
The International Virus Bioinformatics Meeting 2023
 , , , , , , ,
, , , , , , ,  , , , , , , , , , , , , , , , , , , , , and add
Show full author list
, , , , , , , , , , , , , , , , , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Scientific Program
2.1. Phages
Jaeger: A Deep Learning Approach for Predicting Bacteriophage Sequences in Metagenomic Data (by Rajitha Yasas Wijesekara)
2.2. Virus Discovery and Classification
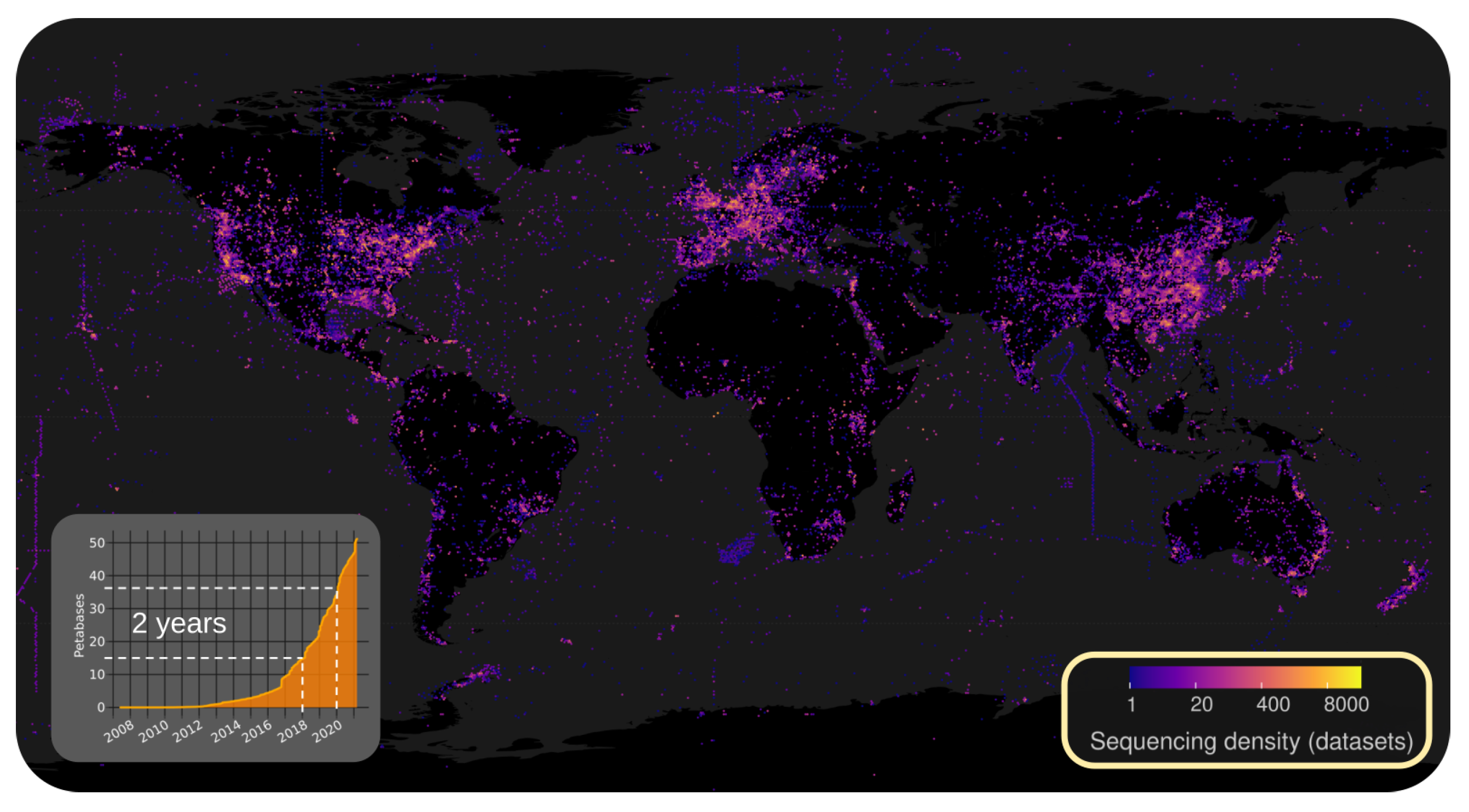
2.2.1. Illuminating the RNA Virome through Ultra-Massive Sequence Analysis (by Artem Babaian)
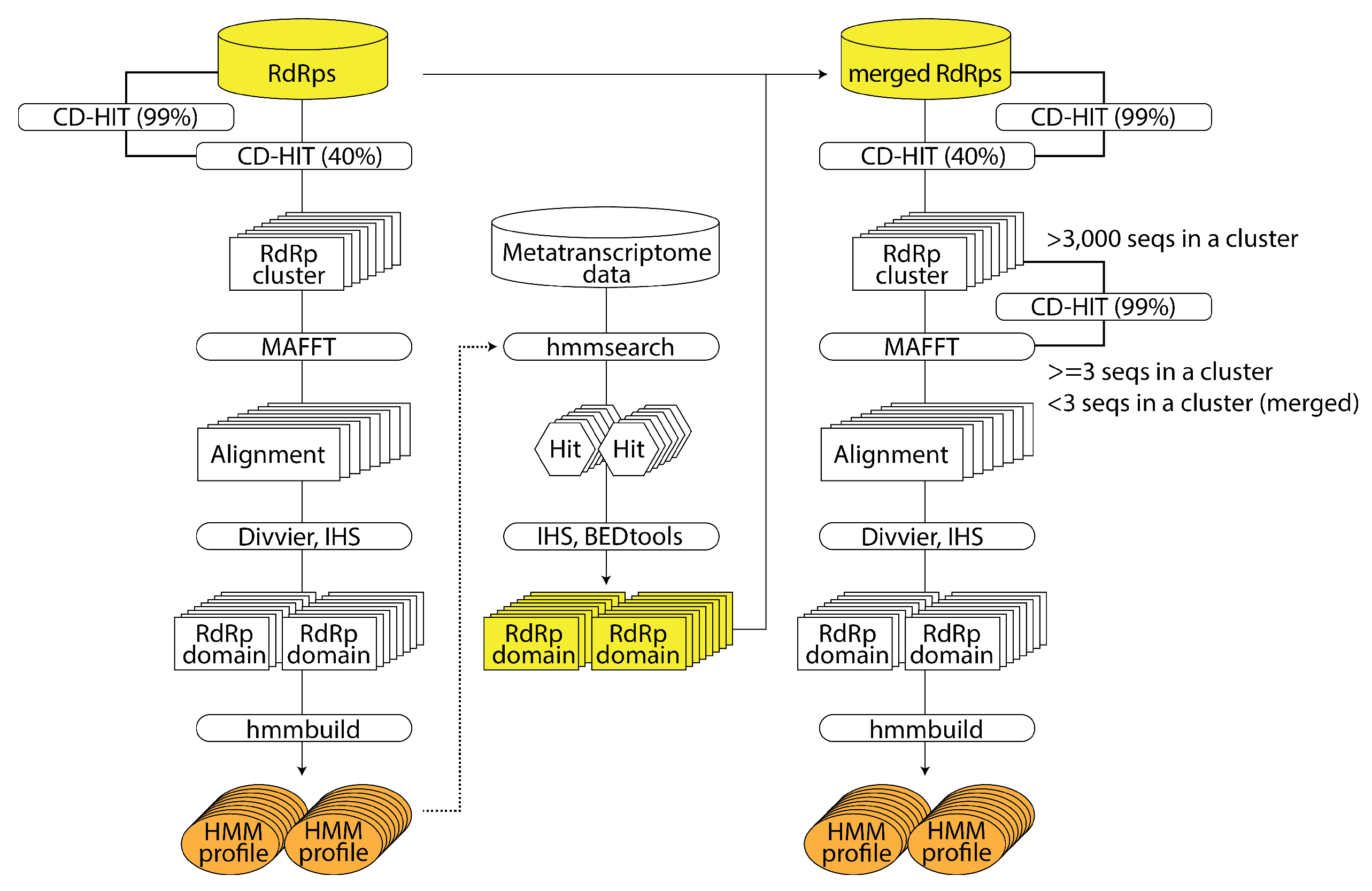
2.2.2. RNA Virus Discovery Using HMM of Large-Scale RNA-Dependent RNA Polymerase Sequence Data: NeoRdRp 2.0 (by Shoichi Sakaguchi)
2.2.3. Automated Classification of Giant Virus Genomes Using Protein Family Barcodes (by Anh Ha)
2.2.4. Using gb2seq to Work with Unannotated Viral Genomes Based on a GenBank Reference (by Terry Jones)
2.3. Virus Visualization
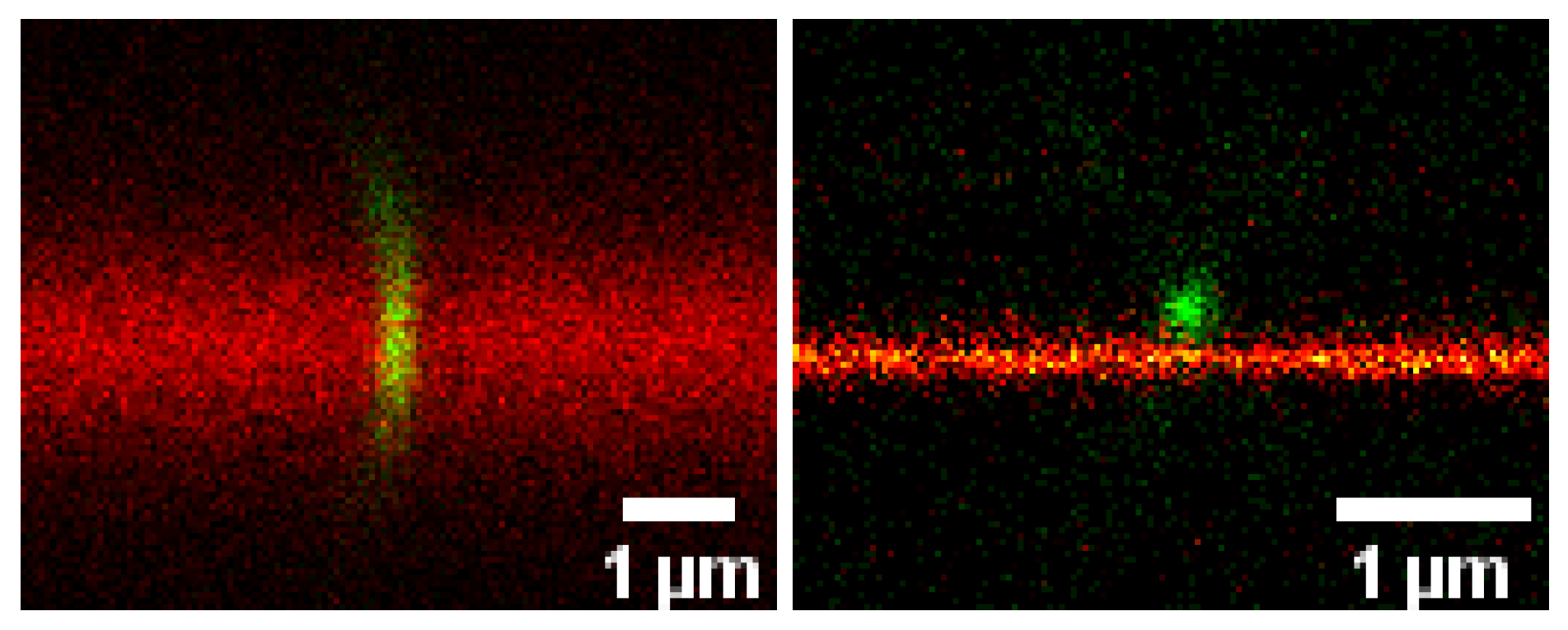
Advanced Optical Microscopy of Virus-Cell Interactions: Challenges and Potentials (by Christian Eggeling)
2.4. Viral Infection
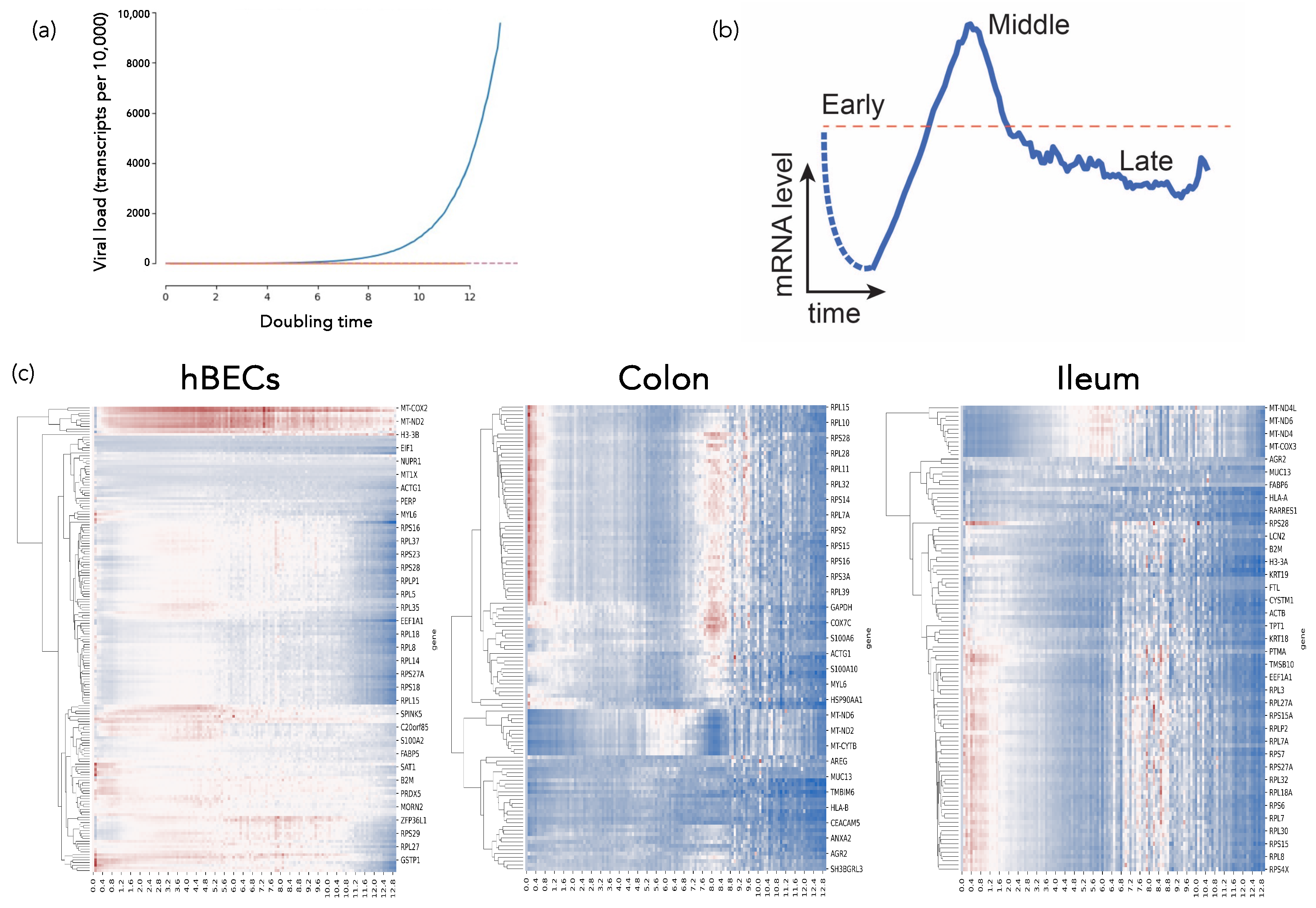
2.4.1. SARS-CoV-2-Host Interactions at the Single-Cell Level: A Dynamical Complex Systems Approach (by Santiago F. Elena)
2.4.2. Metabolic Labeling, Time Series, and Single Cells: A Multifaceted Approach to Studying Infection (by Lygeri Sakellaridi)
2.5. Viromics
