

Virological and Genetic Characterization of the Unusual Avian Influenza H14Nx Viruses in the Northern Asia
 , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
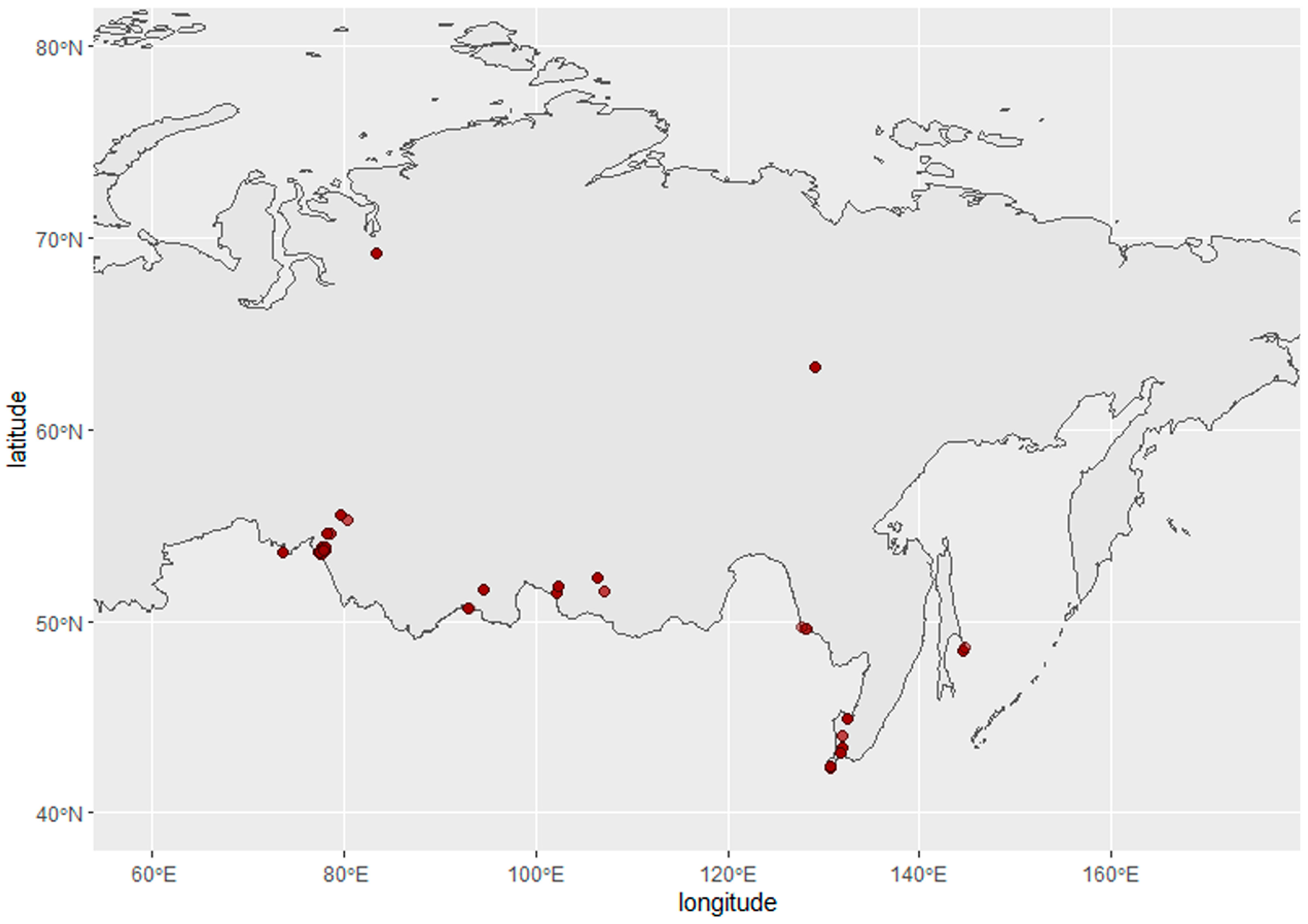
2.1. Sampling
2.2. Virus Detection, Isolation, and RNA Extraction
2.3. Virological Characteristics
2.3.1. Cell Cultures
2.3.2. Hemagglutination Inhibition and Neutralization Assay
2.3.3. Determination of Susceptibility to Neuraminidase Inhibitors
2.3.4. Receptor Specificity
2.4. Sequencing and Genetic Characterization
3. Results
3.1. Virus Isolation
3.2. Virological Characteristics
3.2.1. Growth in Cell Culture, Antigenic Analysis and Neuraminidase Inhibition Tests
3.2.2. Receptor Specificity
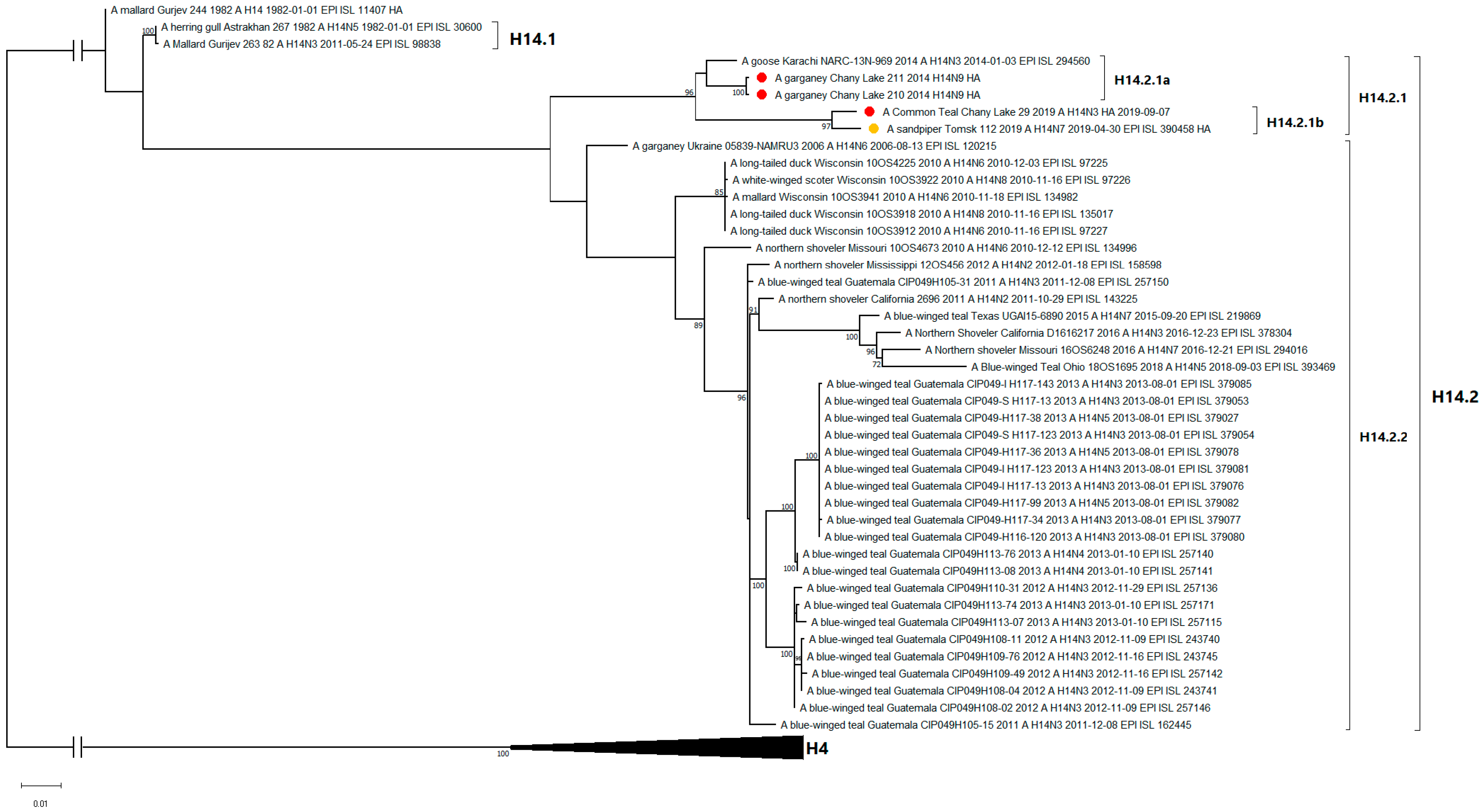
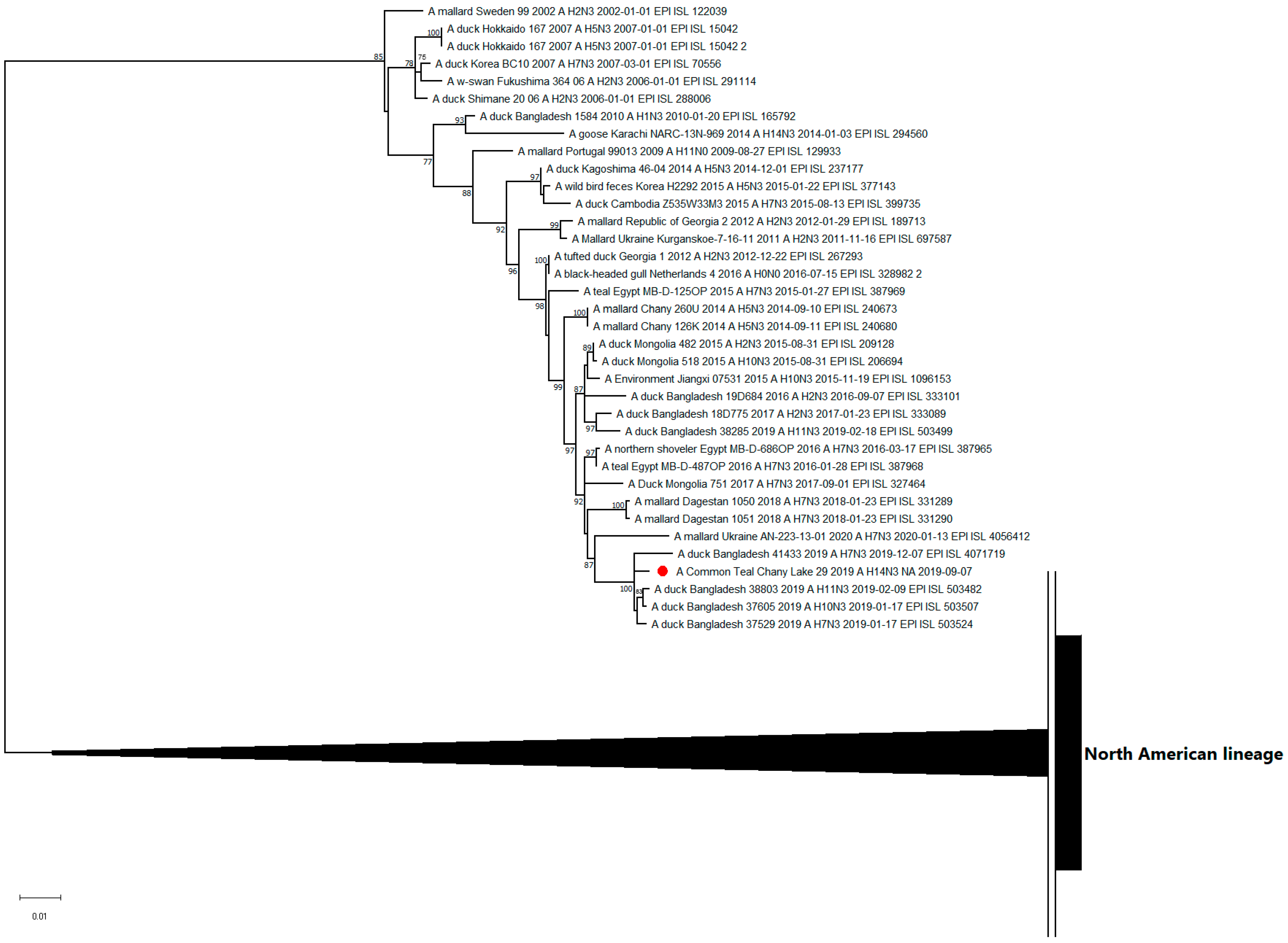
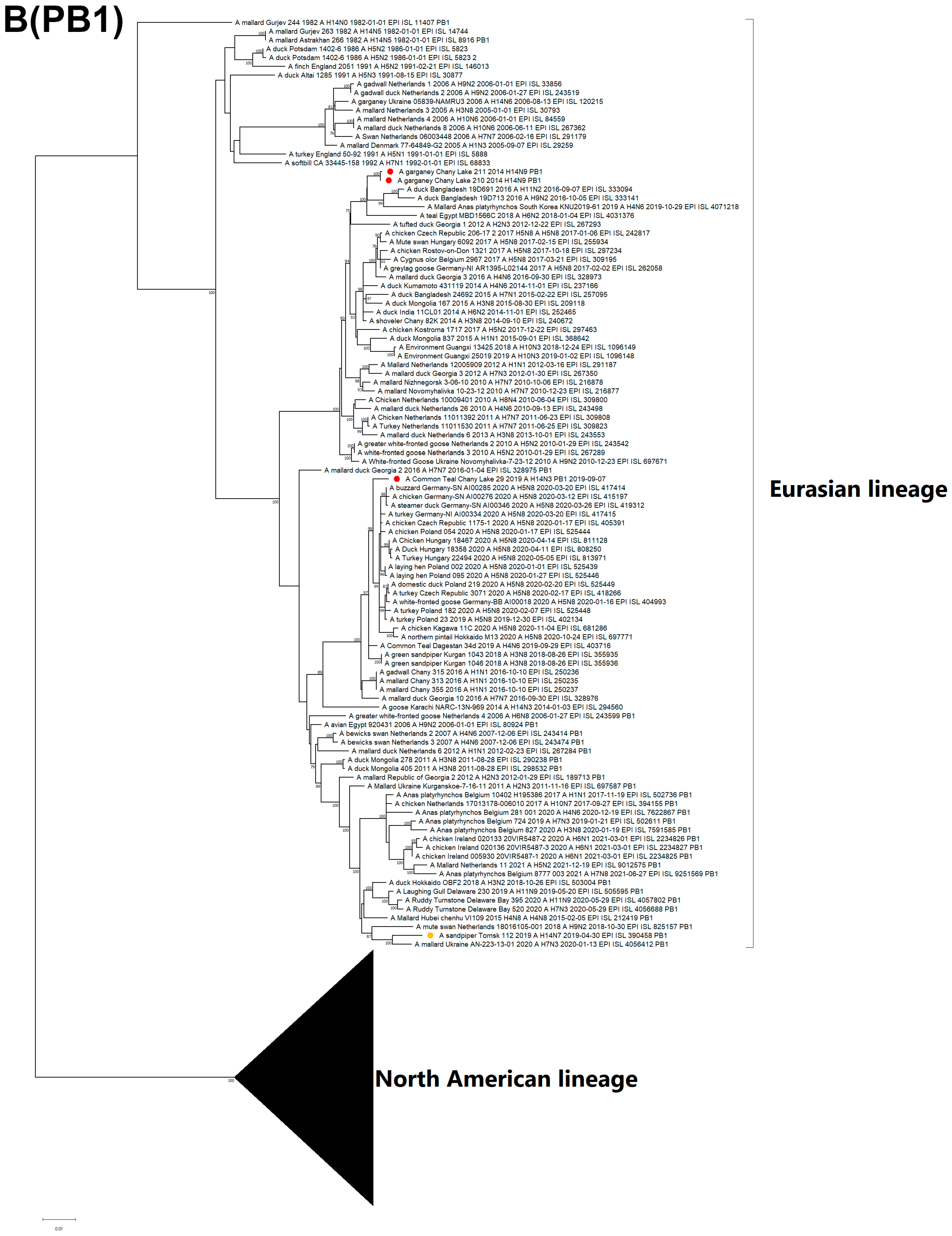
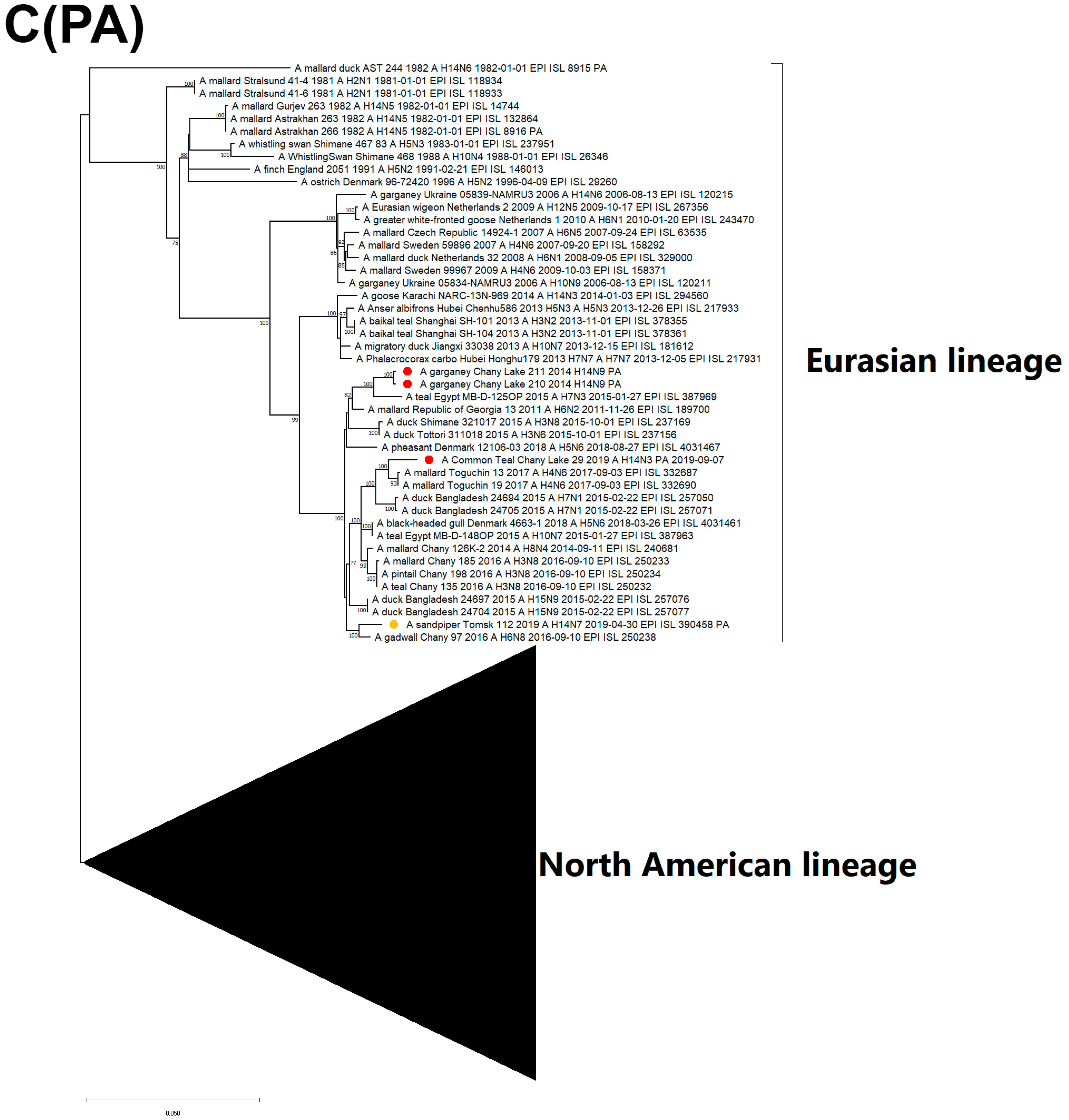
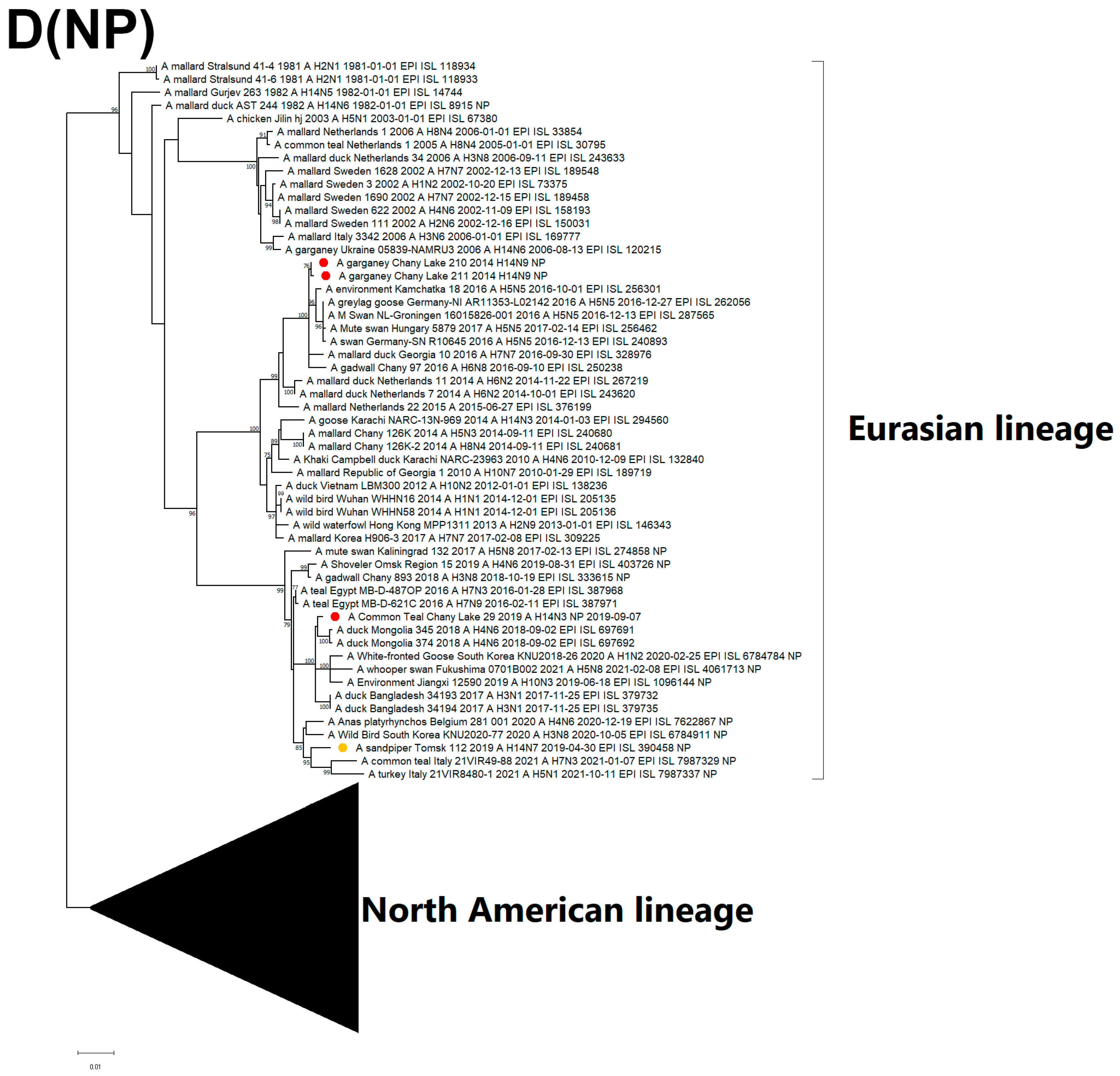
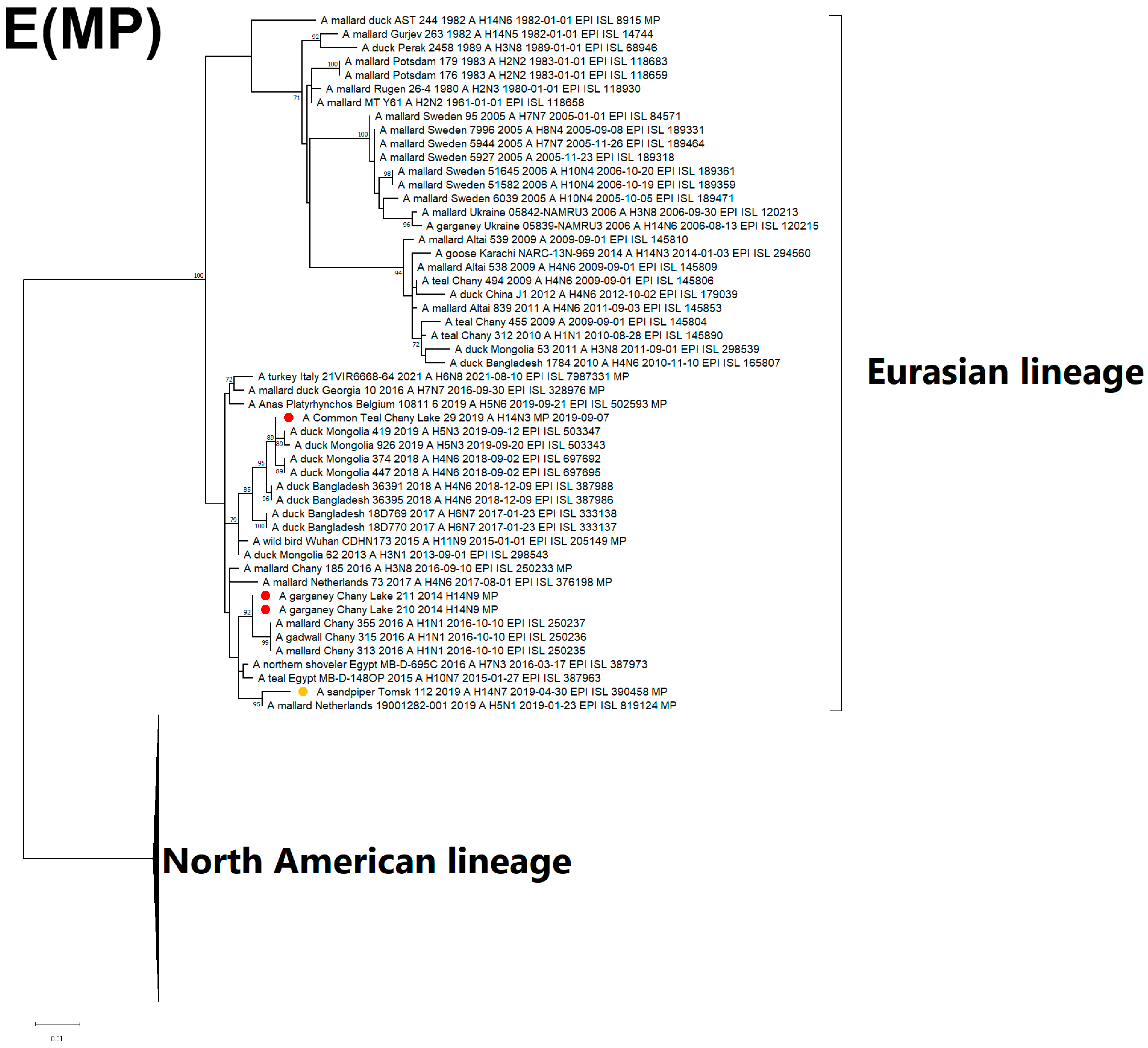
3.3. Genetic Characterization and Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campos, A.C.A.; Góes, L.G.B.; Moreira-Soto, A.; de Carvalho, C.; Ambar, G.; Sander, A.-L.; Fischer, C.; Ruckert da Rosa, A.; Cardoso de Oliveira, D.; Kataoka, A.P.G.; et al. Bat Influenza A(HL18NL11) Virus in Fruit Bats, Brazil. Emerg. Infect. Dis. 2019, 25, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.A.; Chen, L.-M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A Distinct Lineage of Influenza A Virus from Bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhagen, J.H.; Eriksson, P.; Leijten, L.; Blixt, O.; Olsen, B.; Waldenström, J.; Ellström, P.; Kuiken, T. Host Range of Influenza A Virus H1 to H16 in Eurasian Ducks Based on Tissue and Receptor Binding Studies. J. Virol. 