
Infection of Human Endothelial Cells with Lassa Virus Induces Early but Transient Activation and Low Type I IFN Response Compared to the Closely-Related Nonpathogenic Mopeia Virus

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses and Cells
2.2. Titration of Viruses
2.3. Measurement of the Viral Load
2.4. Protein Extraction
2.5. Trypsin Proteolysis and Tandem Mass Spectrometry
2.6. Protein Quantification in Cell Supernatants
2.7. RNA Analysis
2.8. Proteomics Data Analysis
2.9. Functional Enrichment Analysis of Proteomics and Transcriptomics Datasets
2.10. RNA Isolation and Sequencing
2.11. RNA-Seq Mapping and Quantification
2.12. RNA-Seq Statistical Analysis
2.13. Statistical Analysis
3. Results
3.1. Lassa and Mopeia Viruses Productively Infect HUVECs
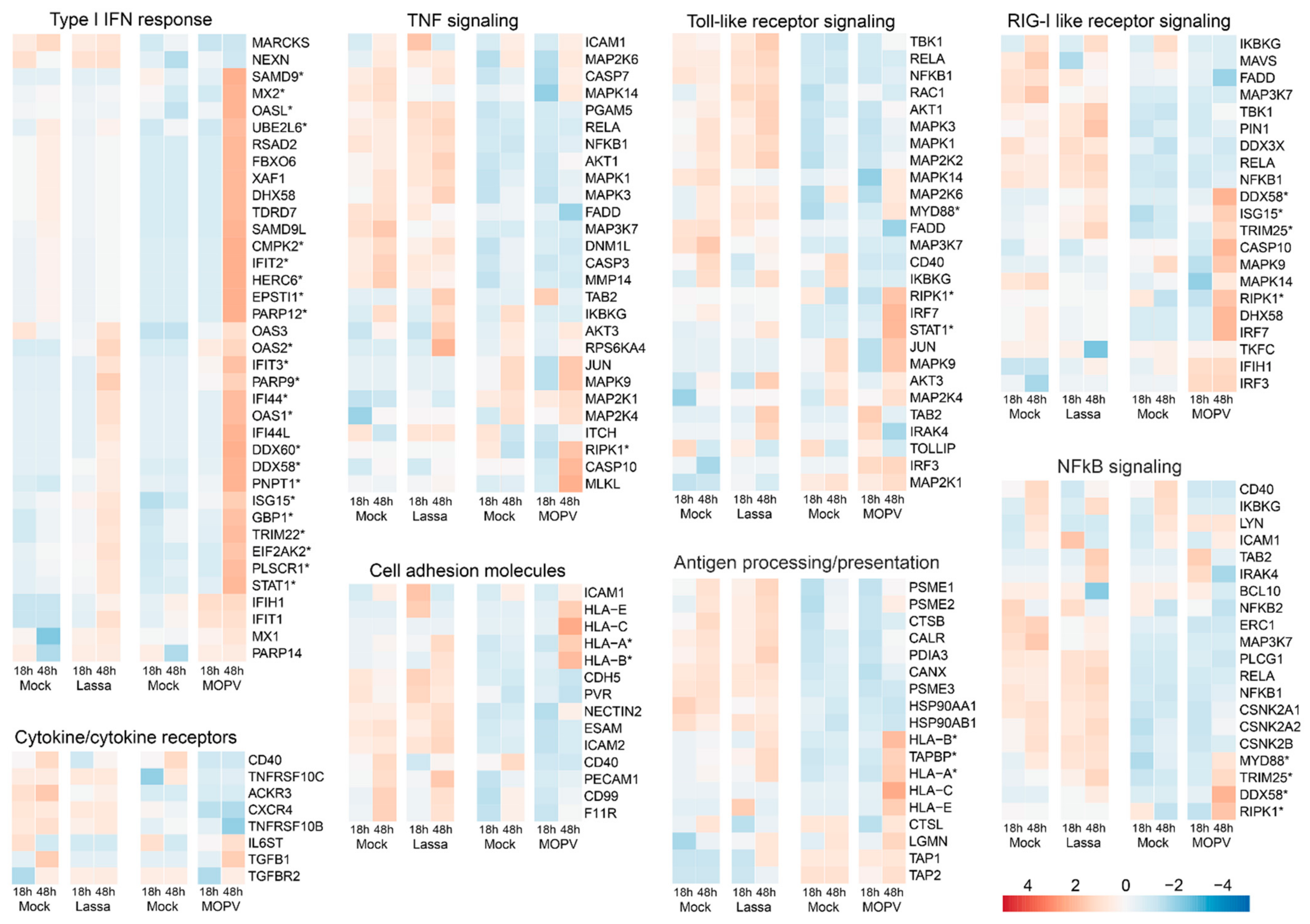
3.2. Lassa and Mopeia Viruses Induce Different Transcriptomic Profiles in HUVECs
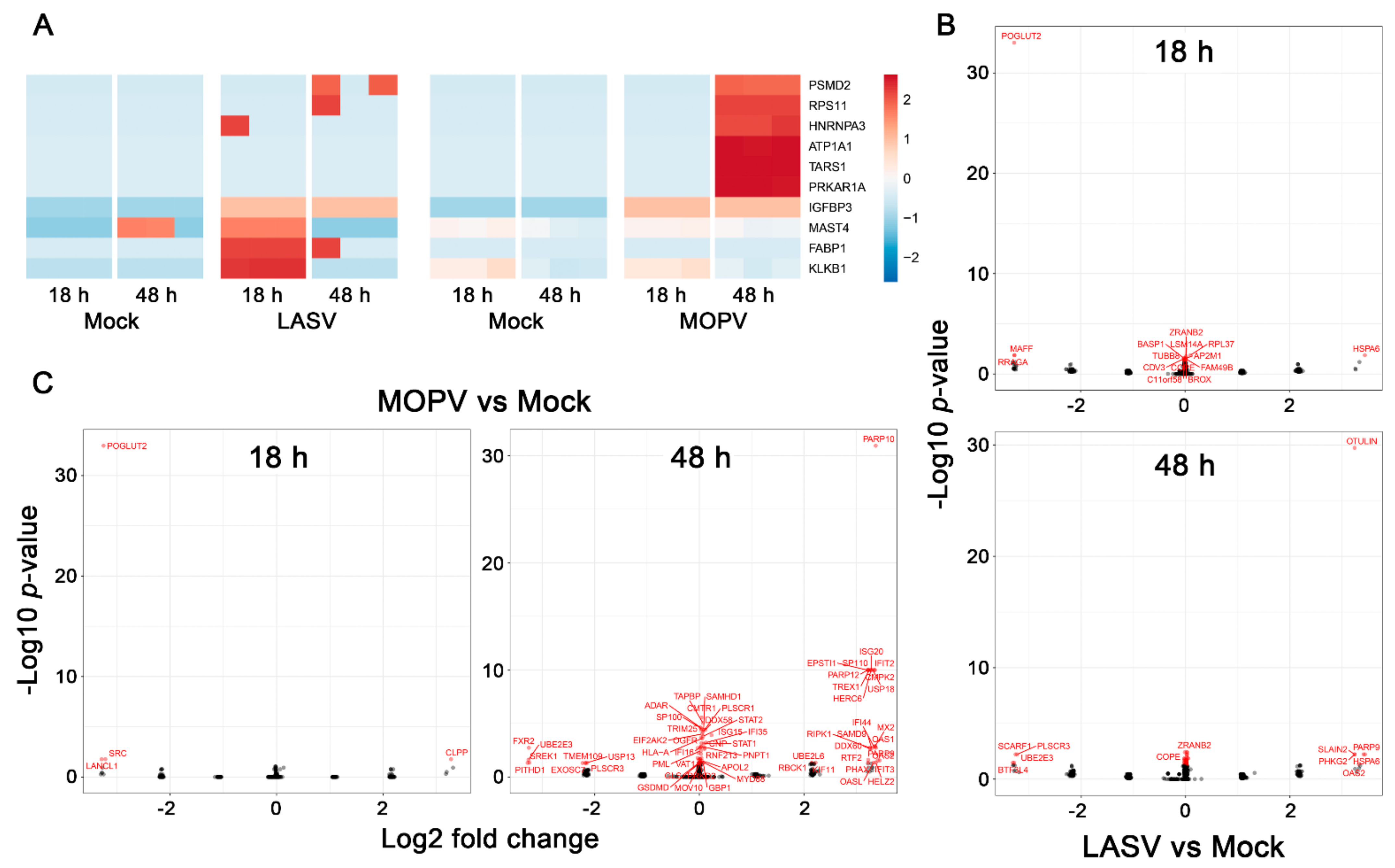
3.3. Different Proteomic Profiles Are Induced in HUVECs by LASV and MOPV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frame, J.D.; Baldwin, J.M.; Gocke, D.J.; Troup, J.M. Lassa Fever, a New Virus Disease of Man from West Africa. Am. J. Trop. Med. Hyg. 1970, 19, 670–676. [Google Scholar] [CrossRef]
- Delgado, S.; Erickson, B.R.; Agudo, R.; Blair, P.J.; Vallejo, E.; Albarino, C.G.; Vargas, J.; Comer, J.A.; Rollin, P.E.; Ksiazek, T.G.; et al. Chapare Virus, a Newly Discovered Arenavirus Isolated from a Fatal Hemorrhagic Fever Case in Bolivia. PLoS Pathog. 2008, 4, e1000047. [Google Scholar] [CrossRef] [PubMed]
- Lisieux, T.; Coimbra, M.; Nassar, E.; de Souza, L.T.M.; Ferreira, I.B.; Rocco, I.M.; Burattini, M.N. New arenavirus isolated in Brazil. Lancet 1994, 343, 391–392. [Google Scholar] [PubMed] [Green Version]
- de Mello Malta, F.; Amgarten, D.; Nastri, A.; Ho, Y.L.; Boas Casadio, L.V.; Basqueira, M.; Selegatto, G.; Cervato, M.C.; Duarte-Neto, A.N.; Higashino, H.R.; et al. Sabiá Virus-Like Mammarenavirus in Patient with Fatal Hemorrhagic Fever, Brazil, 2020. Emerg Infect. Dis 2020, 26, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Salas, R.; Pacheco, M.E.; Ramos, B.; Taibo, M.E.; Jaimes, E.; Vasquez, C.; Querales, J.; de Manzione, N.; Godoy, O.; Betancourt, A.; et al. Venezuelan haemorrhagic fever. Lancet 1991, 338, 1033–1036. [Google Scholar] [CrossRef]
- Bonthius, D.J.; Wright, R.; Tseng, B.; Barton, L.; Marco, E.; Karacay, B.; Larsen, P.D. Congenital lymphocytic choriomeningitis virus infection: Spectrum of disease. Ann. Neurol. 2007, 62, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Paweska, J.T.; Sewlall, N.H.; Ksiazek, T.G.; Blumberg, L.H.; Hale, M.J.; Lipkin, W.I.; al, e. Nosocomial outbreak of novel arenavirus infection, Southern Africa. Emerg. Infect. Dis. 2009, 15, 1598–1602. [Google Scholar] [CrossRef]