2.5.1. Ancient Virome Analyses Using Metagenomic Data from Ancient Individuals (by Luca Nishimura)
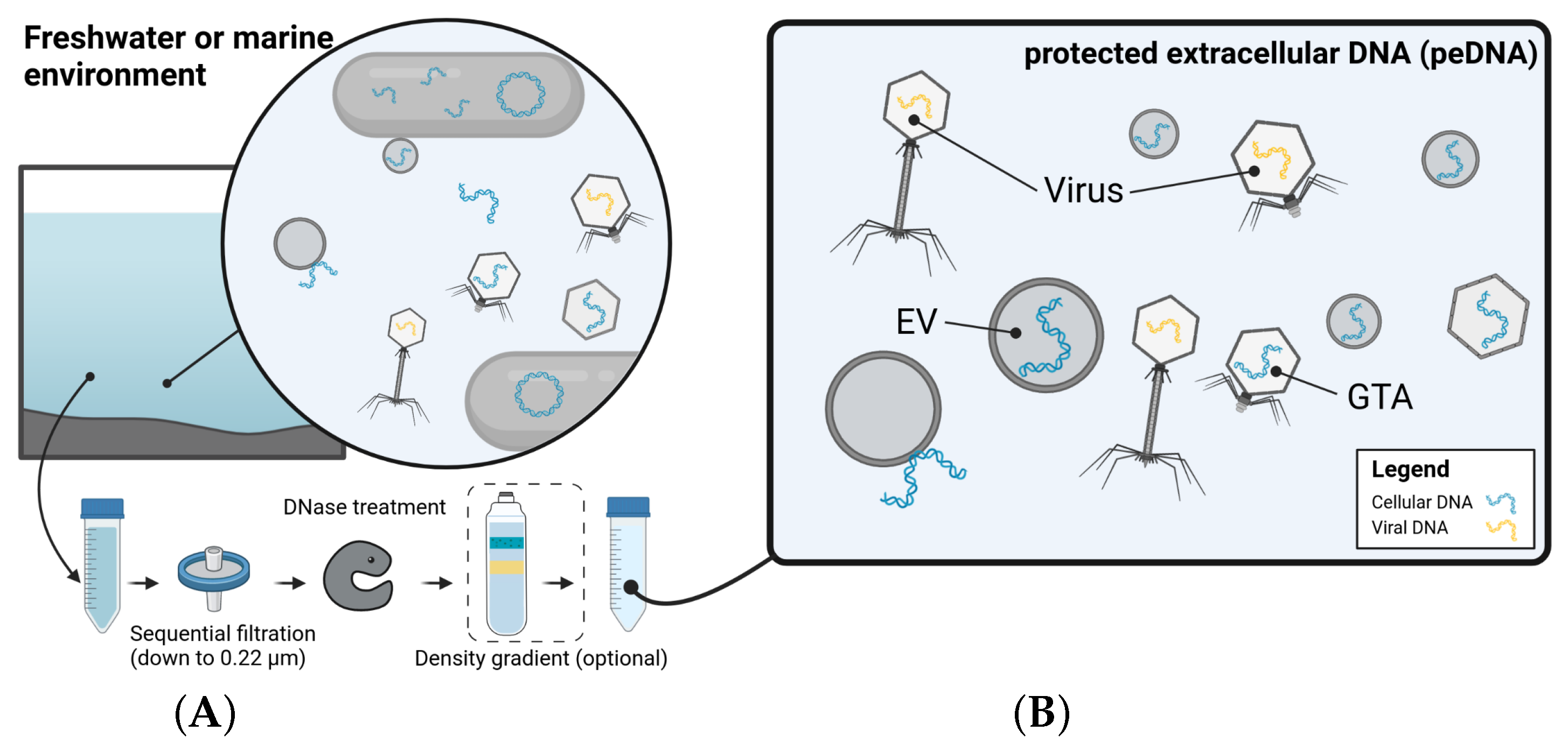
2.5.2. One’s Trash Is Another’s Treasure—Mining Viromics Datasets for Traces of EV Mediated Horizontal Gene Transfer (by Dominik Lücking)
2.6. Molecular Epidemiology and Phylodynamic Analyses
2.6.1. HIV-1 Transmission Studies Using Phylogenetics: Can Evolution Help Guide Public Health Decisions? (by Ana Abecasis)
2.6.2. Molecular Epidemiological Approaches to Investigate the Dispersal Dynamic of Viruses and the Environmental Factors Impacting It (by Simon Dellicour)
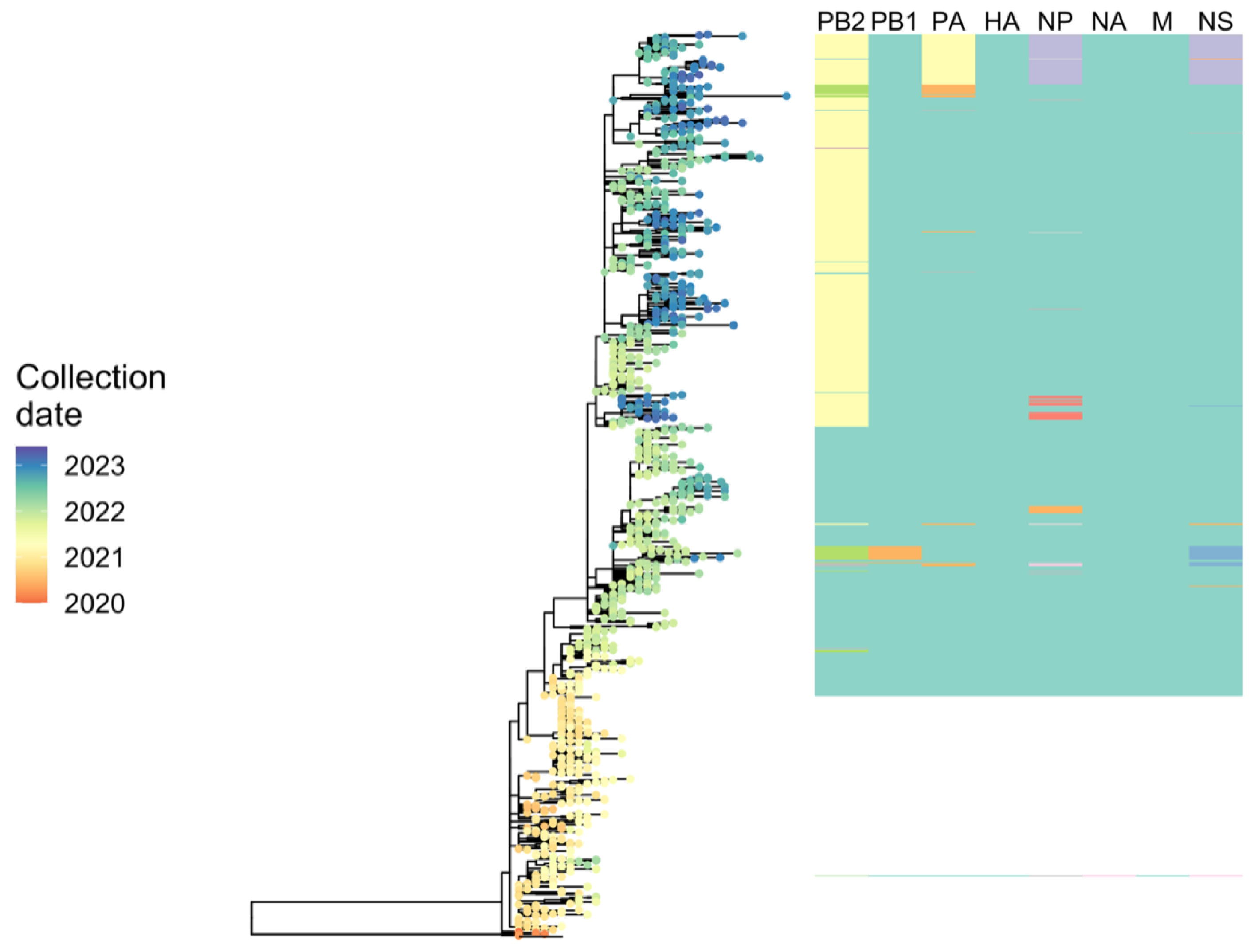
2.6.3. Phylodynamic Analysis of A(H5N1) Highly Pathogenic Avian Influenza Viruses Provides Insight into Movement Dynamics and Host Specificity (by Will Harvey)
2.7. RNA Viruses: Structure and Evolution
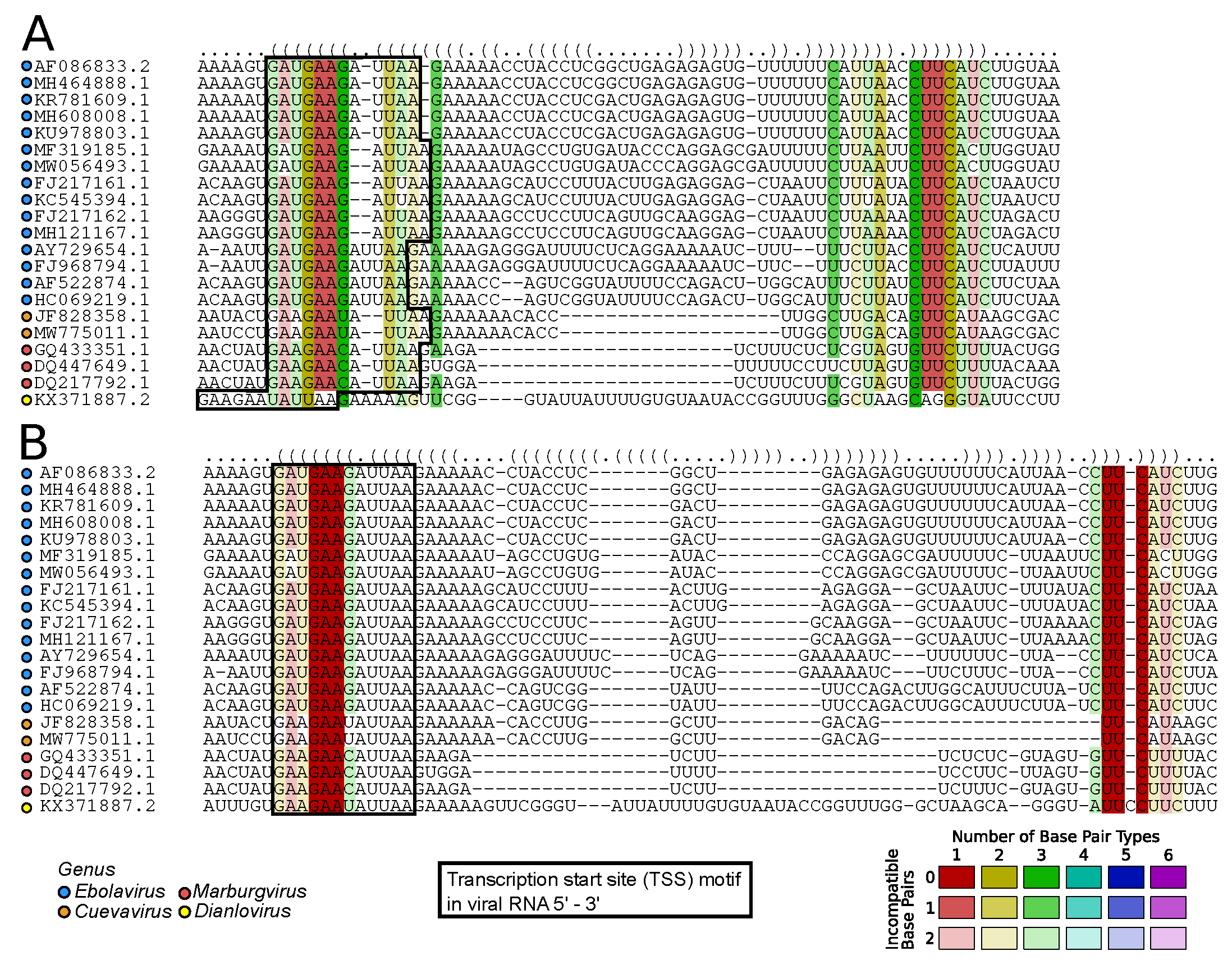
2.7.1. Viral RNA Secondary Structures: Canonical and Beyond (by Kevin Lamkiewicz & Sandra Triebel)
2.7.2. Recombination and Modular Evolution of Positive-Strand RNA Viruses: Similar, but Not the Same (by Yulia Vakulenko)
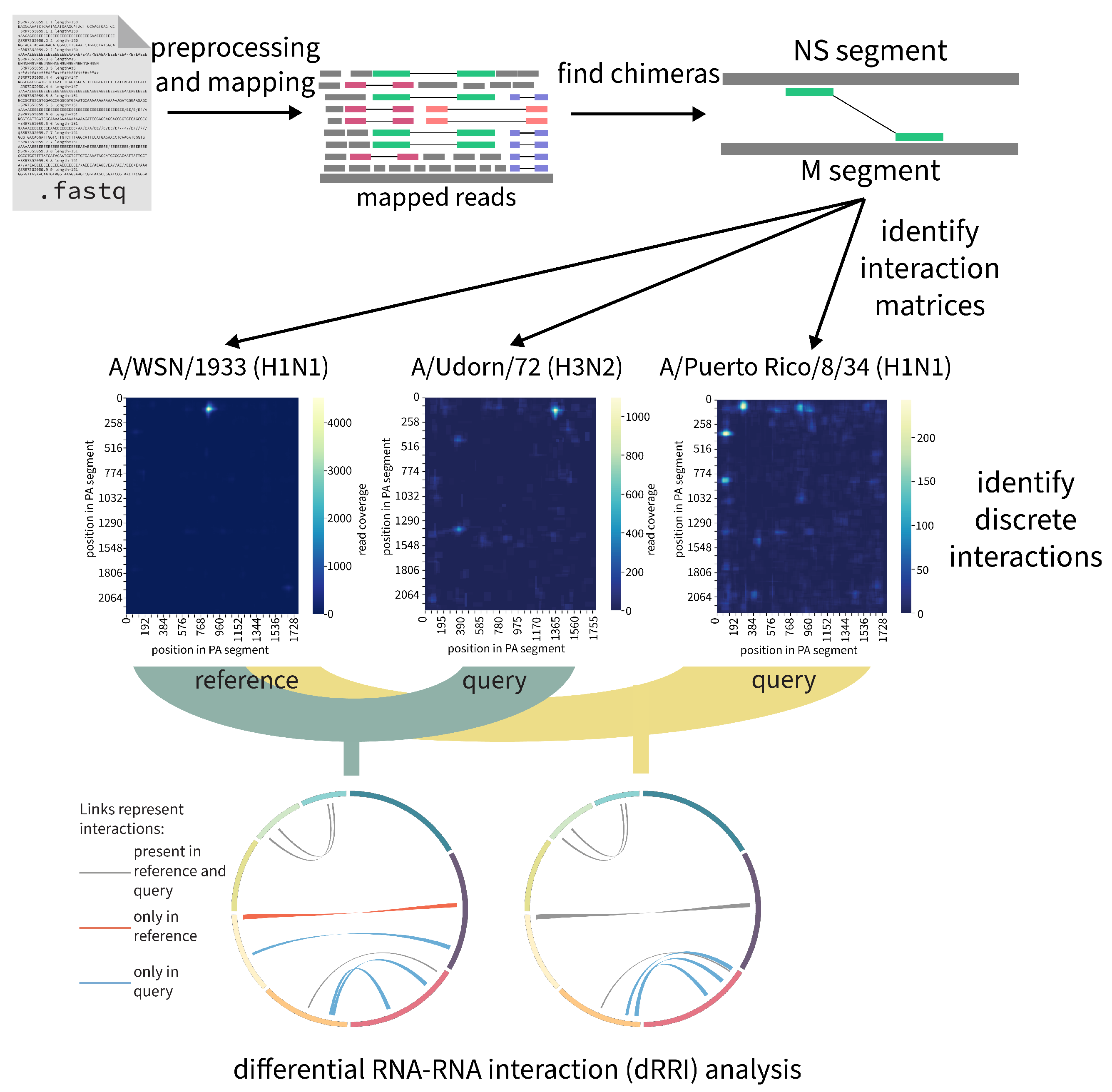
2.7.3. RNAswarm: A Modular Pipeline for Differential RRI Analysis in Influenza a Virus (by Gabriel Lencioni Lovate)
2.8. Viral Sequence Analysis
2.8.1. Embedding Segmented Viral Genomes for Visualisation, Search, and Clustering (by Udo Gieraths)