2021, 95, e01873-20. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.J.; Yabsley, M.J.; Hernandez, S.M. A Review of Pathogen Transmission at the Backyard Chicken–Wild Bird Interface. Front. Vet. Sci. 2020, 7, 539925. [Google Scholar] [CrossRef]
- Caliendo, V.; Lewis, N.S.; Pohlmann, A.; Baillie, S.R.; Banyard, A.C.; Beer, M.; Brown, I.H.; Fouchier, R.A.M.; Hansen, R.D.E.; Lameris, T.K.; et al. Transatlantic Spread of Highly Pathogenic Avian Influenza H5N1 by Wild Birds from Europe to North America in 2021. Sci. Rep. 2022, 12, 11729. [Google Scholar] [CrossRef] [PubMed]
- Kawaoka, Y.; Yamnikova, S.; Chambers, T.M.; Lvov, D.K.; Webster, R.G. Molecular Characterization of a New Hemagglutinin, Subtype H14, of Influenza a Virus. Virology 1990, 179, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, V.Y.; Goncharova, N.I.; Tran, T.N.; Trinh, K.S.; Nguyen, N.Q.; Gavrilova, E.V.; Maksyutov, R.A.; Ryzhikov, A.B. Overview of the Epizootiological Situation on Highly Pathogenic Avian Influenza Virus in Russia in 2019. Probl. Part. Danger. Infect. 2020, 2, 31–37. [Google Scholar] [CrossRef]
- Fries, A.C.; Nolting, J.M.; Danner, A.; Webster, R.G.; Bowman, A.S.; Krauss, S.; Slemons, R.D. Evidence for the Circulation and Inter-Hemispheric Movement of the H14 Subtype Influenza A Virus. PLoS ONE 2013, 8, e59216. [Google Scholar] [CrossRef] [Green Version]
