
Influenza A(H1N1)pdm09 Virus Alters Expression of Endothelial Factors in Pulmonary Vascular Endothelium in Rats
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Virus
2.3. Experimental Influenza Virus Infection
2.4. Immunohistochemistry
2.5. Determination of PAI-1 and tPA Concentration in Rat Plasma
2.6. Statistics
3. Results
3.1. Body Weights; Clinical Observations
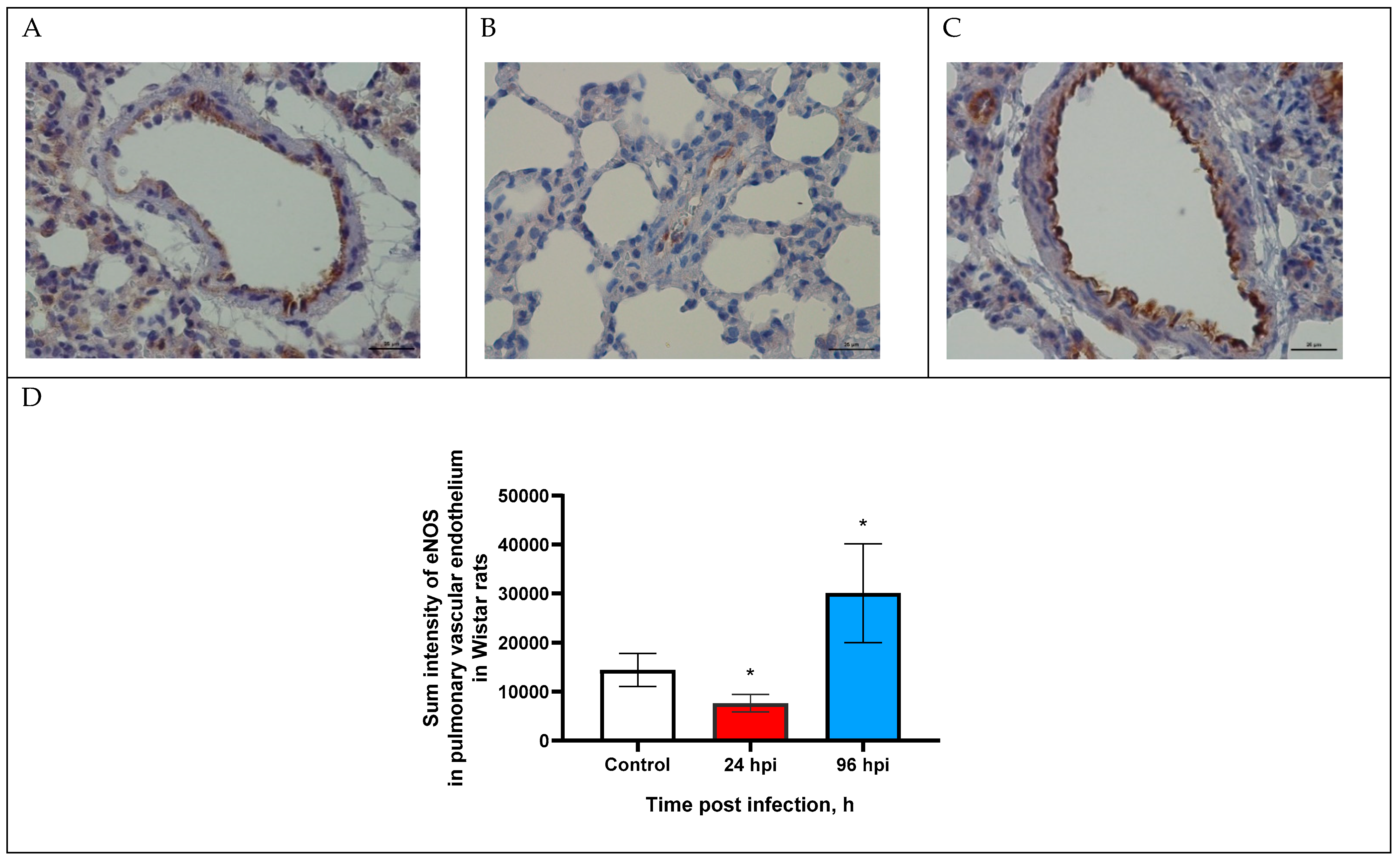
3.2. eNOS Expression in Pulmonary Vascular Endothelium
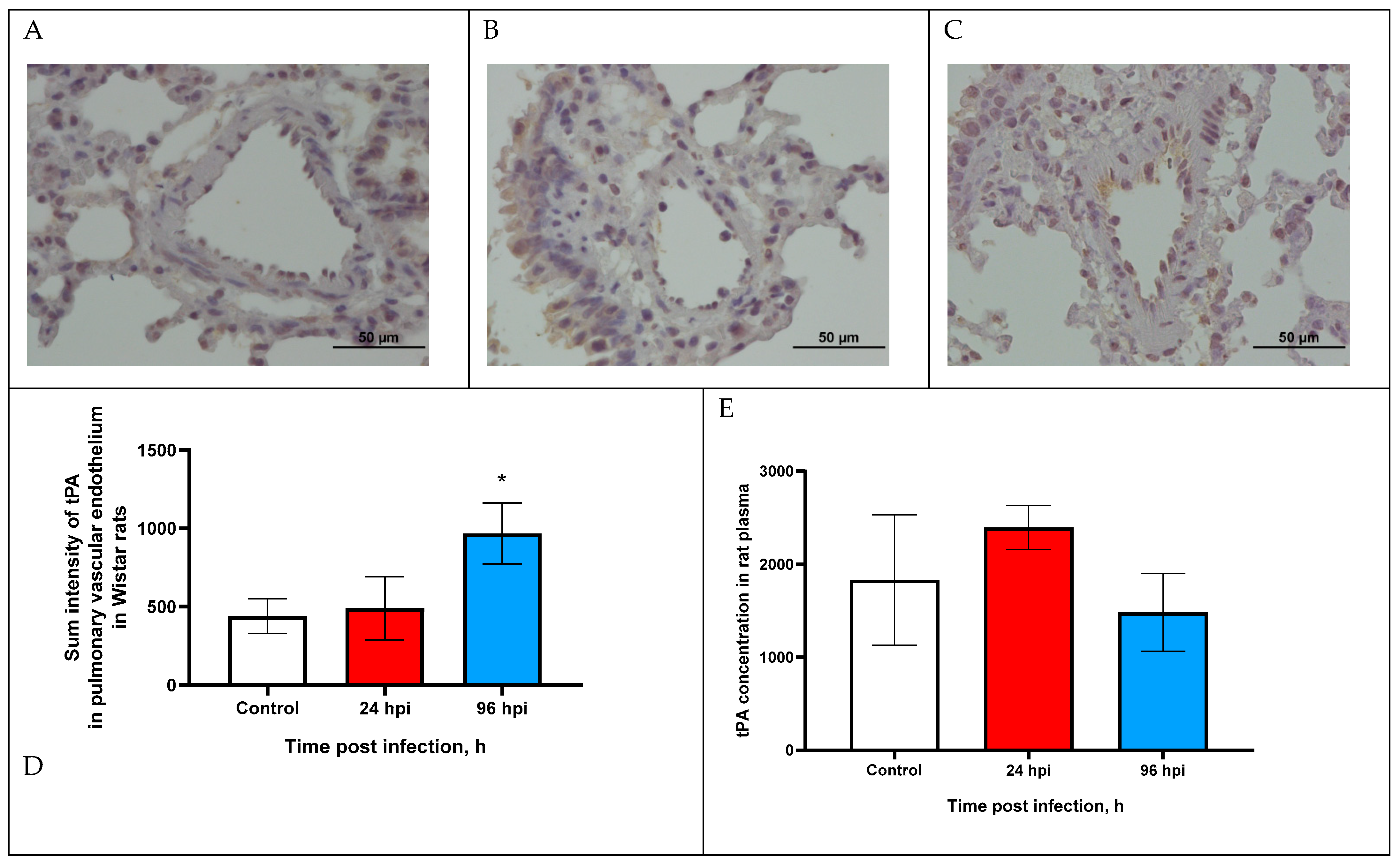
3.3. tPA Expression in Pulmonary Vascular Endothelium and in Blood Plasma of Rats
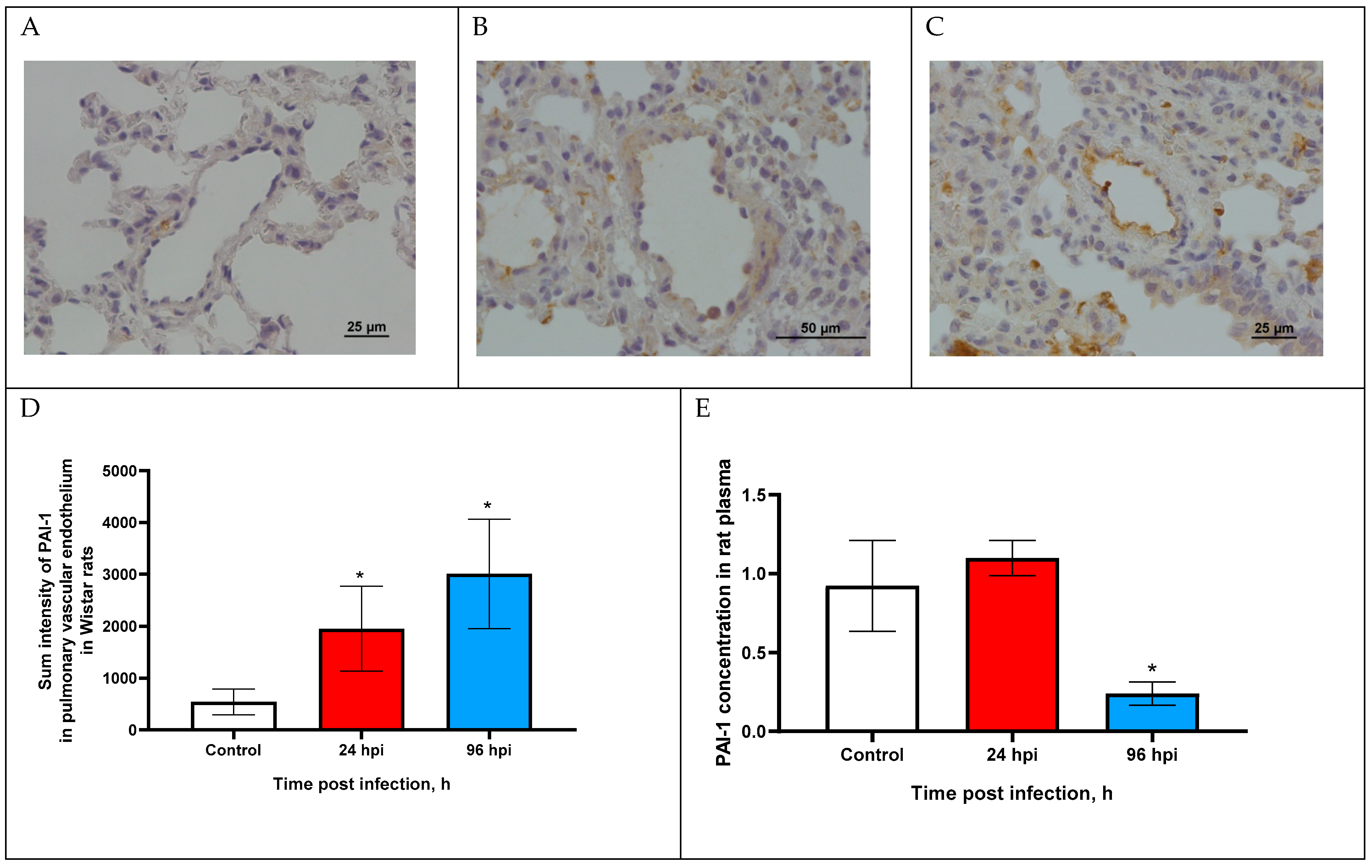
3.4. PAI-1 Expression in Pulmonary Vascular Endothelium and in Blood Plasma of Rats
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Programmes and Projects. Immunization, Vaccines and Biologicals: Influenza. 2008. Available online: http://www.who.int/immunization/topics/influenza/en/index.html (accessed on 13 August 2012).
