
Alterations of Nuclear Architecture and Epigenetic Signatures during African Swine Fever Virus Infection



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
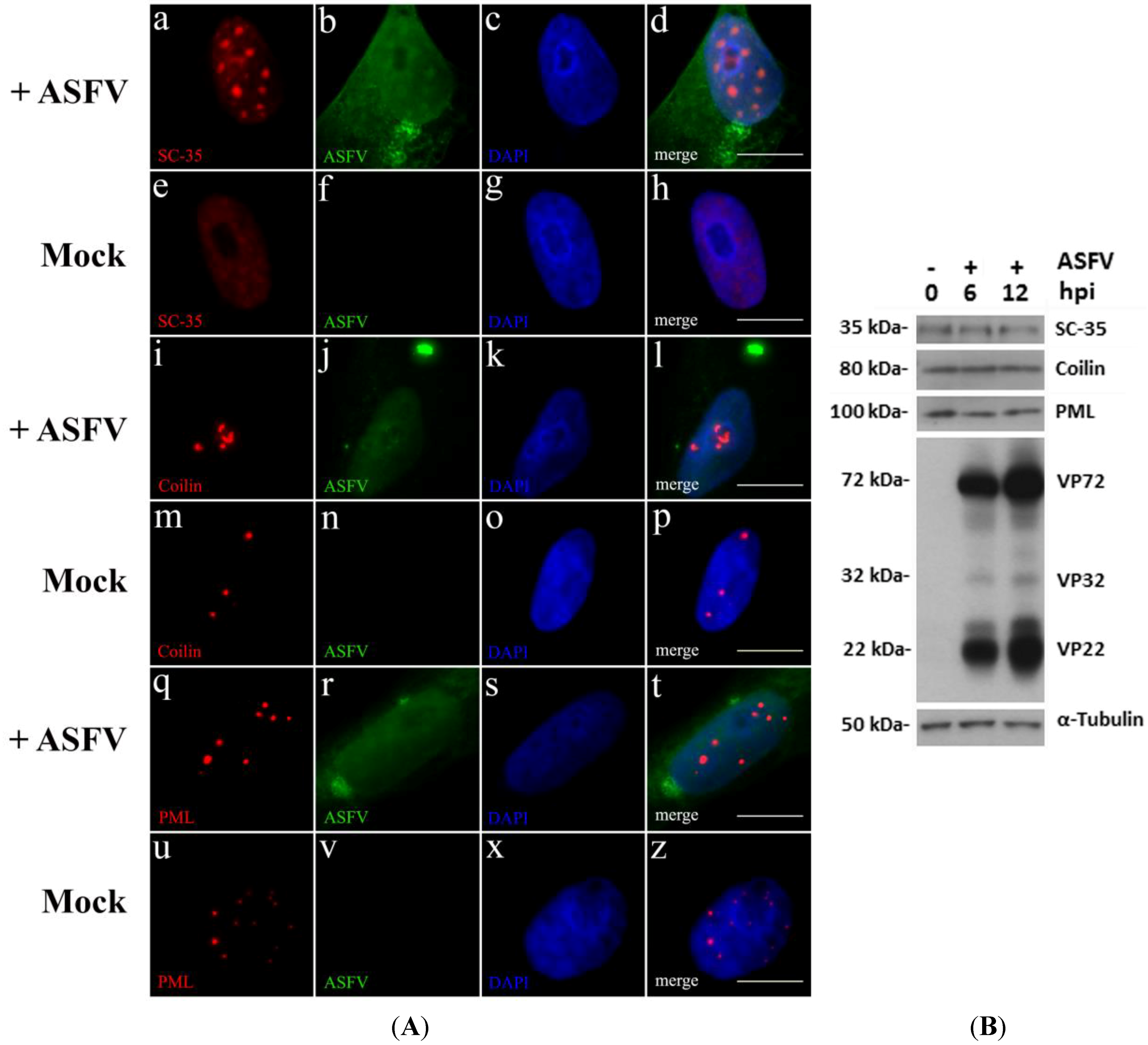
2.1. ASFV Disrupts Host Subnuclear Domains
2.2. ATR-Related Factors Accumulate near PML-NBs during ASFV Infection


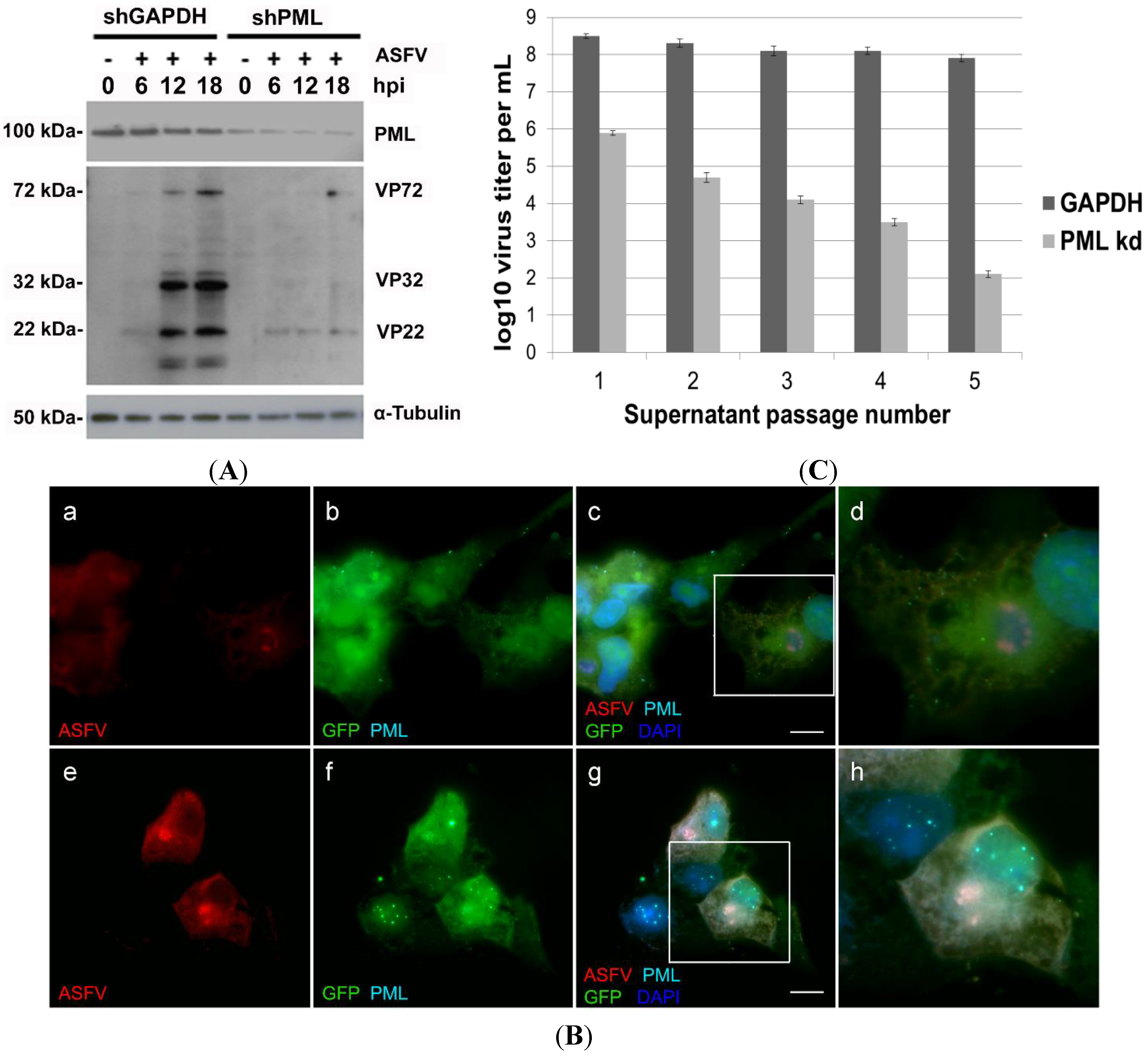