- Del Rosario, J.M.M.; da Costa, K.A.S.; Asbach, B.; Ferrara, F.; Ferrari, M.; Wells, D.A.; Mann, G.S.; Ameh, V.O.; Sabeta, C.T.; Banyard, A.C.; et al. Exploiting Pan Influenza A and Pan Influenza B Pseudotype Libraries for Efficient Vaccine Antigen Selection. Vaccines 2021, 9, 741. [Google Scholar] [CrossRef]
- Ramey, A.M.; Poulson, R.L.; González-Reiche, A.S.; Perez, D.R.; Stallknecht, D.E.; Brown, J.D. Genomic Characterization of H14 Subtype Influenza A Viruses in New World Waterfowl and Experimental Infectivity in Mallards (Anas Platyrhynchos). PLoS ONE 2014, 9, e95620. [Google Scholar] [CrossRef] [Green Version]
- Brauer, R.; Chen, P. Influenza Virus Propagation in Embryonated Chicken Eggs. J. Vis. Exp. JoVE 2015, 97, e52421. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. WHO Manual on Animal Influenza Diagnosis and Surveillance; World Health Organization: Geneva, Switzerland, 2002. [Google Scholar]
- Killian, M.L. Hemagglutination Assay for Influenza Virus. In Animal Influenza Virus; Spackman, E., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; pp. 3–9. ISBN 978-1-4939-0758-8. [Google Scholar]
- Ashmarin, I.; Vorob’ev, A. Statisticheskie Metody v Mikrobiologicheskikh Issledovaniyakh; Medgiz: Leningrad, Russia, 1962; 180p. [Google Scholar]
- World Health Organization. WHO global influenza surveillance network: Manual for the laboratory diagnosis and virological surveillance of influenza. In Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Potier, M.; Mameli, L.; Bélisle, M.; Dallaire, L.; Melançon, S.B. Fluorometric Assay of Neuraminidase with a Sodium (4-Methylumbelliferyl-Alpha-D-N-Acetylneuraminate) Substrate. Anal. Biochem. 