- Buchmeier, M.J.; de la Torre, J.-C.; Peters, C.J. Arenaviridae: The viruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1791–1827. [Google Scholar]
- Monath, T.P.; Newhouse, V.F.; Kemp, G.E.; Setzer, H.W.; Cacciapuoti, A. Lassa Virus Isolation from Mastomys natalensis Rodents during an Epidemic in Sierra Leone. Science 1974, 185, 263–265. [Google Scholar] [CrossRef]
- Kafetzopoulou, L.E.; Pullan, S.T.; Lemey, P.; Suchard, M.A.; Ehichioya, D.U.; Pahlmann, M.; Thielebein, A.; Hinzmann, J.; Oestereich, L.; Wozniak, D.M.; et al. Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak. Science 2019, 363, 74–77. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.G.; Shapiro, B.J.; Matranga, C.; Sealfon, R.; Lin, A.; Moses, L.M.; Folarin, O.A.; Goba, A.; Odia, I.; Ehiane, P.E.; et al. Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus. Cell 2015, 162, 738–750. [Google Scholar] [CrossRef] [Green Version]
- McCormick, J.B.; Webb, P.A.; Krebs, J.W.; Johnson, K.M.; Smith, E.S. A prospective study of the epidemiology and ecology of Lassa fever. J. Infect. Dis. 1987, 155, 437–444. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; Tomori, O.; Nasidi, A.; Perez-Oronoz, G.; Fakile, Y.; Hutwagner, L.; McCormick, J.B. Review of cases of nosocomial Lassa fever in Nigeria: The high price of poor medical practice. Br. Med. J. 1995, 311, 857–859. [Google Scholar] [CrossRef] [Green Version]
- Wolf, T.; Ellwanger, R.; Goetsch, U.; Wetzstein, N.; Gottschalk, R. Fifty years of imported Lassa fever: A systematic review of primary and secondary cases. J. Travel Med. 2020, 27, 15–20. [Google Scholar] [CrossRef]
- Eberhardt, K.A.; Mischlinger, J.; Jordan, S.; Groger, M.; Günther, S.; Ramharter, M. Ribavirin for the treatment of Lassa fever: A systematic review and meta-analysis. Int. J. Infect. Dis. 2019, 87, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Wulff, H.; McIntosh, B.; Hamner, D.; Johnson, K. Isolation of an arenavirus closely related to Lassa virus from Mastomys natalensis in south-east Africa. Bull. World Health Organ. 1977, 55, 441–444. [Google Scholar]
- Wilson, S.M.; Clegg, J.C.S. Sequence analysis of the S RNA of the African Arenavirus Mopeia: An unusual secondary structure feature in the intergenic region. Virology 1991, 180, 543–552. [Google Scholar] [CrossRef]
- Kiley, M.P.; Lange, J.V.; Johnson, K.M. Protection of rhesus monkeys from Lassa virus by immunisation with closely related arenavirus. Lancet 1979, 2, 738–745. [Google Scholar] [CrossRef]
