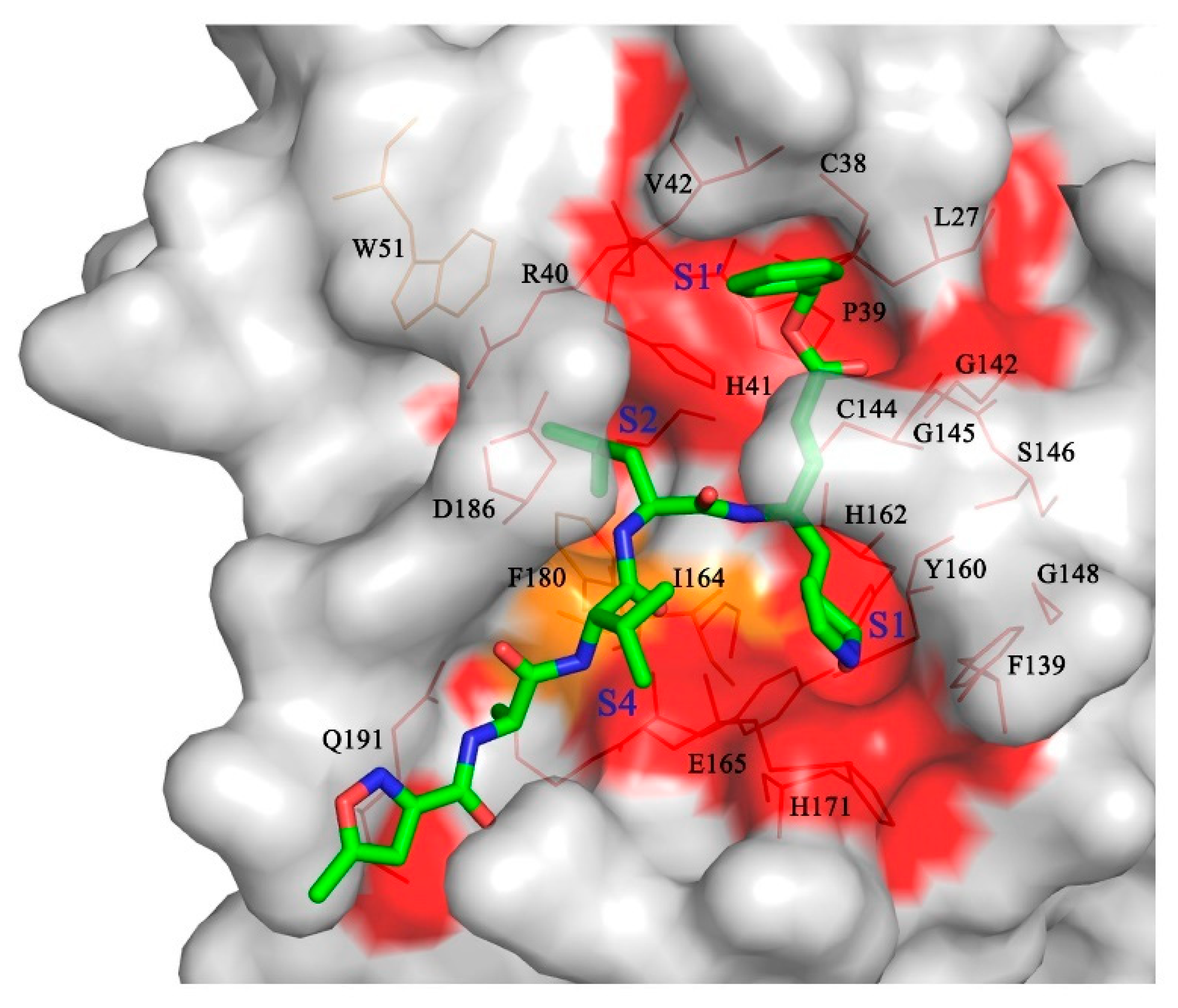
3.5. The P1′ Position May Play an Important Role in the Interaction between PDCoV Mpro and Inhibitors
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.R.; Leibowitz, J.L. Coronavirus pathogenesis. Adv. Virus Res. 2011, 81, 85–164. [Google Scholar] [CrossRef]
- Delaplace, M.; Huet, H.; Gambino, A.; Le Poder, S. Feline coronavirus antivirals: A review. Pathogens 2021, 10, 1150. [Google Scholar] [CrossRef]
- Jackwood, M.W. Review of infectious bronchitis virus around the world. Avian Dis. 2012, 56, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphac oronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosten, C.; Gunther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Kuiken, T.; Fouchier, R.A.; Schutten, M.; Rimmelzwaan, G.F.; van Amerongen, G.; van Riel, D.; Laman, J.D.; de Jong, T.; van Doornum, G.; Lim, W.; et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 2003, 362, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Peiris, J.S.; Lai, S.T.; Poon, L.L.; Guan, Y.; Yam, L.Y.; Lim, W.; Nicholls, J.; Yee, W.K.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- WHO. Novel Coronavirus (SARS-CoV-2) Situation Report. 2022. Available online: https://covid19.who.int/ (accessed on 17 January 2022).