2.8.2. Hyper-EINS: A Tool for Automated Identification of Insertions in the Hepatitis E Virus Hypervariable Region (by Maximilian Nocke)
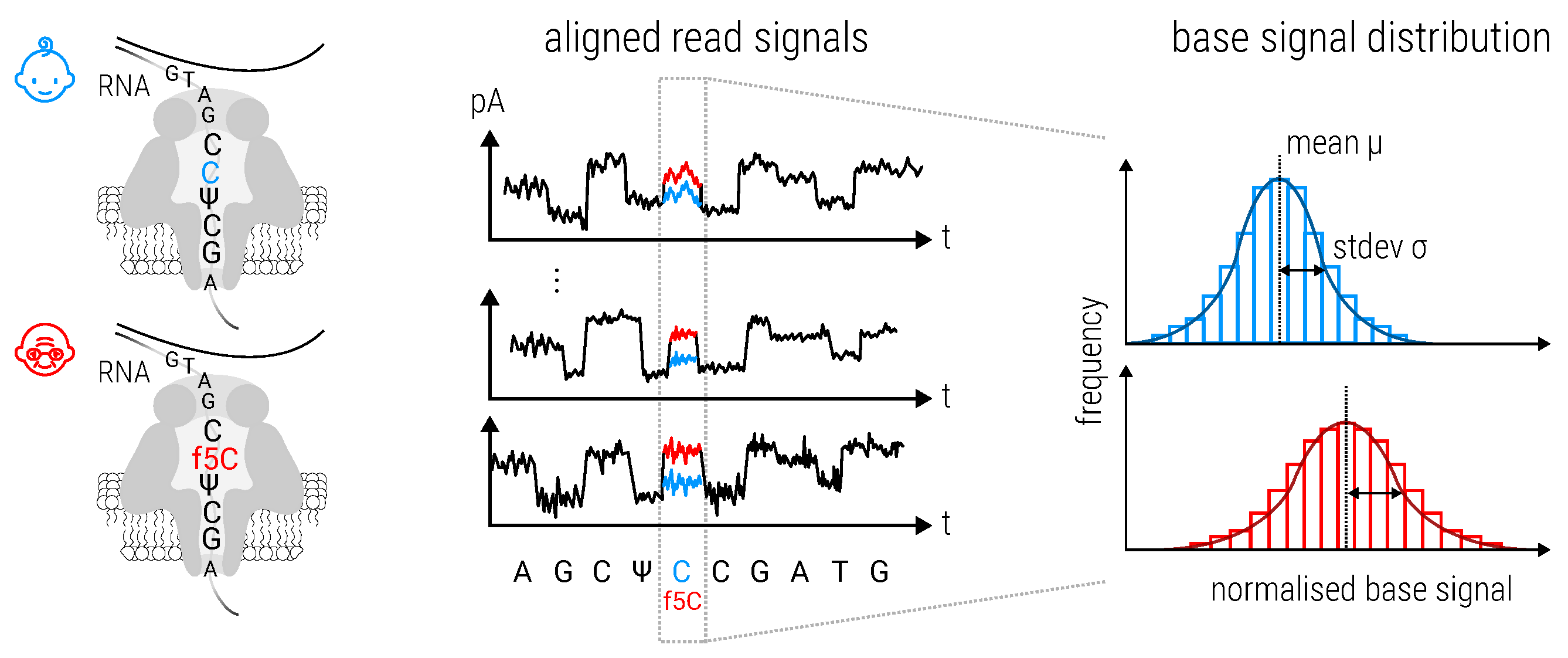
2.8.3. Magnipore: Predicting Differential Single Nucleotide Changes in Oxford Nanopore Technologies Sequencing Signal in SARS-CoV-2 (by Jannes Spangenberg)
2.9. Machine Learning in Viral Surveillance
2.9.1. From High-Throughput Testing to Genomic Surveillance and Public Health Data Integration (by Bernhard Renard)
2.9.2. BLOODVIR: Virus Surveillance System for Plasma Pools Based on High-Throughput Sequencing and Machine Learning (by Martin Machyna)
2.9.3. Modelling the Zoonotic Capabilities of Avian Influenza via Genomic Machine Learning (by Liam Brierley)
2.10. Viral Pathogenesis
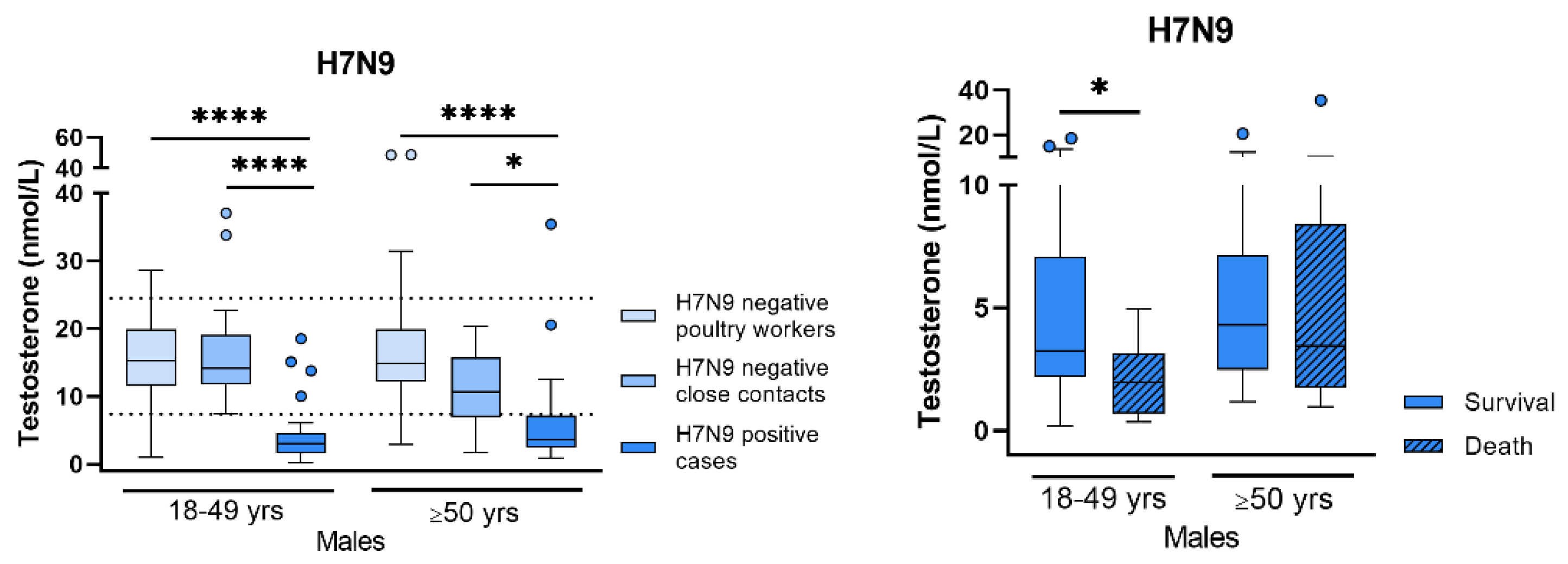
Sex Differences in Respiratory Virus Infections (by Sebastian Beck)
2.11. Metagenomics for Identifying and Tracking Potential Zoonotic Viruses
Discovering and Tracking Potential Zoonotic Species from Metagenomic Samples with a Capture-Based Oriented Pipeline (by Maria Tarradas-Alemany)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ibrahim, B.; McMahon, D.P.; Hufsky, F.; Beer, M.; Deng, L.; Mercier, P.L.; Palmarini, M.; Thiel, V.; Marz, M. A new era of virus bioinformatics. Virus Res. 2018, 251, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Hufsky, F.; Ibrahim, B.; Beer, M.; Deng, L.; Mercier, P.L.; McMahon, D.P.; Palmarini, M.; Thiel, V.; Marz, M. Virologists-Heroes need weapons. PLoS Pathog. 2018, 14, e1006771. [Google Scholar] [CrossRef] [PubMed]
