
Genomic Epidemiology of SARS-CoV-2 in Seychelles, 2020–2021

 , , , , , add
Show full author list
, , , , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Study Site and Samples
2.3. Laboratory Procedures
2.3.1. RNA Extraction, cDNA Synthesis and Amplification
2.3.2. Oxford Nanopore Library Preparation and Sequencing
2.3.3. SARS-CoV-2 Genome Consensus Assembly
2.3.4. Lineage and VOC Assignment
2.3.5. Global Comparison Sequences
2.3.6. Phylogenetic Reconstruction
2.3.7. Estimation of Virus Importation and Exportation into Seychelles
2.3.8. Statistical Analysis
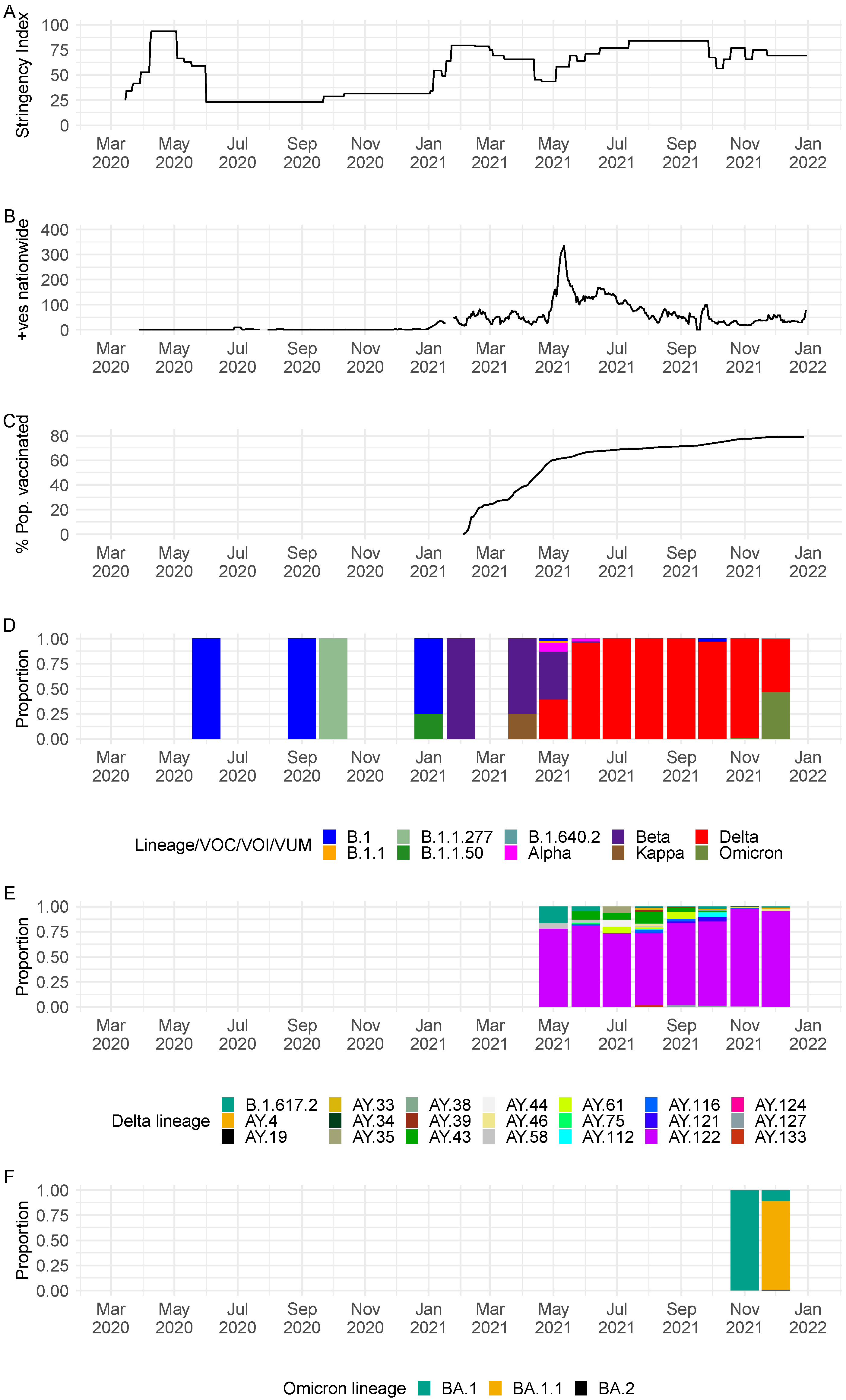
3. Results
3.1. Sequenced COVID-19 Cases in Seychelles