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses-drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, 1742–1752. [Google Scholar] [CrossRef]
- Jung, K.; Hu, H.; Saif, L.J. Porcine deltacoronavirus infection: Etiology, cell culture for virus isolation and propagation, molecular epidemiology and pathogenesis. Virus Res. 2016, 226, 50–59. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Bayer, S.; Cowley, J.A.; Gorbalenya, A.E. The 3C-like proteinase of an invertebrate nidovirus links coronavirus and potyvirus homologs. J. Virol. 2003, 77, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. Embo. J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Cui, W.; Cui, S.S.; Chen, C.; Chen, X.; Wang, Z.F.; Yang, H.T.; Zhang, L. The crystal structure of main protease from mouse hepatitis virus A59 in complex with an inhibitor. Biochem Bioph. Res. Co. 2019, 511, 794–799. [Google Scholar] [CrossRef]
- Ho, B.L.; Cheng, S.C.; Shi, L.; Wang, T.Y.; Ho, K.I.; Chou, C.Y. Critical assessment of the important residues involved in the dimerization and catalysis of MERS coronavirus main protease. PLoS ONE 2015, 10, e0144865. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.C.; Kuo, C.J.; Ko, T.P.; Hsu, M.F.; Tsui, Y.C.; Chang, S.C.; Yang, S.; Chen, S.J.; Chen, H.C.; Hsu, M.C.; et al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem. 2009, 284, 7646–7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Chen, C.; Liu, X.; Yang, K.; Xu, X.; Yang, H. Crystal structure of feline infectious peritonitis virus main protease in complex with synergetic dual inhibitors. J. Virol. 2016, 90, 1910–1917. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Chen, C.; Tan, W.; Yang, K.; Yang, H. Structure of main protease from human coronavirus NL63: Insights for wide spectrum anti-coronavirus drug design. Sci. Rep. 2016, 6, 2267710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Chen, C.; Yang, K.; Xu, Y.; Liu, X.; Gao, F.; Liu, H.; Chen, X.; Zhao, Q.; Liu, X.; et al. Michael acceptor-based peptidomimetic inhibitor of main protease from porcine epidemic diarrhea virus. J. Med. Chem. 2017, 60, 3212–3216. [Google Scholar] [CrossRef]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Li, S.; Xue, F.; Zou, Y.L.; Chen, C.; Bartlam, M.; Rao, Z. Structure of the main protease from a global infectious human coronavirus, HCoV-HKU1. J. Virol. 2008, 82, 8647–8655. [Google Scholar] [CrossRef] [Green Version]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Elbadry, M.A.; Alam, M.M.; Stephenson, C.J.; Bonny, T.S.; Loeb, J.C.; Telisma, T.; Chavannes, S.; et al. Independent infections of porcine deltacoronavirus among Haitian children. Nature 2021, 600, 133–137. [Google Scholar] [CrossRef]
- Li, W.T.; Hulswit, R.J.; Kenney, S.P.; Widjaja, I.; Jung, K.; Alhamo, M.A.; van Dieren, B.; van Kuppeveld, F.J.; Saif, L.J.; Bosch, B.J. Broad receptor engagement of an emerging global coronavirus may potentiate its diverse cross-species transmissibility. Proc. Natl. Acad. Sci. USA 2018, 115, E5135–E5143. [Google Scholar] [CrossRef] [Green Version]