- Baize, S.; Kaplon, J.; Faure, C.; Pannetier, D.; Georges-Courbot, M.C.; Deubel, V. Lassa virus infection of dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 2004, 172, 2861–2869. [Google Scholar] [CrossRef]
- Pannetier, D.; Faure, C.; Georges-Courbot, M.C.; Deubel, V.; Baize, S. Human macrophages, but not dendritic cells, are activated and produce type I interferons in response to Mopeia virus infection. J. Virol. 2004, 78, 10516–10524. [Google Scholar] [CrossRef] [Green Version]
- Pannetier, D.; Reynard, S.; Russier, M.; Journeaux, A.; Tordo, N.; Deubel, V.; Baize, S. Human dendritic cells infected with the non-pathogenic Mopeia virus induce stronger T-cell responses than with Lassa virus. J. Virol. 2011, 85, 8293–8306. [Google Scholar] [CrossRef] [Green Version]
- Hensley, L.; Smith, M.; Geisbert, J.; Fritz, E.; Daddario-DiCaprio, K.; Larsen, T.; Geisbert, T. Pathogenesis of lassa fever in cynomolgus macaques. Virol. J. 2011, 8, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashevich, I.S.; Maryankova, R.; Vladyko, A.S.; Nashkevic, N.; Koleda, S.; Djavani, M.; Horejsh, D.; Voitenok, N.N.; Salvato, M.S. Lassa and Mopeia virus replication in human monocytes/macrophages and in endothelial cells: Different effects on IL-8 and TNF-α gene expression. J. Med. Virol. 1999, 59, 552–560. [Google Scholar] [CrossRef]
- Baillet, N.; Reynard, S.; Perthame, E.; Hortion, J.; Journeaux, A.; Mateo, M.; Carnec, X.; Schaeffer, J.; Picard, C.; Barrot, L.; et al. Systemic viral spreading and defective host responses are associated with fatal Lassa fever in macaques. Commun. Biol. 2021, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.H.; McCormick, J.B.; Johnson, K.M.; Webb, P.A.; Komba-Kono, G.; Elliott, L.H.; Gardner, J.J. Pathologic and virologic study of fatal Lassa fever in man. Am. J. Pathol. 1982, 107, 349–356. [Google Scholar]
- Horton, L.E.; Cross, R.W.; Hartnett, J.N.; Engel, E.J.; Sakabe, S.; Goba, A.; Momoh, M.; Sandi, J.D.; Geisbert, T.W.; Garry, R.F.; et al. Endotheliopathy and Platelet Dysfunction as Hallmarks of Fatal Lassa Fever. Emerg. Infect. Dis. 2020, 26, 2625–2637. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Josse, J.; Husson, F. missMDA: A Package for Handling Missing Values in Multivariate Data Analysis. J. Stat. Softw. 2016, 70, 1–31. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wu, D.; Smyth, G.K. Camera: A competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012, 40, e133. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Gentleman, R.; Carey, V.; Bates, D.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [Green Version]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, H.; Hagan, T.; Duraisingham, S.S.; Lee, E.K.; Kwissa, M.; Rouphael, N.; Frasca, D.; Gersten, M.; Mehta, A.K.; Gaujoux, R.; et al. Systems Analysis of Immunity to Influenza Vaccination across Multiple Years and in Diverse Populations Reveals Shared Molecular Signatures. Immunity 2015, 43, 1186–1198. [Google Scholar] [CrossRef] [Green Version]