- Hufsky, F.; Beslic, D.; Boeckaerts, D.; Duchene, S.; González-Tortuero, E.; Gruber, A.J.; Guo, J.; Jansen, D.; Juma, J.; Kongkitimanon, K.; et al. The International Virus Bioinformatics Meeting 2022. Viruses 2022, 14, 973. [Google Scholar] [CrossRef]
- Hendrix, R.W.; Smith, M.C.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Brockhurst, M.A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Cobián Güemes, A.G.; Youle, M.; Cantú, V.A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as winners in the game of life. Annu. Rev. Virol. 2016, 3, 197–214. [Google Scholar] [CrossRef]
- Howard-Varona, C.; Lindback, M.M.; Bastien, G.E.; Solonenko, N.; Zayed, A.A.; Jang, H.; Andreopoulos, B.; Brewer, H.M.; Glavina Del Rio, T.; Adkins, J.N.; et al. Phage-specific metabolic reprogramming of virocells. ISME J. 2020, 14, 881–895. [Google Scholar] [CrossRef]
- Suttle, C.A. Marine viruses–major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Li, M.; Wang, C.; Guo, Q.; Xu, C.; Xie, Z.; Tan, J.; Wu, S.; Wang, P.; Guo, J.; Fang, Z.; et al. More Positive or More Negative? Metagenomic Analysis Reveals Roles of Virome in Human Disease-Related Gut Microbiome. Front. Cell. Infect. Microbiol. 