- Dong, N.; Fang, L.; Zeng, S.; Sun, Q.; Chen, H.; Xiao, S. Porcine deltacoronavirus in mainland China. Emerg. Infect. Dis. 2015, 21, 2254–2255. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, C. Complete genome characterization of Korean porcine deltacoronavirus strain KOR/KNU14-04/2014. Genome Announc. 2014, 2, e01191-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thachil, A.; Gerber, P.F.; Xiao, C.T.; Huang, Y.W.; Opriessnig, T. Development and application of an ELISA for the detection of porcine deltacoronavirus IgG antibodies. PLoS ONE 2015, 10, e0124363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Byrum, B.; Zhang, Y. Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerg Infect. Dis. 2014, 20, 1227–1230. [Google Scholar] [CrossRef]
- Zhang, Q.Z.; Yoo, D.W. Immune evasion of porcine enteric coronaviruses and viral modulation of antiviral innate signaling. Virus Res. 2016, 226, 128–141. [Google Scholar] [CrossRef]
- Gerdts, V.; Zakhartchouk, A. Vaccines for porcine epidemic diarrhea virus and other swine coronaviruses. Vet. Microbiol. 2017, 206, 45–51. [Google Scholar] [CrossRef]
- Jung, K.; Hu, H.; Eyerly, B.; Lu, Z.Y.; Chepngeno, J.; Saif, L.J. Pathogenicity of 2 Porcine Deltacoronavirus Strains in Gnotobiotic Pigs. Emerg. Infect. Dis. 2015, 21, 650–654. [Google Scholar] [CrossRef]
- Mccoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Cryst. D 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Cryst. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Tan, S.L. Discovery of small-molecule inhibitors of HCVNS3-4A protease as potential therapeutic agents against HCV infection. Curr. Med. Chem. 2005, 12, 2317–2342. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, A.; Samuelsson, B.; Johansson, P.O.; Cummings, M.D.; Lenz, O.; Raboisson, P.; Simmen, K.; Vendeville, S.; de Kock, H.; Nilsson, M.; et al. Discovery and development of simeprevir (TMC435), a HCV NS3/4A protease inhibitor. J. Med. Chem. 2014, 57, 1673–1693. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, S. Discovery of boceprevir, a direct-acting NS3/4A protease inhibitor for treatment of chronic hepatitis C infections. Trends Pharm. Sci. 2012, 33, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Arias, L.; Tozser, J. HIV-1 protease inhibitors: Effects on HIV-2 replication and resistance. Trends Pharm. Sci. 2008, 29, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Mudgal, M.M.; Birudukota, N.; Doke, M.A. Applications of click chemistry in the development of HIV protease inhibitors. Int. J. Med. Chem. 2018, 2018, 2946730. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Zhou, X.; Peng, Q.; Chen, Y.; Zhang, F.; Huang, T.; Zhang, T.; Li, A.; Huang, D.; Wu, Q.; et al. Newly emerged porcine deltacoronavirus associated with diarrhoea in swine in China: Identification, prevalence and full-length genome sequence analysis. Transbound. Emerg. Dis. 2015, 62, 575–580. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics | Value for the Porcine deltacoronavirus (PDCoV) main protease (Mpro)-N3 Complex |
|---|---|
| Data collection | |
| Wavelength (Å) | 0.97923 |
| Resolution limit (Å) | 50.00–2.60 (2.64–2.60) |
| Space group | P61 |
| Cell parameters | |
| a (Å) | 122.29 |
| b (Å) | 122.29 |
| c (Å) | 289.75 |
| α, β, γ (°) | 90, 90, 120 |
| Total no. of reflections | 75,173 (3733) |
| No. of unique reflections | 74,737 (7392) |
| Completeness (%) | 99.6 (99.3) |
| Redundancy | 20.5 (20.3) |