- Querec, T.D.; Akondy, R.S.; Lee, E.K.; Cao, W.; Nakaya, H.I.; Teuwen, D.; Pirani, A.; Gernert, K.; Deng, J.; Marzolf, B.; et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 2008, 10, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Fisher-Hoch, S.P.; McCormick, J.B.; Sasso, D.; Craven, R.B. Hematologic dysfunction in Lassa fever. J. Med. Virol. 1988, 26, 127–135. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; Mitchell, S.W.; Sasso, D.R.; Lange, J.V.; Ramsey, R.; McCormick, J.B. Physiological and immunologic disturbances associated with shock in a primate model of Lassa fever. J. Infect. Dis. 1987, 155, 465–474. [Google Scholar] [CrossRef]
- Tang, H.; Abouleila, Y.; Mashaghi, A. Lassa hemorrhagic shock syndrome-on-a-chip. Biotechnol. Bioeng. 2021, 118, 1405–1410. [Google Scholar] [CrossRef]
- Lander, H.M.; Grant, A.M.; Albrecht, T.; Hill, T.; Peters, C.J. Endothelial cell permeability and adherens junction disruption induced by junín virus infection. Am. J. Trop. Med. Hyg. 2014, 90, 993–1002. [Google Scholar] [CrossRef] [Green Version]
- Baccala, R.; Welch, M.J.; Gonzalez-Quintial, R.; Walsh, K.B.; Teijaro, J.R.; Nguyen, A.; Ng, C.T.; Sullivan, B.M.; Zarpellon, A.; Ruggeri, Z.M.; et al. Type I interferon is a therapeutic target for virus-induced lethal vascular damage. Proc. Natl. Acad. Sci. USA 2014, 111, 8925–8930. [Google Scholar] [CrossRef] [Green Version]
- Gowen, B.; Julander, J.; London, N.; Wong, M.-H.; Larson, D.; Morrey, J.; Li, D.; Bray, M. Assessing changes in vascular permeability in a hamster model of viral hemorrhagic fever. Virol. J. 2010, 7, 240. [Google Scholar] [CrossRef] [Green Version]
- Oestereich, L.; Lüdtke, A.; Ruibal, P.; Pallasch, E.; Kerber, R.; Rieger, T.; Wurr, S.; Bockholt, S.; Pérez-Girón, J.V.; Krasemann, S.; et al. Chimeric Mice with Competent Hematopoietic Immunity Reproduce Key Features of Severe Lassa Fever. PLoS Pathog. 2016, 12, e1005656. [Google Scholar] [CrossRef]
- Downs, I.; Shaia, C.; Zeng, X.; Johnson, J.; Hensley, L.; Saunders, D.; Rossi, F.; Kashman, K.; Esham, H.; Gregory, M.; et al. Natural History of Aerosol Induced Lassa Fever in Non-Human Primates. Viruses 2020, 12, 593. [Google Scholar] [CrossRef]