1979, 94, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Gubareva, L.V.; Webster, R.G.; Hayden, F.G. Detection of Influenza Virus Resistance to Neuraminidase Inhibitors by an Enzyme Inhibition Assay. Antivir. Res. 2002, 53, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St. George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-Reaction Genomic Amplification Accelerates Sequencing and Vaccine Production for Classical and Swine Origin Human Influenza A Viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mine, J.; Tsunekuni, R.; Tanikawa, T.; Uchida, Y.; Dubovitskiy, N.; Derko, A.; Sobolev, I.; Shestopalov, A.; Sharshov, K.; Saito, T. Genetics of Japanese H5N8 High Pathogenicity Avian Influenza Viruses Isolated in Winter 2020–2021 and Their Genetic Relationship with Avian Influenza Viruses in Siberia. Transbound. Emerg. Dis. 2022, 69, e2195–e2213. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; Maio, N.D.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Gambaryan, A.S.; Matrosovich, T.Y.; Boravleva, E.Y.; Lomakina, N.F.; Yamnikova, S.S.; Tuzikov, A.B.; Pazynina, G.V.; Bovin, N.V.; Fouchier, R.A.M.; Klenk, H.-D.; et al. Receptor-Binding Properties of Influenza Viruses Isolated from Gulls. Virology 2018, 522, 37–45. [Google Scholar] [CrossRef]
- Li, Y.; Li, M.; Tian, J.; Zhang, Y.; Bai, X.; Wang, X.; Shi, J.; Wang, Y.; Ma, L.; Yang, C.; et al. Characteristics of the First H16N3 Subtype Influenza A Viruses Isolated in Western China. Transbound. Emerg. Dis. 2020, 67, 1677–1687. [Google Scholar] [CrossRef]