| Rmerge (%) | 17.2 (>100) |
| Sigma cutoff | 0 |
| I/σ(I) | 12.4 (1.1) |
| Refinement | |
| Resolution range (Å) | 49.74–2.60 |
| Rwork (%) | 19.21 |
| Rfree a (%) | 24.14 |
| No. atoms | |
| Protein | 14,122 |
| Water | 461 |
| Ligands | 294 |
| B-factors (Å2) | |
| Protein | 57.29 |
| Water | 47.07 |
| Ligands | 69.01 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.72 |
| Ramachandran | |
| Favored (%) | 96.05 |
| Allowed (%) | 3.95 |
| Outliers (%) | 0.00 |
| Genus | Coronaviruses | RMSD (Å) (Domain I and Domain II) | RMSD (Å) (Domain III) | PDB ID | References |
|---|---|---|---|---|---|
| Alphacoronavirus | HCoV-NL63 | 1.6 | 2.1 | 5GWY | [26] |
| HCoV-229E | 1.5 | 1.7 | 2ZU2 | [24] | |
| FIPV | 1.5 | 1.6 | 5EU8 | [25] | |
| PEDV | 1.4 | 1.7 | 5GWZ | [27] | |
| TGEV | 1.7 | 1.6 | 2AMP | [15] | |
| Betacoronavirus | SARS-CoV-2 | 1.3 | 1.9 | 6LU7 | [23] |
| SARS-CoV | 1.3 | 2.1 | 2AMQ | [15] | |
| MERS-CoV | 1.3 | 1.6 | 5C3N | [22] | |
| HCoV-HKU1 | 1.4 | 1.6 | 3D23 | [29] | |
| MHV-A59 | 1.3 | 1.5 | 6JIJ | [21] | |
| Gammacoronavirus | IBV | 1.1 | 1.4 | 2Q6F | [28] |
| Genus | Coronaviruses | Km (µM) | kcat (s−1) | References |
|---|---|---|---|---|
| Deltacoronavirus | PDCoV | 56.6 ± 1.9 | 0.030 ± 0.009 | In this study |
| Alphacoronavirus | HCoV-NL63 | 50.8± 3.4 | 0.098 ± 0.004 | [26] |
| HCoV-229E | 29.8 ± 0.9 | 1.27 ± 0.09 | [15] | |
| FIPV | 13.5 ± 1.8 | 0.6 ± 0.06 | [15] | |
| TGEV | 61 ± 5 | 1.39 ± 0.09 | [15] | |
| Betacoronavirus | SARS-CoV | 129 ± 7 | 0.14 ± 0.01 | [15] |
| HCoV-HKU1 | 83.2 ± 13.3 | 1.1 ± 0.12 | [29] | |
| MHV-A59 | 77 ± 5 | 0.083 ± 0.006 | [15] | |
| Gammacoronavirus | IBV | 139 ± 15 | 0.22 ± 0.03 | [15] |
| Compound | Inhibition Ratio (Ir) | Inhibitory Activity a |
|---|---|---|
| N3 | 62% | + + + |
| M1 c | 37% | + + |
| M2 c | 53% | + + + |
| M3 c | 21% | + |
| M5 c | - | N/A b |
| M6 c | - | N/A |
| M7 c | 30% | + + |
| M8 c | 20% | + |
| M10 c | 62% | + + + |
| M11 c | 23% | + |
| M12 c | 27% | + |
| M13 c | 21% | + |
| M14 c | 82% | + + + + + |
| M17 c | 39% | + + |
| M18 c | 50% | + + + |
| M19 c | 53% | + + + |
| M25 c | 74% | + + + + |
| Compound | Structure | Ki (µM) | k3 (10−3s−1) | k3/Ki |
|---|---|---|---|---|
| N3 |  | 11.98 ± 0.13 | 72.9 ± 7.1 | 6.1 |
| M14 |  | 7.56 ± 0.26 | 104.4 ± 2.3 | 13.8 |
| M25 |  | 8.80 ± 0.15 | 86.8 ± 5.1 | 9.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Chen, C.; Wang, Z.; Han, X.; Shi, P.; Zhou, K.; Liu, X.; Xiao, Y.; Cai, Y.; Huang, J.; et al. The Structure of the Porcine Deltacoronavirus Main Protease Reveals a Conserved Target for the Design of Antivirals. Viruses 2022, 14, 486. https://doi.org/10.3390/v14030486
Wang F, Chen C, Wang Z, Han X, Shi P, Zhou K, Liu X, Xiao Y, Cai Y, Huang J, et al. The Structure of the Porcine Deltacoronavirus Main Protease Reveals a Conserved Target for the Design of Antivirals. Viruses. 2022; 14(3):486. https://doi.org/10.3390/v14030486
Chicago/Turabian StyleWang, Fenghua, Cheng Chen, Zefang Wang, Xu Han, Peidian Shi, Kaixuan Zhou, Xiaomei Liu, Yunjie Xiao, Yan Cai, Jinhai Huang, and et al. 2022. "The Structure of the Porcine Deltacoronavirus Main Protease Reveals a Conserved Target for the Design of Antivirals" Viruses 14, no. 3: 486. https://doi.org/10.3390/v14030486