- Zeng, H.; Goldsmith, C.S.; Maines, T.R.; Belser, J.A.; Gustin, K.M.; Pekosz, A.; Zaki, S.R.; Katz, J.M.; Tumpey, T.M. Tropism and infectivity of influenza virus, including highly pathogenic avian H5N1 virus, in ferret tracheal differentiated primary epithelial cell cultures. J. Virol. 2013, 87, 2597–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Smith, C.W.; Katkin, J.P.; Thurmon, L.M.; Xu, X.; Mendoza, L.H.; Ballantyne, C.M. Endothelial alpha 2,6-linked sialic acid inhibits VCAM-1-dependent adhesion under flow conditions. J. Immunol. 1999, 163, 2867–2876. [Google Scholar] [PubMed]
- Cioffi, D.L.; Pandey, S.; Alvarez, D.F.; Cioffi, E.A. Terminal sialic acids are an important determinant of pulmonary endothelial barrier integrity. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L1067–L1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.M.; Darwish, I.; Lee, W.L. Endothelial activation and dysfunction in the pathogenesis of influenza A virus infection. Virulence 2013, 4, 537–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, K.R.; Veldhuis Kroeze, E.J.; Reperant, L.A.; Richard, M.; Kuiken, T. Influenza virus and endothelial cells: A species specific relationship. Front. Microbiol. 2014, 5, 653. [Google Scholar] [CrossRef] [Green Version]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef]
- van Asten, L.; Harmsen, C.N.; Stoeldraijer, L.; Klinkenberg, D.; Teirlinck, A.C.; De Lange, M.M.; van de Kassteele, J.; van Gageldonk-Lafeber, A.B.; van den Hof, S.; van der Hoek, W. Excess Deaths during Influenza and Coronavirus Disease and Infection-Fatality Rate for Severe Acute Respiratory Syndrome Coronavirus 2, the Netherlands. Emerg. Infect. Dis. 2021, 27, 411–420. [Google Scholar] [CrossRef]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, N.; Gran, J.M.; Schanzer, D.; Cowling, P.B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Vallance, P. Nitric oxide in the human cardiovascular system—SKB lecture 1997. Br. J. Clin. Pharmacol. 1998, 45, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Alvarez, R.A.; Berra, L.; Gladwin, M.T. Home Nitric Oxide Therapy for COVID-19. Am. J. Respir. Crit. Care Med. 2020, 202, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Pahor, M.; Incalzi, R.A. Plasminogen activator inhibitor-1 (PAI-1): A key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Vaughan, D.E.; Parikh, S.H.; Grodzinsky, A.J.; Libby, P.; Lark, M.W.; Lee, R.T. Regulation of matrix metalloproteinases and plasminogen activator inhibitor-1 synthesis by plasminogen in cultured human vascular smooth muscle cells. Circ. Res. 1996, 78, 44–49. [Google Scholar] [CrossRef]
- Tjärnlund-Wolf, A.; Brogren, H.; Lo, E.H.; Wang, X. Plasminogen activator inhibitor-1 and thrombotic cerebrovascular diseases. Stroke 2012, 43, 2833–2839. [Google Scholar] [CrossRef]
- Dittmann, M.; Hoffmann, H.H.; Scull, M.A.; Gilmore, R.H.; Bell, K.L.; Ciancanelli, M.; Wilson, S.J.; Crotta, S.; Yu, Y.; Flatley, B.; et al. A serpin shapes the extracellular environment to prevent influenza A virus maturation. Cell 2015, 160, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.; Lin, L.; Hu, K. NF-κB and tPA Signaling in Kidney and Other Diseases. Cells 2020, 9, 1348. [Google Scholar] [CrossRef]