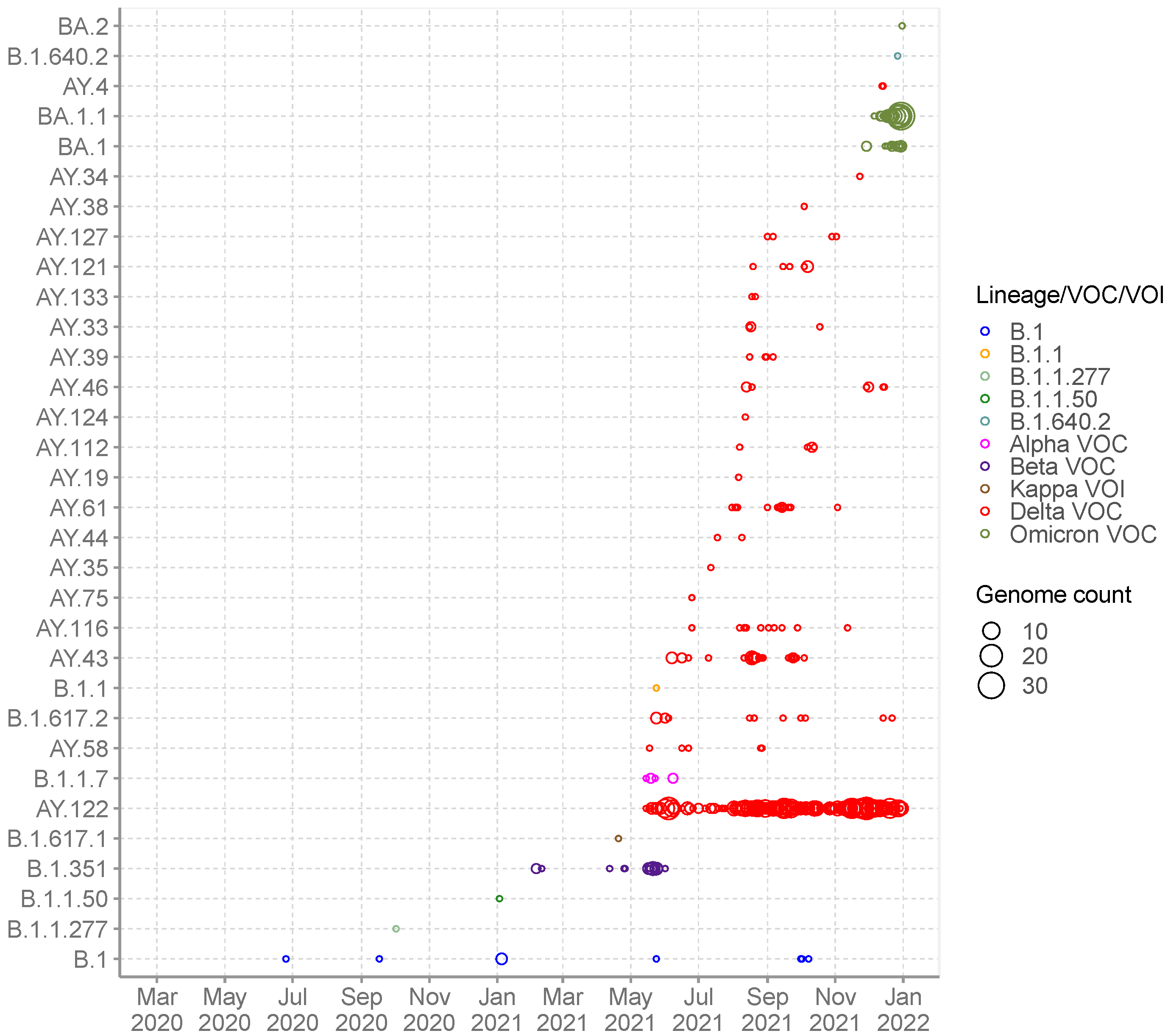
3.2. SARS-CoV-2 Lineages Circulating in Seychelles
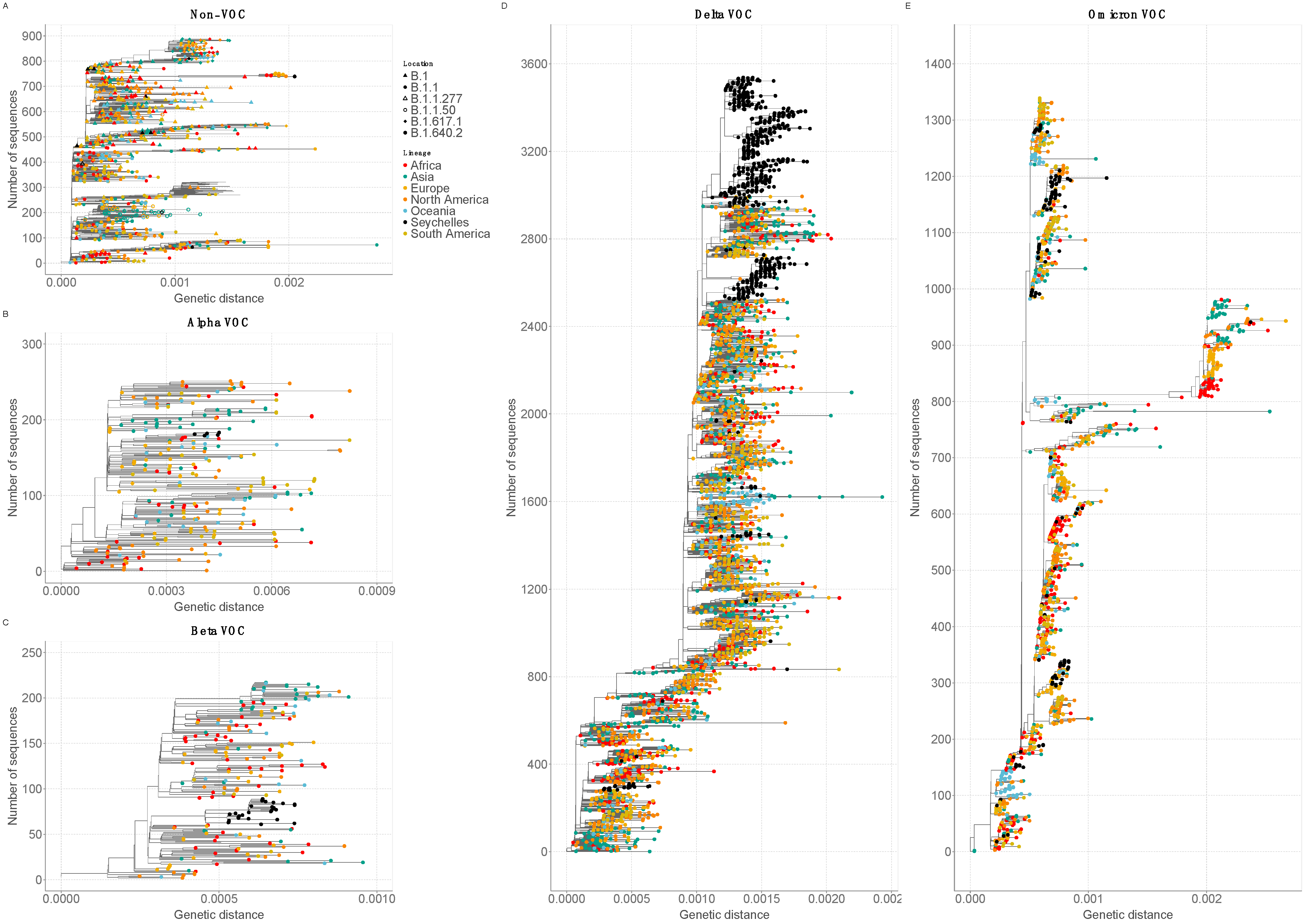
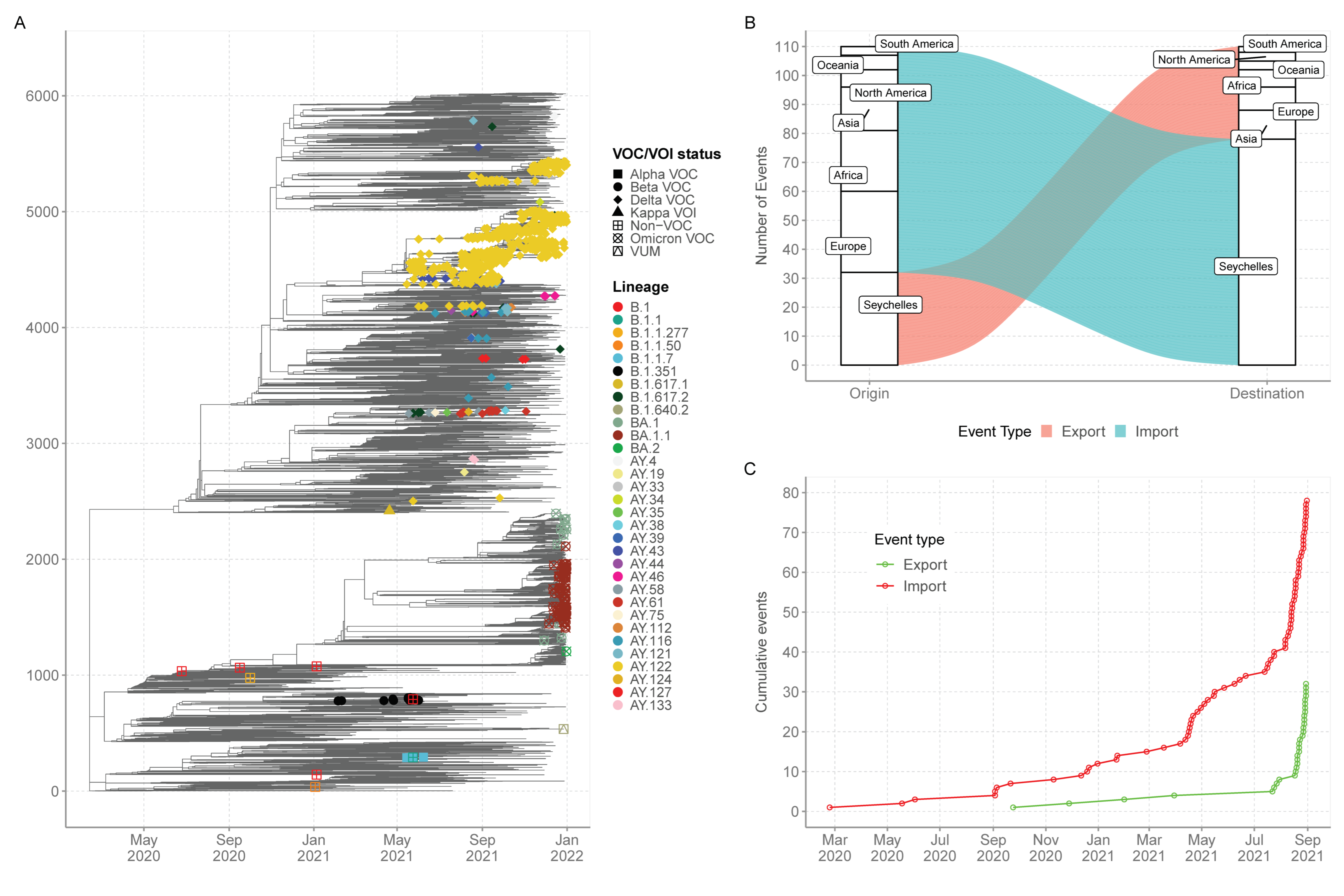
3.3. Phylogenetic Clustering of Seychelles Sequences
3.4. SARS-CoV-2 Diversity and Mutational Analysis in Seychelles Genomes
3.5. Export and Import of SARS-CoV-2 Lineages in Seychelles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Bureau of Statistics Seychelles. Population and Vital Statistics. Mid 2021 Population Estimates. 31 August 2021. Available online: https://www.nbs.gov.sc/downloads/data-acquisition-census/population-and-vital-statistics/2021 (accessed on 29 March 2022).