2022, 12, 846063. [Google Scholar] [CrossRef]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying viruses from metagenomic data using deep learning. Quant. Biol. 2020, 8, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Auslander, N.; Gussow, A.B.; Benler, S.; Wolf, Y.I.; Koonin, E.V. Seeker: Alignment-free identification of bacteriophage genomes by deep learning. BioRxiv 2020. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.P.; Nayfach, S.; Chen, I.M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Ritter, S.J.; Reddy, T.B.K.; Mukherjee, S.; Schulz, F.; et al. IMG/VR v4: An expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic Acids Res. 2023, 51, D733–D743. [Google Scholar] [CrossRef]
- Wu, L.Y.; Pappas, N.; Wijesekara, Y.; Piedade, G.J.; Brussaard, C.P.; Dutilh, B.E. Benchmarking Bioinformatic Virus Identification Tools Using Real-World Metagenomic Data across Biomes. BioRxiv 2023. [Google Scholar] [CrossRef]
- Katz, K.; Shutov, O.; Lapoint, R.; Kimelman, M.; Brister, J.; O’Sullivan, C. The Sequence Read Archive: A decade more of explosive growth. Nucleic Acids Res. 2022, 50, D387–D390. [Google Scholar] [CrossRef]
- Courtot, M.; Cherubin, L.; Faulconbridge, A.; Vaughan, D.; Green, M.; Richardson, D.; Harrison, P.; Whetzel, P.L.; Parkinson, H.; Burdett, T. BioSamples database: An updated sample metadata hub. Nucleic Acids Res. 2019, 47, D1172–D1178. [Google Scholar] [CrossRef]
- The Cost of Sequencing a Human Genome. 2022. Available online: https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost (accessed on 9 July 2023).