- Tse, L.V.; Marcano, V.C.; Huang, W.; Pocwierz, M.S.; Whittaker, G.R. Plasmin-mediated activation of pandemic H1N1 influenza virus hemagglutinin is independent of the viral neuraminidase. J. Virol. 2013, 87, 5161–5169. [Google Scholar] [CrossRef] [Green Version]
- Tsinzerling, V.A.; Vorob’ev, S.L.; Zarubaev, V.V.; Dedov, V.A.; Belyaevskaya, S.V.; Esaulenko, E.V.; Dedov, V.A.; Grudinin, M.P.; Buzitskaia, Z.V.; Pisareva, M.M.; et al. Pathogenic aspects of influenza during the epidemics caused by H1N1v virus in 2009–2010. Arkhiv. Patologii. 2011, 73, 21–25. (In Russian) [Google Scholar]
- Marchenko, V.A.; Barashkova, S.V.; Zelinskaya, I.A.; Toropova, Y.G.; Sorokin, E.V.; Zhilinskaya, I.N. Modeling influenza virus infection in matureWistar rats. Probl. Virol. 2020, 65, 159–166. (In Russian) [Google Scholar] [CrossRef]
- Mary, J.; Selgrade, D.; Doerfler, D.; Gilmour, M.I. Kinetic profile of influenza virus infection in three rat strains. Comp. Med. 2003, 53, 293–298. [Google Scholar]
- Ibanes, J.D.; Morgan, K.T.; Burleson, G.R. Histopathological Changes in the Upper Respiratory Tract of F344 Rats Following Infection with a Rat-adapted Influenza Virus. Vet. Pathol. 1996, 33, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, V.; Zelinskaya, I.; Toropova, Y.; Shmakova, T.; Podyacheva, E.; Lioznov, D.; Zhilinskaya, I.N. Influenza A Virus Causes Histopathological Changes and Impairment in Functional Activity of Blood Vessels in Different Vascular Beds. Viruses 2022, 14, 396. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef] [Green Version]
- Niethamer, T.K.; Stabler, C.T.; Leach, J.P.; Zepp, J.A.; Morley, M.P.; Babu, A.; Zhou, S.; Morrisey, E.E. Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. Elife 2020, 9, e53072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latreille, E.; Lee, W.L. Interactions of Influenza and SARS-CoV-2 with the Lung Endothelium: Similarities, Differences, and Implications for Therapy. Viruses 2021, 13, 161. [Google Scholar] [CrossRef]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta,, A.; Paone, S.; et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef] [Green Version]
- Stroes, E.; Hijmering, M.; van Zandvoort, M.; Wever, R.; Rabelink, T.J.; van Faassen, E.E. Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett. 1998, 438, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases--the role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Hekman, C.M.; Loskutoff, D.J. Kinetic analysis of the interactions between plasminogen activator inhibitor 1 and both urokinase and tissue plasminogen activator. Arch. Biochem. Biophys. 1988, 262, 199–210. [Google Scholar] [CrossRef]
- Boudier, C.; Gils, A.; Declerck, P.J.; Bieth, J.G. The conversion of active to latent plasminogen activator inhibitor-1 is an energetically silent event. Biophys J. 2005, 88, 2848–2854. [Google Scholar] [CrossRef]
- Vaughan, D.E. PAI-1 and atherothrombosis. J. Thromb. Haemost. 2005, 3, 1879–1883. [Google Scholar] [CrossRef] [PubMed]
- Stals, M.; Grootenboers, M.; van Guldener, C.; Kaptein, F.H.; Braken, S.J.; Chen, Q.; Chu, G.; van Driel, E.M.; del Sol, A.I.; de Jonge, E.; et al. Risk of thrombotic complications in influenza versus COVID-19 hospitalized patients. Res. Pract. Thromb. Haemost. 2021, 5, 412–420. [Google Scholar] [CrossRef]
- McCarthy, Z.; Xu, S.; Rahman, A.; Bragazzi, N.; Corrales-Medina, V.F.; Lee, J.; Seet, B.T.; Neame, D.; Thommes, E.; Heffernan, J.; et al. Modelling the linkage between influenza infection and cardiovascular events via thrombosis. Sci. Rep. 2020, 10, 14264. [Google Scholar] [CrossRef]