- WHO. WHO Coronavirus (COVID-19) Dashboard. 2021. Available online: https://covid19.who.int/ (accessed on 16 June 2021).
- Centres for Disease Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions. 16 March 2022. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html (accessed on 6 June 2022).
- WHO. Tracking SARS-CoV-2 Variants. 2021. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 27 August 2021).
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2013. Available online: http://www.R-project.org/ (accessed on 9 November 2021).
- Wilkinson, E.; Giovanetti, M.; Tegally, H.; San, J.E.; Lessells, R.; Cuadros, D.; Martin, D.P.; Rasmussen, D.A.; Zekri, A.R.N.; Sangare, A.K.; et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Science 2021, 374, 423–431. [Google Scholar] [CrossRef]
- Agoti, C.; Ochola-Oyier, L.I.; Mohammed, K.S.; Lambisia, A.; de Laurent, Z.; Morobe, J.; Mburu, M.; Omuoyo, D.; Ongera, E.; Ndwiga, L.; et al. Transmission networks of SARS-CoV-2 in coastal Kenya during the first two waves: A retrospective genomic study. medRxiv 2021. [Google Scholar] [CrossRef]
- Ritchie, H.; Ortiz-Ospina, E.; Beltekian, D.; Mathieu, E.; Hasell, J.; Macdonald, B.; Giattino, C.; Appel, C.; LRG and MR. Coronavirus Pandemic (COVID-19). Our World in Data. 2020. Available online: https://ourworldindata.org/coronavirus (accessed on 1 March 2022).
- Yamasoba, D.; Kimura, I.; Nasser, H.; Morioka, Y.; Nao, N.; Ito, J.; Uriu, K.; Tsuda, M.; Zahradnik, J.; Shirakawa, K.; et al. Virological characteristics of SARS-CoV-2 BA.2 variant. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lyngse, F.P.; Kirkeby, C.T.; Denwood, M.; Christiansen, L.E.; Mølbak, K.; Møller, C.H.; Skov, R.L.; Krause, T.G.; Rasmussen, M.; Sieber, R.N.; et al. Transmission of SARS-CoV-2 Omicron VOC subvariants BA.1 and BA.2: Evidence from Danish Households. medRxiv 2022. [Google Scholar] [CrossRef]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Yin, M.T.; Sobieszczyk, M.E.; et al. Antibody Evasion Properties of SARS-CoV-2 Omicron Sublineages. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef] [PubMed]
- McCormick, K.D.; Jacobs, J.L.; Mellors, J.W. The emerging plasticity of SARS-CoV-2. Science 2021, 371, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Harari, D.; Chiaravalli, J.; Meyer, B.; Rudich, Y.; Li, C.; Marton, I.; et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 2021, 6, 1188–1198. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef]