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucía-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and evolution of the global RNA virome. MBio 2018, 9, e02329-18. [Google Scholar] [CrossRef]
- Zayed, A.A.; Wainaina, J.M.; Dominguez-Huerta, G.; Pelletier, E.; Guo, J.; Mohssen, M.; Tian, F.; Pratama, A.A.; Bolduc, B.; Zablocki, O.; et al. Cryptic and abundant marine viruses at the evolutionary origins of Earth’s RNA virome. Science 2022, 376, 156–162. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Urayama, S.I.; Takaki, Y.; Hirosuna, K.; Wu, H.; Suzuki, Y.; Nunoura, T.; Nakano, T.; Nakagawa, S. NeoRdRp: A Comprehensive Dataset for Identifying RNA-dependent RNA Polymerases of Various RNA Viruses from Metatranscriptomic Data. Microbes Environ. 2022, 37, ME22001. [Google Scholar] [CrossRef] [PubMed]
- Aylward, F.O.; Moniruzzaman, M.; Ha, A.D.; Koonin, E.V. A phylogenomic framework for charting the diversity and evolution of giant viruses. PLoS Biol. 2021, 19, e3001430. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Weinheimer, A.R.; Martinez-Gutierrez, C.A.; Aylward, F.O. Widespread endogenization of giant viruses shapes genomes of green algae. Nature 2020, 588, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Ha, A.D.; Moniruzzaman, M.; Aylward, F.O. High Transcriptional Activity and Diverse Functional Repertoires of Hundreds of Giant Viruses in a Coastal Marine System. mSystems 2021, 6, e0029321. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Martinez-Gutierrez, C.A.; Weinheimer, A.R.; Aylward, F.O. Dynamic genome evolution and complex virocell metabolism of globally-distributed giant viruses. Nat. Commun. 2020, 11, 1710. [Google Scholar] [CrossRef]
- Ha, A.D.; Moniruzzaman, M.; Aylward, F.O. Assessing the biogeography of marine giant viruses in four oceanic transects. ISME Commun. 2023, 3, 43. [Google Scholar] [CrossRef]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Müller, B.; Hell, S.W.; Usslich, H.G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef]
- Chojnacki, J.; Waithe, D.; Carravilla, P.; Huarte, N.; Galiani, S.; Enderlein, J.; Eggeling, C. Envelope glycoprotein mobility on HIV-1 particles depends on the virus maturation state. Nat. Commun. 2017, 8, 545. [Google Scholar] [CrossRef]
- Chojnacki, J.; Eggeling, C. Super-resolution fluorescence microscopy studies of human immunodeficiency virus. Retrovirology 2018, 15, 41. [Google Scholar] [CrossRef]
- Chojnacki, J.; Eggeling, C. Zooming in on virus surface protein mobility. Future Virol. 2018, 13, 225–227. [Google Scholar] [CrossRef]
- Gutierrez, P.A.; Elena, S.F. Single-cell RNA-sequencing data analysis reveals a highly correlated triphasic transcriptional response to SARS-CoV-2 infection. Commun. Biol. 2022, 5, 1302. [Google Scholar] [CrossRef] [PubMed]
- Ravindra, N.G.; Alfajaro, M.M.; Gasque, V.; Huston, N.C.; Wan, H.; Szigeti-Buck, K.; Yasumoto, Y.; Greaney, A.M.; Habet, V.; Chow, R.D.; et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium identifies target cells, alterations in gene expression, and cell state changes. PLoS Biol. 2021, 19, e3001143. [Google Scholar] [CrossRef] [PubMed]
- Triana, S.; Metz-Zumaran, C.; Ramirez, C.; Kee, C.; Doldan, P.; Shahraz, M.; Schraivogel, D.; Gschwind, A.R.; Sharma, A.K.; Steinmetz, L.M.; et al. Single-cell analyses reveal SARS-CoV-2 interference with intrinsic immune response in the human gut. Mol. Syst. Biol. 2021, 17, e10232. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.M.; St Clair, L.A.; Perera, R.; Parker, R. SARS-CoV-2 infection triggers widespread host mRNA decay leading to an mRNA export block. RNA 2021, 27, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Finkel, Y.; Gluck, A.; Nachshon, A.; Winkler, R.; Fisher, T.; Rozman, B.; Mizrahi, O.; Lubelsky, Y.; Zuckerman, B.; Slobodin, B.; et al. SARS-CoV-2 uses a multipronged strategy to impede host protein synthesis. Nature 2021, 594, 240–245. [Google Scholar] [CrossRef]
- Herzog, V.A.; Reichholf, B.; Neumann, T.; Rescheneder, P.; Bhat, P.; Burkard, T.R.; Wlotzka, W.; von Haeseler, A.; Zuber, J.; Ameres, S.L. Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 2017, 14, 1198–1204. [Google Scholar] [CrossRef]
- Schofield, J.A.; Duffy, E.E.; Kiefer, L.; Sullivan, M.C.; Simon, M.D. TimeLapse-seq: Adding a temporal dimension to RNA sequencing through nucleoside recoding. Nat. Methods 2018, 15, 221–225. [Google Scholar] [CrossRef]
- Riml, C.; Amort, T.; Rieder, D.; Gasser, C.; Lusser, A.; Micura, R. Osmium-Mediated Transformation of 4-Thiouridine to Cytidine as Key To Study RNA Dynamics by Sequencing. Angew. Chem. Int. Ed. Engl. 2017, 56, 13479–13483. [Google Scholar] [CrossRef]
- Jürges, C.; Dölken, L.; Erhard, F. Dissecting newly transcribed and old RNA using GRAND-SLAM. Bioinformatics 2018, 34, i218–i226. [Google Scholar] [CrossRef]
- Rummel, T.; Sakellaridi, L.; Erhard, F. grandR: A comprehensive package for nucleotide conversion RNA-seq data analysis. Nat. Commun. 2023, 14, 3559. [Google Scholar] [CrossRef]
- Goldberg, A.V. An Efficient Implementation of a Scaling Minimum-Cost Flow Algorithm. J. Algorithms 1997, 2, 1–29. [Google Scholar] [CrossRef]
- Nishimura, L.; Fujito, N.; Sugimoto, R.; Inoue, I. Detection of Ancient Viruses and Long-Term Viral Evolution. Viruses 2022, 14, 1336. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, L.; Sugimoto, R.; Inoue, J.; Nakaoka, H.; Kanzawa-Kiriyama, H.; Shinoda, K.I.; Inoue, I. Identification of ancient viruses from metagenomic data of the Jomon people. J. Hum. Genet. 2021, 66, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Biller, S.J.; Schubotz, F.; Roggensack, S.E.; Thompson, A.W.; Summons, R.E.; Chisholm, S.W. Bacterial vesicles in marine ecosystems. Science 2014, 343, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Linney, M.D.; Schvarcz, C.R.; Steward, G.F.; DeLong, E.F.; Karl, D.M. A method for characterizing dissolved DNA and its application to the North Pacific Subtropical Gyre. Limnol. Oceanogr. Methods 2021, 19, 210–221. [Google Scholar] [CrossRef]
- Abecasis, A.B.; Geretti, A.M.; Albert, J.; Power, L.; Weait, M.; Vandamme, A.M. Science in court: The myth of HIV fingerprinting. Lancet Infect. Dis. 2011, 11, 78–79. [Google Scholar] [CrossRef]
- Pineda-Peña, A.C.; Theys, K.; Stylianou, D.C.; Demetriades, I.; SPREAD/ESAR Program; Abecasis, A.B.; Kostrikis, L.G. HIV-1 infection in Cyprus, the Eastern Mediterranean European frontier: A densely sampled transmission dynamics analysis from 1986 to 2012. Sci. Rep. 2018, 8, 1702. [Google Scholar] [CrossRef]
- Pimentel, V.F.; Pingarilho, M.; Sole, G.; Alves, D.; Miranda, M.; Diogo, I.; Fernandes, S.; Pineda-Pena, A.; Martins, M.R.O.; Camacho, R.; et al. Differential patterns of postmigration HIV-1 infection acquisition among Portuguese immigrants of different geographical origins. AIDS 2022, 36, 997–1005. [Google Scholar] [CrossRef]
- Pingarilho, M.; Pimentel, V.; Miranda, M.N.S.; Silva, A.R.; Diniz, A.; Ascenção, B.B.; Piñeiro, C.; Koch, C.; Rodrigues, C.; Caldas, C.; et al. HIV-1-transmitted drug resistance and transmission clusters in newly diagnosed patients in Portugal between 2014 and 2019. Front. Microbiol. 2022, 13, 823208. [Google Scholar] [CrossRef]