- Walker, D.H.; Johnson, K.M.; Lange, J.V.; Gardner, J.J.; Kiley, M.P.; McCormick, J.B. Experimental infection of Rhesus monkeys with Lassa virus and a closely related Arenavirus, Mozambique virus. J. Infect. Dis. 1982, 146, 360–368. [Google Scholar] [CrossRef]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 2396–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynard, S.; Russier, M.; Fizet, A.; Carnec, X.; Baize, S. Exonuclease Domain of the Lassa Virus Nucleoprotein Is Critical to Avoid Retinoic Acid-Inducible Gene I Signaling and To Inhibit the Innate Immune Response. J. Virol 2014, 88, 13923–13927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnec, X.; Baize, S.; Reynard, S.; Diancourt, L.; Caro, V.; Tordo, N.; Bouloy, M. Lassa virus nucleoprotein mutants generated by reverse genetics induce robust type I IFN response in human dendritic cells and macrophages. J. Virol. 2011, 85, 12093–12097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baize, S.; Pannetier, D.; Faure, C.; Marianneau, P.; Marendat, I.; Georges-Courbot, M.C.; Deubel, V. Role of interferons in the control of Lassa virus replication in human dendritic cells and macrophages. Microbes Infect. 2006, 8, 1193–1422. [Google Scholar] [CrossRef]
- Carnec, X.; Mateo, M.; Page, A.; Reynard, S.; Hortion, J.; Picard, C.; Yekwa, E.; Barrot, L.; Barron, S.; Vallve, A.; et al. A Vaccine Platform against Arenaviruses Based on a Recombinant Hyperattenuated Mopeia Virus Expressing Heterologous Glycoproteins. J. Virol. 2018, 92, e02230-17. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, J.; Carnec, X.; Reynard, S.; Mateo, M.; Picard, C.; Pietrosemoli, N.; Dillies, M.-A.; Baize, S. Lassa virus activates myeloid dendritic cells but suppresses their ability to stimulate T cells. PLoS Pathog. 2018, 14, e1007430. [Google Scholar] [CrossRef]
- Schaeffer, J.; Reynard, S.; Carnec, X.; Pietrosemoli, N.; Dillies, M.A.; Baize, S. Non-Pathogenic Mopeia Virus Induces More Robust Activation of Plasmacytoid Dendritic Cells than Lassa Virus. Viruses 2019, 11, 287. [Google Scholar] [CrossRef] [Green Version]
- Pannetier, D.; Reynard, S.; Russier, M.; Carnec, X.; Baize, S. Production of CXC and CC chemokines by human antigen-presenting cells in response to Lassa virus or closely related immunogenic viruses, and in cynomolgus monkeys with Lassa fever. PLoS Neglected Trop. Dis. 2014, 8, e2637. [Google Scholar] [CrossRef]
- Christensen, J.E.; de Lemos, C.; Moos, T.; Christensen, J.P.; Thomsen, A.R. CXCL10 is the key ligand for CXCR3 on CD8+ effector T cells involved in immune surveillance of the lymphocytic choriomeningitis virus-infected central nervous system. J. Immunol. 2006, 176, 4235–4243. [Google Scholar] [CrossRef] [Green Version]