- Beerens, N.; Heutink, R.; Harders, F.; Roose, M.; Pritz-Verschuren, S.B.E.; Germeraad, E.A.; Engelsma, M. Incursion of Novel Highly Pathogenic Avian Influenza A(H5N8) Virus, The Netherlands, October 2020. Emerg. Infect. Dis. 2021, 27, 1750. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, I.; Sharshov, K.; Dubovitskiy, N.; Kurskaya, O.; Alekseev, A.; Leonov, S.; Yushkov, Y.; Irza, V.; Komissarov, A.; Fadeev, A.; et al. Highly Pathogenic Avian Influenza A(H5N8) Virus Clade 2.3.4.4b, Western Siberia, Russia, 2020. Emerg. Infect. Dis. 2021, 27, 2224–2227. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Choi, W.-S.; Antigua, K.J.C.; Choi, Y.K.; Govorkova, E.A.; Webby, R.J.; Baek, Y.H.; Song, M.-S. In Vitro Profiling of Laninamivir-Resistant Substitutions in N3 to N9 Avian Influenza Virus Neuraminidase Subtypes and Their Association with In Vivo Susceptibility. J. Virol. 2020, 95, e01679-20. [Google Scholar] [CrossRef]
- WHO Director-General’s Opening Remarks at the Media Briefing—8 February 2023. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing---8-february-2023 (accessed on 28 February 2023).
- Paulson, J.C. 5-Interactions of Animal Viruses with Cell Surface Receptors. In The Receptors; Conn, P.M., Ed.; Academic Press: Cambridge, MA, USA, 1985; pp. 131–219. ISBN 978-0-12-185202-3. [Google Scholar]
- Ito, T.; Suzuki, Y.; Suzuki, T.; Takada, A.; Horimoto, T.; Wells, K.; Kida, H.; Otsuki, K.; Kiso, M.; Ishida, H.; et al. Recognition of N-Glycolylneuraminic Acid Linked to Galactose by the A2,3 Linkage Is Associated with Intestinal Replication of Influenza A Virus in Ducks. J. Virol. 