- Gong, S.Y.; Chatterjee, D.; Richard, J.; Prévost, J.; Tauzin, A.; Gasser, R.; Bo, Y.; Vézina, D.; Goyette, G.; Gendron-Lepage, G.; et al. Contribution of single mutations to selected SARS-CoV-2 emerging variants Spike antigenicity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Ren, X.; Storey, M.; Hadfield, J.; Jelley, L.; Jefferies, S.; Sherwood, J.; Paine, S.; Huang, S.; Douglas, J.; et al. Genomic epidemiology reveals transmission patterns and dynamics of SARS-CoV-2 in Aotearoa New Zealand. Nat. Commun. 2020, 11, 6351. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.A.; Lebarbenchon, C.; Atyame, C.; Hafsia, S.; Jaffar-Bandjee, M.C.; Yemadje-Menudier, L.; Tanaka, S.; Meilhac, O.; Mavingui, P. Genomic insights into early SARS-CoV-2 strains isolated in Reunion Island. medRxiv 2021. [Google Scholar] [CrossRef]
- Agoti, C.N.; Githinji, G.; Mohammed, K.S.; Lambisia, A.W.; de Laurent, Z.R.; Mburu, M.W.; Ong’era, E.M.; Morobe, J.M.; Otieno, E.; Azali, H.A.; et al. Detection of SARS-CoV-2 variant 501Y.V2 in Comoros Islands in January 2021. Wellcome Open Res. 2021, 6, 192. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Sequenced | Samples Not Sequenced | Total | |||
|---|---|---|---|---|---|
| Number (n = 1056) | Population Proportion (%) | Number (n = 242) | Population Proportion (%) | ||
| Sex | |||||
| Female | 560 | 53.0 | 120 | 49.6 | 680 |
| Male | 473 | 44.8 | 102 | 42.1 | 575 |
| Unknown | 23 | 2.2 | 20 | 8.3 | 43 |
| Age | |||||
| Mean | 33.4 (18.3) | - | 34.3 (20.4) | - | |
| Median | 32 | - | 34 | - | |
| Min, Max | 0, 98 | - | 0, 89 | - | |
| Missing | 18 | 1.7 | 9 | 3.7 | 27 |
| Age distribution | |||||
| 0–9 | 106 | 10.0 | 37 | 15.3 | 143 |
| 10–19 | 132 | 12.5 | 28 | 11.6 | 160 |
| 20–29 | 215 | 20.4 | 30 | 12.4 | 245 |
| 30–39 | 225 | 21.3 | 42 | 17.4 | 267 |
| 40–49 | 146 | 13.8 | 45 | 18.6 | 191 |
| 50–59 | 127 | 12.0 | 21 | 8.7 | 148 |
| 60–69 | 58 | 5.5 | 19 | 7.9 | 77 |
| 70–79 | 14 | 1.3 | 6 | 2.5 | 20 |
| >80 | 15 | 1.4 | 5 | 2.1 | 20 |
| Travel information | |||||
| Yes | 3 | 0.3 | 8 | 3.3 | 11 |
| No | 1053 | 99.7 | 234 | 96.7 | 1287 |
| Symptoms | |||||
| Asymptomatic | 37 | 3.5 | 31 | 12.8 | 68 |
| Symptomatic | 273 | 25.9 | 52 | 21.5 | 325 |
| Deceased | 3 | 0.3 | 4 | 1.7 | 7 |
| Missing | 738 | 69.9 | 155 | 64.0 | 893 |
| Non-VOC/VOI/VOC/VUM | Lineage | Freq | Proportion (%) | Earliest Date | Description |
|---|---|---|---|---|---|
| Non-VOC/VOI | B.1 | 9 | 0.9 | 1 January 2020 | Predominantly found in Europe |
| B.1.1 | 1 | 0.1 | 1 January 2020 | Predominantly found in Europe | |
| B.1.1.277 | 1 | 0.1 | 7 March 2020 | Predominantly found in Europe | |
| B.1.1.50 | 1 | 0.1 | 29 March 2020 | Predominantly found in Israel and Palestine | |
| VUM | B.1.640.2 | 1 | 0.1 | 15 October 2021 | Predominantly found in France |
| Kappa VOI | B.1.617.1 | 1 | 0.1 | 3 March 2020 | Kappa variant of interest, predominantly found in India lineage with 484Q |
| Alpha VOC | B.1.1.7 | 5 | 0.5 | 7 February 2020 | Alpha variant of concern, first identified in UK |