- Foley, J.F.; Yu, C.-R.; Solow, R.; Yacobucci, M.; Peden, K.W.C.; Farber, J.M. Roles for CXC chemokine ligand 10 and 11 in recruiting CD4+ T cells to HIV-1-infected monocyte-derived macrophages, dendritic cells, and lymph nodes. J. Immunol. 2005, 174, 4892–4900. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.-F.; Lai, S.-L.; Chen, J.-P.; Sung, J.-M.; Lin, Y.-L.; Wu-Hsieh, B.A.; Gerard, C.; Luster, A.; Liao, F. Both CXCR3 and CXCL10/IFN-inducible protein 10 are required for resistance to primary infection by dengue virus. J. Immunol. 2006, 177, 1855–1863. [Google Scholar] [CrossRef] [Green Version]
- Mahanty, S.; Bausch, D.G.; Thomas, R.L.; Goba, A.; Bah, A.; Peters, C.J.; Rollin, P.E. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute Lassa fever. J. Infect. Dis. 2001, 183, 1713–1721. [Google Scholar] [CrossRef] [Green Version]
- Harryvan, T.J.; de Lange, S.; Hawinkels, L.; Verdegaal, E.M.E. The ABCs of Antigen Presentation by Stromal Non-Professional Antigen-Presenting Cells. Int. J. Mol. Sci. 2021, 23, 137. [Google Scholar] [CrossRef]
- Baize, S.; Marianneau, P.; Loth, P.; Reynard, S.; Journeaux, A.; Chevallier, M.; Tordo, N.; Deubel, V.; Contamin, H. Early and strong immune responses are associated with control of viral replication and recovery in Lassa virus-infected cynomolgus monkeys. J. Virol. 2009, 83, 5890–5903. [Google Scholar] [CrossRef] [Green Version]
- Strampe, J.; Asogun, D.A.; Speranza, E.; Pahlmann, M.; Soucy, A.; Bockholt, S.; Pallasch, E.; Becker-Ziaja, B.; Duraffour, S.; Bhadelia, N.; et al. Factors associated with progression to death in patients with Lassa fever in Nigeria: An observational study. Lancet Infect. Dis. 2021, 21, 876–886. [Google Scholar] [CrossRef]
- Hendel, A.; Hsu, I.; Granville, D.J. Granzyme B releases vascular endothelial growth factor from extracellular matrix and induces vascular permeability. Lab. Investig. A J. Tech. Methods Pathol. 2014, 94, 716–725. [Google Scholar] [CrossRef] [Green Version]
- Lawler, P.R.; Lawler, J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2012, 2, a006627. [Google Scholar] [CrossRef]
- Li, Q.; Fu, X.; Yuan, J.; Han, S. Contribution of Thrombospondin-1 and -2 to Lipopolysaccharide-Induced Acute Respiratory Distress Syndrome. Mediat. Inflamm. 2021, 2021, 8876484. [Google Scholar] [CrossRef]
- Mariappan, V.; Adikari, S.; Shanmugam, L.; Easow, J.M.; Balakrishna Pillai, A. Expression dynamics of vascular endothelial markers: Endoglin and syndecan-1 in predicting dengue disease outcome. Transl. Res. J. Lab. Clin. Med. 2021, 232, 121–141. [Google Scholar] [CrossRef]