2000, 74, 9300–9305. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Ito, T.; Suzuki, T.; Holland, R.E.; Chambers, T.M.; Kiso, M.; Ishida, H.; Kawaoka, Y. Sialic Acid Species as a Determinant of the Host Range of Influenza A Viruses. J. Virol. 2000, 74, 11825–11831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veen, J.; Yurlov, A.; Delany, S.; Mihantiev, A.I.; Selivanova, M.; Boere, G.C. An Atlas of Movements of Southwest Siberian Waterbirds; Wetlands International: Wageningen, The Netherlands, 2005. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Lab ID | Accession ID |
|---|---|---|
| A/common_teal/Chany_Lake/29/2019 (H14N3) | A/29 | 400267 |
| A/garganey/Chany_Lake/210/2014 (H14N9) | A/210 | 14854178 |
| A/garganey/Chany_Lake/211/2014 (H14N9) | A/211 | 14853905 |
| Viruses | MEM with 2% FBS, log10TCID50/mL | MEM with 0.2% BSA and 2 µg/mL of Trypsin, log10TCID50/mL | Oseltamivir (Tamiflu) | |
|---|---|---|---|---|
| Mean IC50 a (nM) ± SD | Fold Change b | |||
| A/29 (H14N3) | 6.55 ± 0.3 | 7.67 ± 0.3 | 0.58 ± 0.03 | 1.42 |
| A/210 (H14N9) | 7.17 ± 0.3 | 8.17 ± 0.2 | 0.67 ± 0.15 | 1.63 |
| A/15O (H4N6) | 6.30 ± 0.2 | 7.30 ± 0.4 | 0.51 ± 0.04 | 1.24 |
| A(H1N1)pdm09 | n/a | 7.25 ± 0.3 | 0.41 ± 0.04 | 1.0 |
| Quail Post Infectious Sera | Antigens | ||
|---|---|---|---|
| A/29 (H14N3) | A/210 (H14N9) | A/15O (H4N6) | |
| A/29 (H14N3) | 160 (80) | 160 (80) | 5 (5) |
| A/210 (H14N9) | 160 (160) | 160 (160) | 5 (5) |
| A/15O (H4N6) | 5 (5) | 5 (5) | 160 (320) |
| Virus Strain | Oligosaccharides | RFU |
|---|---|---|