| Beta VOC | B.1.351 | 29 | 2.7 | 27 March 2020 | Beta variant of concern, first identified in South Africa |
| Delta VOC | B.1.617.2 | 13 | 1.2 | 15 April 2020 | Predominantly found in India |
| AY.4 | 2 | 0.2 | 3 August 2020 | Predominantly found in UK | |
| AY.19 | 1 | 0.1 | 7 April 2021 | Predominantly found in South Africa | |
| AY.33 | 4 | 0.4 | 13 June 2020 | Lineage circulating mostly in Belgium, Denmark, France, Netherlands, Germany | |
| AY.34 | 1 | 0.1 | 18 November 2020 | Predominantly found in UK | |
| AY.35 | 1 | 0.1 | 21 August 2020 | Predominantly found in lineage with spike E484Q circulating in USA | |
| AY.38 | 1 | 0.1 | 27 March 2021 | Predominantly found in in South Africa | |
| AY.39 | 4 | 0.4 | 14 January 2021 | Predominantly found in USA | |
| AY.43 | 33 | 3.1 | 21 August 2021 | Predominantly found in European | |
| AY.44 | 2 | 0.2 | 11 May 2020 | Predominantly found in USA | |
| AY.46 | 8 | 0.8 | 15 October 2021 | Predominantly found in Africa | |
| AY.58 | 5 | 0.5 | 16 March 2021 | Predominantly found in Italy | |
| AY.61 | 15 | 1.4 | 7 January 2021 | Predominantly found in Italy | |
| AY.75 | 1 | 0.1 | 6 January 2021 | Predominantly found in USA | |
| AY.112 | 5 | 0.5 | 5 December 2020 | Predominantly found in India | |
| AY.116 | 11 | 1.0 | 21 January 2021 | Africa lineage | |
| AY.121 | 7 | 0.7 | 24 January 2021 | Predominantly found in Turkey | |
| AY.122 | 742 | 70.3 | 7 September 2020 | European lineage | |
| AY.124 | 1 | 0.1 | 9 January 2021 | Predominantly found in Portugal and other European countries | |
| AY.127 | 4 | 0.4 | 10 December 2020 | Predominantly found in India | |
| AY.133 | 2 | 0.2 | 10 February 2021 | Predominantly found in France | |
| Omicron VOC | BA.1 | 18 | 1.7 | 10 September 2021 | Predominantly found in UK |
| BA.1.1 | 126 | 11.9 | 13 September 2021 | Predominantly found in USA | |
| BA.2 | 1 | 0.1 | 17 November 2021 | Predominantly found in UK |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morobe, J.M.; Pool, B.; Marie, L.; Didon, D.; Lambisia, A.W.; Makori, T.; Mohammed, K.S.; de Laurent, Z.R.; Ndwiga, L.; Mburu, M.W.; et al. Genomic Epidemiology of SARS-CoV-2 in Seychelles, 2020–2021. Viruses 2022, 14, 1318. https://doi.org/10.3390/v14061318
Morobe JM, Pool B, Marie L, Didon D, Lambisia AW, Makori T, Mohammed KS, de Laurent ZR, Ndwiga L, Mburu MW, et al. Genomic Epidemiology of SARS-CoV-2 in Seychelles, 2020–2021. Viruses. 2022; 14(6):1318. https://doi.org/10.3390/v14061318
Chicago/Turabian StyleMorobe, John Mwita, Brigitte Pool, Lina Marie, Dwayne Didon, Arnold W. Lambisia, Timothy Makori, Khadija Said Mohammed, Zaydah R. de Laurent, Leonard Ndwiga, Maureen W. Mburu, and et al. 2022. "Genomic Epidemiology of SARS-CoV-2 in Seychelles, 2020–2021" Viruses 14, no. 6: 1318. https://doi.org/10.3390/v14061318