- Varejckova, M.; Gallardo-Vara, E.; Vicen, M.; Vitverova, B.; Fikrova, P.; Dolezelova, E.; Rathouska, J.; Prasnicka, A.; Blazickova, K.; Micuda, S.; et al. Soluble endoglin modulates the pro-inflammatory mediators NF-κB and IL-6 in cultured human endothelial cells. Life Sci. 2017, 175, 52–60. [Google Scholar] [CrossRef]
- Jerkic, M.; Letarte, M. Increased endothelial cell permeability in endoglin-deficient cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 3678–3688. [Google Scholar] [CrossRef] [Green Version]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- Garland, P.; Quraishe, S.; French, P.; O’Connor, V. Expression of the MAST family of serine/threonine kinases. Brain Res. 2008, 1195, 12–19. [Google Scholar] [CrossRef]
- Tanaka, M.; Furuhashi, M.; Moniwa, N.; Maeda, T.; Takizawa, H.; Matsumoto, M.; Sakai, A.; Higashiura, Y.; Gocho, Y.; Koyama, M.; et al. Significance of urinary fatty acid-binding protein 4 level as a possible biomarker for the identification of minimal change disease in patents with nephrotic-range proteinuria. BMC Nephrol. 2020, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yayama, K.; Kunimatsu, N.; Teranishi, Y.; Takano, M.; Okamoto, H. Tissue kallikrein is synthesized and secreted by human vascular endothelial cells. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2003, 1593, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Golias, C.; Charalabopoulos, A.; Stagikas, D.; Charalabopoulos, K.; Batistatou, A. The kinin system--bradykinin: Biological effects and clinical implications. Multiple role of the kinin system--bradykinin. Hippokratia 2007, 11, 124–128. [Google Scholar] [PubMed]
- Taylor, S.L.; Wahl-Jensen, V.; Copeland, A.M.; Jahrling, P.B.; Schmaljohn, C.S. Endothelial cell permeability during hantavirus infection involves factor XII-dependent increased activation of the kallikrein-kinin system. PLoS Pathog. 2013, 9, e1003470. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. eLife 2020, 9, e57555. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Zhang, A.; Du, Y.; Fang, M.; Minze, L.J.; Liu, Y.J.; Li, X.C.; Zhang, Z. Identification of poly(ADP-ribose) polymerase 9 (PARP9) as a noncanonical sensor for RNA virus in dendritic cells. Nat. Commun 2021, 12, 2681. [Google Scholar] [CrossRef] [PubMed]