| A/garganey/Chany lake/210/2014 (H14N9) | Neu5Aca2-3Galb1-4(2-O-Su-Fuca1-3)(6-O-Su)GlcNAcb-sp3 (Neu5Aca3′(2-suFuca3)(6-su)LN-C3) | 17,793 |
| Neu5Aca2-3(6-O-Su)Galb1-4(6-O-Su)GlcNAcb-sp3 (Neu5Aca3′(6,6′-su2)LN-C3) | 17,396 | |
| A/common teal/Chany Lake/29/2019 (H14N3) | Neu5Acα2-3Galβ1-4(Fucβ1-3)GlcNAcβ-sp3 (SiaLeX) | 64,732 |
| Neu5Acα2-3Galβ1-4(2-O-Su-Fucα1-3)GlcNAcβ-sp3 | 61,445 | |
| (SiaLeX2‴Su) | ||
| Neu5Acα2-3Galβ1-4-(6-O-Su)GlcNAcβ-sp3 | 57,287 | |
| (3′SLN6Su) | ||
| Neu5Acα2-3Galβ1-3(Fucα1-4)GlcNAcβ-sp3(SiaLea) | 56,604 | |
| Neu5Acα2-3Galβ1-3GlcNAcβ-sp3 (3′SiaLeC) | 51,191 | |
| Neu5Acα2-3Galβ1-4GlcNAcβ-sp3(3′SLN) | 48,444 | |
| Neu5Acα2-3GalNAcα-sp3 (3-SiaTn) | 29,289 | |
| A/little tern/Guriev/779/83 (H16N3) | Neu5Acα2-6(Galβ1-3)GalNAcα-sp3 (6SiaTF) | 43,606 |
| Neu5Acα2-3Galβ1-4(Fucβ1-3)GlcNAcβ-sp3 (SiaLeX) | 29,451 | |
| Neu5Acα2-6Galβ1-4GlcNAcβ-sp3 (6′SLN) | 28,472 | |
| Neu5Acα2-6Galβ1-4-(6-O-Su)GlcNAcβ-sp3 (6′SLN6Su) | 27,473 | |
| Neu5Acα2-3Galβ1-4GlcNAcβ-sp3 (3′SLN) | 15,806 |
| Strain | Related Strain | Country | HA Identity, % |
|---|---|---|---|
| A/29 | A/sandpiper/Tomsk/112/2019 (H14N7) | Russia | 98.65 |
| A/210 | A/goose/Karachi/NARC-13N-969/2014 (H14N3) | Pakistan | 98.24 |
| A/211 | A/goose/Karachi/NARC-13N-969/2014 (H14N3) | Pakistan | 98.24 |
| Strain | 6 | 58 | 62 | 77 | 127 | 143 | 145 | 189 | 211 | 226 | 228 | 281 | 301 | 326 | 329 | 441 | 515 | 523 | Cleavage Site Amino Acid Sequence |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A/goose/Karachi/NARC-13N-969/2014 (H14N3) | I | I | N | H | G | R | G | Q | I | Q | G | P | I | D | A | T | I | M | NIPGKQAK/G |