- Klitting, R.; Kafetzopoulou, L.E.; Thiery, W.; Dudas, G.; Gryseels, S.; Kotamarthi, A.; Vrancken, B.; Gangavarapu, K.; Momoh, M.; Sandi, J.D.; et al. Predicting the evolution of the Lassa virus endemic area and population at risk over the next decades. Nat. Commun. 2022, 13, 5596. [Google Scholar] [CrossRef]
- Dellicour, S.; Baele, G.; Dudas, G.; Faria, N.R.; Pybus, O.G.; Suchard, M.A.; Rambaut, A.; Lemey, P. Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak. Nat. Commun. 2018, 9, 2222. [Google Scholar] [CrossRef] [PubMed]
- Dellicour, S.; Lequime, S.; Vrancken, B.; Gill, M.S.; Bastide, P.; Gangavarapu, K.; Matteson, N.L.; Tan, Y.; du Plessis, L.; Fisher, A.A.; et al. Epidemiological hypothesis testing using a phylogeographic and phylodynamic framework. Nat. Commun. 2020, 11, 5620. [Google Scholar] [CrossRef] [PubMed]
- Tsukiyama-Kohara, K.; Iizuka, N.; Kohara, M.; Nomoto, A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992, 66, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.; Soto-Acosta, R.; Bradrick, S.; Garcia-Blanco, M.; Ooi, E. The 5′ and 3′ Untranslated Regions of the Flaviviral Genome. Viruses 2017, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Ochsenreiter, R.; Hofacker, I.; Wolfinger, M. Functional RNA Structures in the 3′UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses. Viruses 2019, 11, 298. [Google Scholar] [CrossRef]
- Sola, I.; Mateos-Gomez, P.A.; Almazan, F.; Zuñiga, S.; Enjuanes, L. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biol. 2011, 8, 237–248. [Google Scholar] [CrossRef]
- Yang, D.; Leibowitz, J.L. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015, 206, 120–133. [Google Scholar] [CrossRef]
- Madhugiri, R.; Karl, N.; Petersen, D.; Lamkiewicz, K.; Fricke, M.; Wend, U.; Scheuer, R.; Marz, M.; Ziebuhr, J. Structural and functional conservation of cis-acting RNA elements in coronavirus 5’-terminal genome regions. Virology 2018, 517, 44–55. [Google Scholar] [CrossRef]
- Dadonaite, B.; Gilbertson, B.; Knight, M.L.; Trifkovic, S.; Rockman, S.; Laederach, A.; Brown, L.E.; Fodor, E.; Bauer, D.L.V. The structure of the influenza A virus genome. Nat. Microbiol. 2019, 4, 1781–1789. [Google Scholar] [CrossRef]
- Jones, J.E.; Le Sage, V.; Padovani, G.H.; Calderon, M.; Wright, E.S.; Lakdawala, S.S. Parallel evolution between genomic segments of seasonal human influenza viruses reveals RNA-RNA relationships. eLife 2021, 10, e66525. [Google Scholar] [CrossRef]
- Jakob, C.; Lovate, G.L.; Desirò, D.; Gießler, L.; Smyth, R.; Marquet, R.; Lamkiewicz, K.; Marz, M.; Schwemmle, M.; Bolte, H. Sequential disruption of SPLASH-identified vRNA–vRNA interactions challenges their role in influenza A virus genome packaging. Nucleic Acids Res. 2023, 51, 6479–6494. [Google Scholar] [CrossRef] [PubMed]
- Will, S.; Reiche, K.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. Inferring Noncoding RNA Families and Classes by Means of Genome-Scale Structure-Based Clustering. PLoS Comput. Biol. 2007, 3, e65. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.; Demper, J.C.; Biedenkopf, N.; Becker, S.; Hartmann, R.K. RNA secondary structure at the transcription start site influences EBOV transcription initiation and replication in a length- and stability-dependent manner. RNA Biol. 2021, 18, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Mühlberger, E.; Trommer, S.; Funke, C.; Volchkov, V.; Klenk, H.D.; Becker, S. Termini of All mRNA Species of Marburg Virus: Sequence and Secondary Structure. Virology 1996, 223, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Holmes, E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999, 80 Pt 10, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Recombination and selection in the evolution of picornaviruses and other Mammalian positive-stranded RNA viruses. J. Virol. 2006, 80, 11124–11140. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Vakulenko, Y.; Deviatkin, A.; Drexler, J.F.; Lukashev, A. Modular Evolution of Coronavirus Genomes. Viruses 2021, 13, 1270. [Google Scholar] [CrossRef]
- Lukashev, A.N. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef]
- Desselberger, U. Other Than Noroviruses. Viruses 2019, 11, 286. [Google Scholar] [CrossRef]
- Bosch, A.; Pintó, R.M.; Guix, S. Human astroviruses. Clin. Microbiol. Rev. 2014, 27, 1048–1074. [Google Scholar] [CrossRef] [PubMed]
- Vakulenko, Y.A.; Orlov, A.V.; Lukashev, A.N. Patterns and Temporal Dynamics of Natural Recombination in Noroviruses. Viruses 2023, 15, 372. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S.; et al. RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Aw, J.; Shen, Y.; Wilm, A.; Sun, M.; Lim, X.; Boon, K.L.; Tapsin, S.; Chan, Y.S.; Tan, C.P.; Sim, A.; et al. In Vivo Mapping of Eukaryotic RNA Interactomes Reveals Principles of Higher-Order Organization and Regulation. Mol. Cell 2016, 62, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Sage, V.L.; Kanarek, J.P.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S.; Lee, N. Mapping of Influenza Virus RNA-RNA Interactions Reveals a Flexible Network. Cell Rep. 2020, 31, 107823. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Aggarwal, R. Hepatitis E: The endemic perspective. Clin. Liver Dis. 2013, 2, 240–244. [Google Scholar] [CrossRef]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Torian, U.; Faulk, K.; Mather, K.; Engle, R.E.; Thompson, E.; Bonkovsky, H.L.; Emerson, S.U. A naturally occurring human/hepatitis E recombinant virus predominates in serum but not in faeces of a chronic hepatitis E patient and has a growth advantage in cell culture. J. Gen. Virol. 2012, 93, 526–530. [Google Scholar] [CrossRef]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Mansuy, J.M.; Rostaing, L.; Kamar, N.; Izopet, J. Characterization of the polyproline region of the hepatitis E virus in immunocompromised patients. J. Virol. 2014, 88, 12017–12025. [Google Scholar] [CrossRef]
- Biedermann, P.; Klink, P.; Nocke, M.K.; Papp, C.P.; Harms, D.; Kebelmann, M.; Thürmer, A.; Choi, M.; Altmann, B.; Todt, D.; et al. Insertions and deletions in the hypervariable region of the hepatitis E virus genome in individuals with acute and chronic infection. Liver Int. Off. J. Int. Assoc. Study Liver 2023, 43, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Gorris, M.; van der Lecq, B.M.; van Erpecum, K.J.; de Bruijne, J. Treatment for chronic hepatitis E virus infection: A systematic review and meta-analysis. J. Viral Hepat. 2021, 28, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal 2011, 17, 10. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Ontiveros, R.J.; Stoute, J.; Liu, K.F. The chemical diversity of RNA modifications. Biochem. J. 2019, 476, 1227–1245. [Google Scholar] [CrossRef]
- Lichinchi, G.; Zhao, B.S.; Wu, Y.; Lu, Z.; Qin, Y.; He, C.; Rana, T.M. Dynamics of Human and Viral RNA Methylation during Zika Virus Infection. Cell Host Microbe 2016, 20, 666–673. [Google Scholar] [CrossRef]