- Patrassi, G.M.; Sartori, M.T.; Viero, M.L.; Boscaro, M.; Boeri, G.; Girolami, A. Venous thrombosis and tissue plasminogen activator release deficiency: A family study. Blood Coagul. Fibrinolysis 1991, 2, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Binder, B.R.; Christ, G.; Gruber, F.; Grubic, N.; Hufnagl, P.; Krebs, M.; Mihaly, J.; Prager, G.W. Plasminogen activator inhibitor 1: Physiological and pathophysiological roles. News Physiol. Sci. 2002, 17, 56–61. [Google Scholar] [CrossRef]
- Karimi, Z.; Oskouie, A.A.; Rezaei, F.; Ajaminejad, F.; Marashi, S.M.; Azad, T.M. The effect of influenza virus on the metabolism of peripheral blood mononuclear cells with a metabolomics approach. J. Med. Virol. 2022, 94, 4383–4392. [Google Scholar] [CrossRef]
- Huarte, M.; Sanz-Ezquerro, J.J.; Roncal, F.; Ortín, J.; Nieto, A. PA subunit from influenza virus polymerase complex interacts with a cellular protein with homology to a family of transcriptional activators. J. Virol. 2001, 75, 8597–8604. [Google Scholar] [CrossRef] [Green Version]
- Azarenok, A.A.; Eropkina, E.M.; Prochukhanova, A.R.; Shaldzhyan, A.A.; Kozlova, N.M.; Kozeletskaya, K.N.; Zhilinskaya, I.N. The influenza viruses and their surface proteins impact on the metabolism of human blood vessel endothelial cells. Probl. Virol. 2013, 3, 25–27. (In Russian) [Google Scholar]
- Zhilinskaya, I.N.; Prochukhanova, A.R.; Fadeev, A.V.; Kharchenko, E.P. Homology between segments of human hemostatic proteins and proteins of viruses which cause acute respiratory infections or diseases with similar symptoms. J. Infectol. 2017, 9, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Shiota, M.; Kido, H.; Ohtsu YKashiwagi, T.; Iwahashi, J.; Hamada, N.; Mizoue, K.; Tsumura, N.; Kato, H.; Toyoda, T. Influenza virus RNA polymerase PA subunit is a novel serine protease with Ser624 at the active site. Genes Cells 2001, 6, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, P.; Smedje, H.; Eriksson, N.; Kohnke, H.; Daniilidou, M.; Öhman, I.; Yue, Q.-Y.; Cavalli, M.; Wadelius, C.; Magnusson, P.K.; et al. Pandemrix-induced narcolepsy is associated with genes related to immunity and neuronal survival. EBioMedicine 2019, 40, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ambati, A.; Lin, L.; Bonvalet, M.; Partinen, M.; Ji, X.; Maecker, H.T.; Mignot, E.J.-M. Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc. Natl. Acad. Sci. USA 2018, 115, E12323–E12332. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchenko, V.; Mukhametdinova, D.; Amosova, I.; Lioznov, D.; Zhilinskaya, I. Influenza A(H1N1)pdm09 Virus Alters Expression of Endothelial Factors in Pulmonary Vascular Endothelium in Rats. Viruses 2022, 14, 2518. https://doi.org/10.3390/v14112518
Marchenko V, Mukhametdinova D, Amosova I, Lioznov D, Zhilinskaya I. Influenza A(H1N1)pdm09 Virus Alters Expression of Endothelial Factors in Pulmonary Vascular Endothelium in Rats. Viruses. 2022; 14(11):2518. https://doi.org/10.3390/v14112518
Chicago/Turabian StyleMarchenko, Vladimir, Darya Mukhametdinova, Irina Amosova, Dmitry Lioznov, and Irina Zhilinskaya. 2022. "Influenza A(H1N1)pdm09 Virus Alters Expression of Endothelial Factors in Pulmonary Vascular Endothelium in Rats" Viruses 14, no. 11: 2518. https://doi.org/10.3390/v14112518