| A/common teal/Chany Lake/29/2019 (H14N3) | T | . | D | . | N | . | S | T | T | Q | G | S | . | . | T | . | V | . | NIPDKQTK/G |
| A/garganey/Chany Lake/210/2014 (H14N9) | T | V | D | . | S | . | . | . | . | Q | G | . | . | . | . | . | V | . | NIPDKQAK/G |
| A/garganey/Chany Lake/211/2014 (H14N9) | T | V | D | . | S | . | . | . | . | Q | G | . | . | G | . | . | V | . | NIPGKQAK/G |
| A/sandpiper/Tomsk/112/2019 | T | . | D | R | N | H | S | K | T | Q | G | . | M | . | T | A | V | T | NIPDKQTK/G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubovitskiy, N.; Derko, A.; Sobolev, I.; Prokopyeva, E.; Murashkina, T.; Solomatina, M.; Kurskaya, O.; Komissarov, A.; Fadeev, A.; Danilenko, D.; et al. Virological and Genetic Characterization of the Unusual Avian Influenza H14Nx Viruses in the Northern Asia. Viruses 2023, 15, 734. https://doi.org/10.3390/v15030734
Dubovitskiy N, Derko A, Sobolev I, Prokopyeva E, Murashkina T, Solomatina M, Kurskaya O, Komissarov A, Fadeev A, Danilenko D, et al. Virological and Genetic Characterization of the Unusual Avian Influenza H14Nx Viruses in the Northern Asia. Viruses. 2023; 15(3):734. https://doi.org/10.3390/v15030734
Chicago/Turabian StyleDubovitskiy, Nikita, Anastasiya Derko, Ivan Sobolev, Elena Prokopyeva, Tatyana Murashkina, Maria Solomatina, Olga Kurskaya, Andrey Komissarov, Artem Fadeev, Daria Danilenko, and et al. 2023. "Virological and Genetic Characterization of the Unusual Avian Influenza H14Nx Viruses in the Northern Asia" Viruses 15, no. 3: 734. https://doi.org/10.3390/v15030734