- Brocard, M.; Ruggieri, A.; Locker, N. m6A RNA methylation, a new hallmark in virus-host interactions. J. Gen. Virol. 2017, 98, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Hao, S.; Chen, H.; Chen, Z.; Zhang, Y.; Wang, J.; Wang, H.; Zhang, B.; Qiu, J.; Deng, F.; et al. N6-methyladenosine modification and METTL3 modulate enterovirus 71 replication. Nucleic Acids Res. 2018, 47, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.D.; Gokhale, N.S.; Horner, S.M. Regulation of Viral Infection by the RNA Modification N6-Methyladenosine. Annu. Rev. Virol. 2019, 6, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Burgess, H.M.; Depledge, D.P.; Thompson, L.; Srinivas, K.P.; Grande, R.C.; Vink, E.I.; Abebe, J.S.; Blackaby, W.P.; Hendrick, A.; Albertella, M.R.; et al. Targeting the m6A RNA modification pathway blocks SARS-CoV-2 and HCoV-OC43 replication. Genes Dev. 2021, 35, 1005–1019. [Google Scholar] [CrossRef]
- Ulrich, J.U.; Lutfi, A.; Rutzen, K.; Renard, B.Y. ReadBouncer: Precise and scalable adaptive sampling for nanopore sequencing. Bioinformatics 2022, 38, i153–i160. [Google Scholar] [CrossRef]
- Tausch, S.H.; Strauch, B.; Andrusch, A.; Loka, T.P.; Lindner, M.S.; Nitsche, A.; Renard, B.Y. LiveKraken—Real-time metagenomic classification of illumina data. Bioinformatics 2018, 34, 3750–3752. [Google Scholar] [CrossRef]
- Näher, A.F.; Vorisek, C.N.; Klopfenstein, S.A.; Lehne, M.; Thun, S.; Alsalamah, S.; Pujari, S.; Heider, D.; Ahrens, W.; Pigeot, I.; et al. Secondary data for global health digitalisation. Lancet Digit. Health 2023, 5, e93–e101. [Google Scholar] [CrossRef]
- Wittig, A.; Miranda, F.; Hölzer, M.; Altenburg, T.; Bartoszewicz, J.M.; Beyvers, S.; Dieckmann, M.A.; Genske, U.; Giese, S.H.; Nowicka, M.; et al. CovRadar: Continuously tracking and filtering SARS-CoV-2 mutations for genomic surveillance. Bioinformatics 2022, 38, 4223–4225. [Google Scholar] [CrossRef]
- Nasri, F.; Kongkitimanon, K.; Wittig, A.; Cortés, J.; Brinkmann, A.; Nitsche, A.; Schmachtenberg, A.; Renard, B.; Fuchs, S. MpoxRadar: A worldwide MPXV genomic surveillance dashboard. Nucleic Acids Res. 2023, 51, W331–W337. [Google Scholar] [CrossRef]
- Bartoszewicz, J.M.; Seidel, A.; Renard, B.Y. Interpretable detection of novel human viruses from genome sequencing data. NAR Genom. Bioinform. 2021, 3, lqab004. [Google Scholar] [CrossRef]
- Pérot, P.; Fourgeaud, J.; Rouzaud, C.; Regnault, B.; Rocha, N.D.; Fontaine, H.; Pavec, J.L.; Dolidon, S.; Garzaro, M.; Chrétien, D.; et al. Circovirus Hepatitis Infection in Heart-Lung Transplant Patient, France. Emerg. Infect. Dis. 2023, 29, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, D.W.; Zagordi, O.; Geissberger, F.D.; Kufner, V.; Schmutz, S.; Böni, J.; Metzner, K.J.; Trkola, A.; Huber, M. Optimization and validation of sample preparation for metagenomic sequencing of viruses in clinical samples. Microbiome 2017, 5, 94. [Google Scholar] [CrossRef]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome Capture Sequencing Enables Sensitive Viral Diagnosis and Comprehensive Virome Analysis. mBio 2015, 6, e01491-15. [Google Scholar] [CrossRef] [PubMed]
- LaPierre, N.; Mangul, S.; Alser, M.; Mandric, I.; Wu, N.C.; Koslicki, D.; Eskin, E. MiCoP: Microbial community profiling method for detecting viral and fungal organisms in metagenomic samples. BMC Genom. 2019, 20, 423. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3, e00069-18. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Liang, Q.; Bible, P.W.; Liu, Y.; Zou, B.; Wei, L. DeepMicrobes: Taxonomic classification for metagenomics with deep learning. NAR Genom. Bioinform. 2020, 2, lqaa009. [Google Scholar] [CrossRef]
- Sit, T.H.C.; Sun, W.; Tse, A.C.N.; Brackman, C.J.; Cheng, S.M.S.; Tang, A.W.Y.; Cheung, J.T.L.; Peiris, M.; Poon, L.L.M. Novel Zoonotic Avian Influenza A(H3N8) Virus in Chicken, Hong Kong, China. Emerg. Infect. Dis. 2022, 28, 2009–2015. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Mirinaviciute, G.; Niqueux, E.; Stahl, K.; Staubach, C.; Terregino, C.; et al. Avian influenza overview December 2022–March 2023. EFSA J. 2023, 21, e07917. [Google Scholar]
- Mollentze, N.; Babayan, S.A.; Streicker, D.G. Identifying and prioritizing potential human-infecting viruses from their genome sequences. PLoS Biol. 2021, 19, e3001390. [Google Scholar] [CrossRef]
- Steinegger, M.; S"oding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 2018, 9, 2542. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wojtczak, D. Dive into machine learning algorithms for influenza virus host prediction with hemagglutinin sequences. Biosystems 2022, 220, 104740. [Google Scholar] [CrossRef]
- Carlson, C.J.; Farrell, M.J.; Grange, Z.; Han, B.A.; Mollentze, N.; Phelan, A.L.; Rasmussen, A.L.; Albery, G.F.; Bett, B.; Brett-Major, D.M.; et al. The future of zoonotic risk prediction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2021, 376, 20200358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Dong, X.; Liu, G.H.; Gao, Y.D. Risk and protective factors for COVID-19 morbidity, severity, and mortality. Clin. Rev. Allergy Immunol. 2023, 64, 90–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, H.; Wu, P.; Uyeki, T.M.; Feng, L.; Lai, S.; Wang, L.; Huo, X.; Xu, K.; Chen, E.; et al. Epidemiology of avian influenza A H7N9 virus in human beings across five epidemics in mainland China, 2013-17: An epidemiological study of laboratory-confirmed case series. Lancet Infect. Dis. 2017, 17, 822–832. [Google Scholar] [CrossRef]
- Bai, T.; Chen, Y.; Beck, S.; Stanelle-Bertram, S.; Mounogou, N.K.; Chen, T.; Dong, J.; Schneider, B.; Jia, T.; Yang, J.; et al. H7N9 avian influenza virus infection in men is associated with testosterone depletion. Nat. Commun. 2022, 13, 6936. [Google Scholar] [CrossRef]
- Schroeder, M.; Schaumburg, B.; Mueller, Z.; Parplys, A.; Jarczak, D.; Roedl, K.; Nierhaus, A.; de Heer, G.; Grensemann, J.; Schneider, B.; et al. High estradiol and low testosterone levels are associated with critical illness in male but not in female COVID-19 patients: A retrospective cohort study. Emerg. Microbes Infect. 2021, 10, 1807–1818. [Google Scholar] [CrossRef]
- Salonia, A.; Pontillo, M.; Capogrosso, P.; Pozzi, E.; Ferrara, A.M.; Cotelessa, A.; Belladelli, F.; Corsini, C.; Gregori, S.; Rowe, I.; et al. Testosterone in males with COVID-19: A 12-month cohort study. Andrology 2023, 11, 17–23. [Google Scholar] [CrossRef]
- Garner, E.; Davis, B.C.; Milligan, E.; Blair, M.F.; Keenum, I.; Maile-Moskowitz, A.; Pan, J.; Gnegy, M.; Liguori, K.; Gupta, S.; et al. Next generation sequencing approaches to evaluate water and wastewater quality. Water Res. 2021, 194, 116907. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hufsky, F.; Abecasis, A.B.; Babaian, A.; Beck, S.; Brierley, L.; Dellicour, S.; Eggeling, C.; Elena, S.F.; Gieraths, U.; Ha, A.D.; et al. The International Virus Bioinformatics Meeting 2023. Viruses 2023, 15, 2031. https://doi.org/10.3390/v15102031
Hufsky F, Abecasis AB, Babaian A, Beck S, Brierley L, Dellicour S, Eggeling C, Elena SF, Gieraths U, Ha AD, et al. The International Virus Bioinformatics Meeting 2023. Viruses. 2023; 15(10):2031. https://doi.org/10.3390/v15102031
Chicago/Turabian StyleHufsky, Franziska, Ana B. Abecasis, Artem Babaian, Sebastian Beck, Liam Brierley, Simon Dellicour, Christian Eggeling, Santiago F. Elena, Udo Gieraths, Anh D. Ha, and et al. 2023. "The International Virus Bioinformatics Meeting 2023" Viruses 15, no. 10: 2031. https://doi.org/10.3390/v15102031