- Merrick, B.A.; Dhungana, S.; Williams, J.G.; Aloor, J.J.; Peddada, S.; Tomer, K.B.; Fessler, M.B. Proteomic profiling of S-acylated macrophage proteins identifies a role for palmitoylation in mitochondrial targeting of phospholipid scramblase 3. Mol. Cell. Proteom. 2011, 10, M110.006007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Ortiz, Z.G.; Pendergraft, W.F., 3rd; Prasad, A.; Byrne, M.H.; Iram, T.; Blanchette, C.J.; Luster, A.D.; Hacohen, N.; El Khoury, J.; Means, T.K. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat. Immunol. 2013, 14, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Deb, G.; Wingelhofer, B.; Amaral, F.M.R.; Maiques-Diaz, A.; Chadwick, J.A.; Spencer, G.J.; Williams, E.L.; Leong, H.-S.; Maes, T.; Somervaille, T.C.P. Pre-clinical activity of combined LSD1 and mTORC1 inhibition in MLL-translocated acute myeloid leukaemia. Leukemia 2020, 34, 1266–1277. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Schneider, M.; Williamson, D.B.; Ito, A.; Takeuchi, M.; Handford, P.A.; Haltiwanger, R.S. Two novel protein O-glucosyltransferases that modify sites distinct from POGLUT1 and affect Notch trafficking and signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E8395–E8402. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xiao, Q.; Chen, X.; Tong, S.; Sun, J.; Lv, R.; Wang, S.; Gou, Y.; Tan, L.; Xu, J.; et al. LanCL1 protects prostate cancer cells from oxidative stress via suppression of JNK pathway. Cell Death Dis. 2018, 9, 197. [Google Scholar] [CrossRef] [Green Version]
- Kondo, H.; Matsumura, T.; Kaneko, M.; Inoue, K.; Kosako, H.; Ikawa, M.; Takahama, Y.; Ohigashi, I. PITHD1 is a proteasome-interacting protein essential for male fertilization. J. Biol. Chem. 2020, 295, 1658–1672. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merabet, O.; Pietrosemoli, N.; Perthame, E.; Armengaud, J.; Gaillard, J.-C.; Borges-Cardoso, V.; Daniau, M.; Legras-Lachuer, C.; Carnec, X.; Baize, S. Infection of Human Endothelial Cells with Lassa Virus Induces Early but Transient Activation and Low Type I IFN Response Compared to the Closely-Related Nonpathogenic Mopeia Virus. Viruses 2022, 14, 652. https://doi.org/10.3390/v14030652
Merabet O, Pietrosemoli N, Perthame E, Armengaud J, Gaillard J-C, Borges-Cardoso V, Daniau M, Legras-Lachuer C, Carnec X, Baize S. Infection of Human Endothelial Cells with Lassa Virus Induces Early but Transient Activation and Low Type I IFN Response Compared to the Closely-Related Nonpathogenic Mopeia Virus. Viruses. 2022; 14(3):652. https://doi.org/10.3390/v14030652
Chicago/Turabian StyleMerabet, Othmann, Natalia Pietrosemoli, Emeline Perthame, Jean Armengaud, Jean-Charles Gaillard, Virginie Borges-Cardoso, Maïlys Daniau, Catherine Legras-Lachuer, Xavier Carnec, and Sylvain Baize. 2022. "Infection of Human Endothelial Cells with Lassa Virus Induces Early but Transient Activation and Low Type I IFN Response Compared to the Closely-Related Nonpathogenic Mopeia Virus" Viruses 14, no. 3: 652. https://doi.org/10.3390/v14030652