2.3. PML Protein Plays a Proviral Role in ASFV Infection

2.4. ASFV Modifies Host Chromatin Epigenetic State

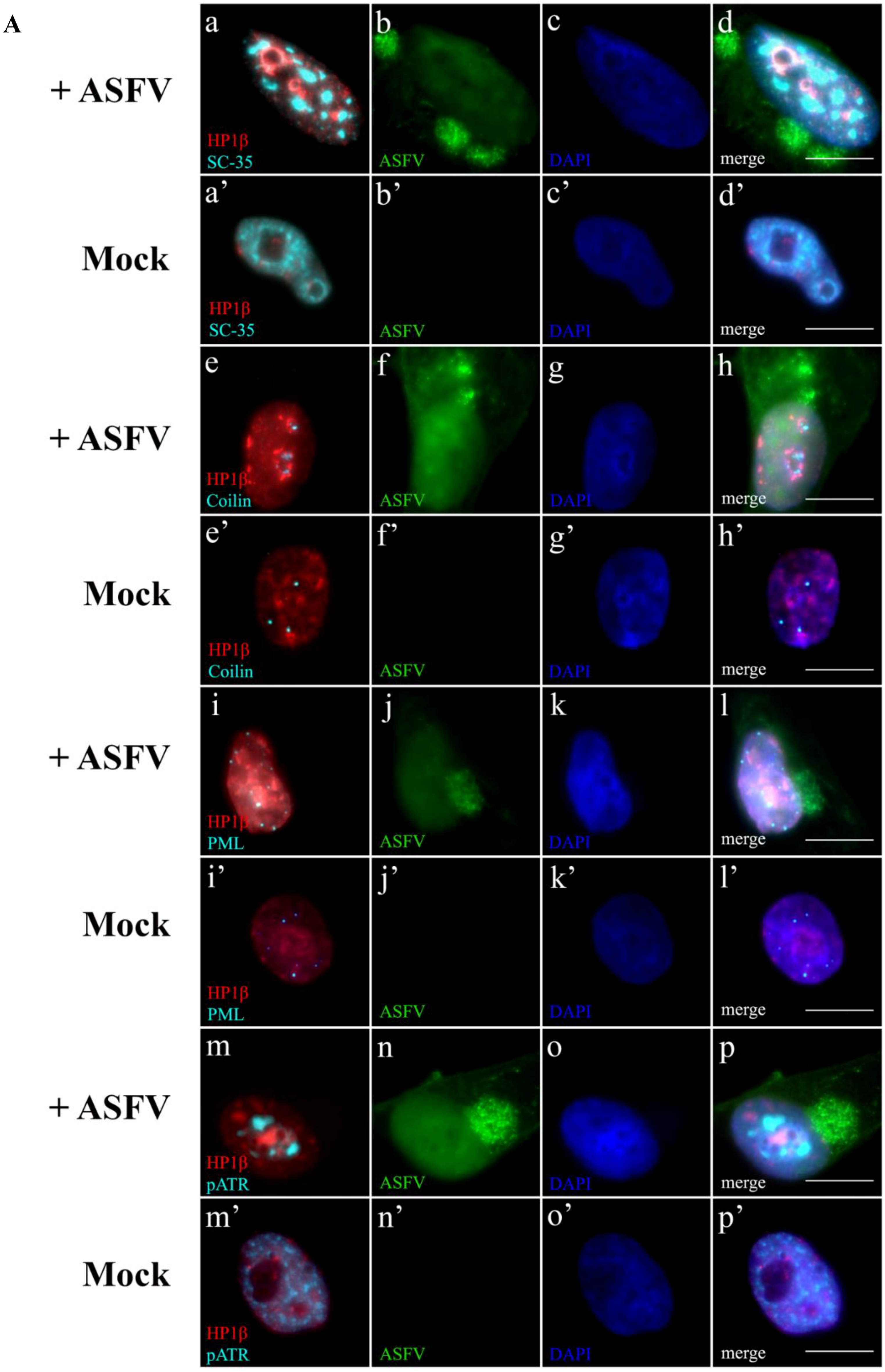
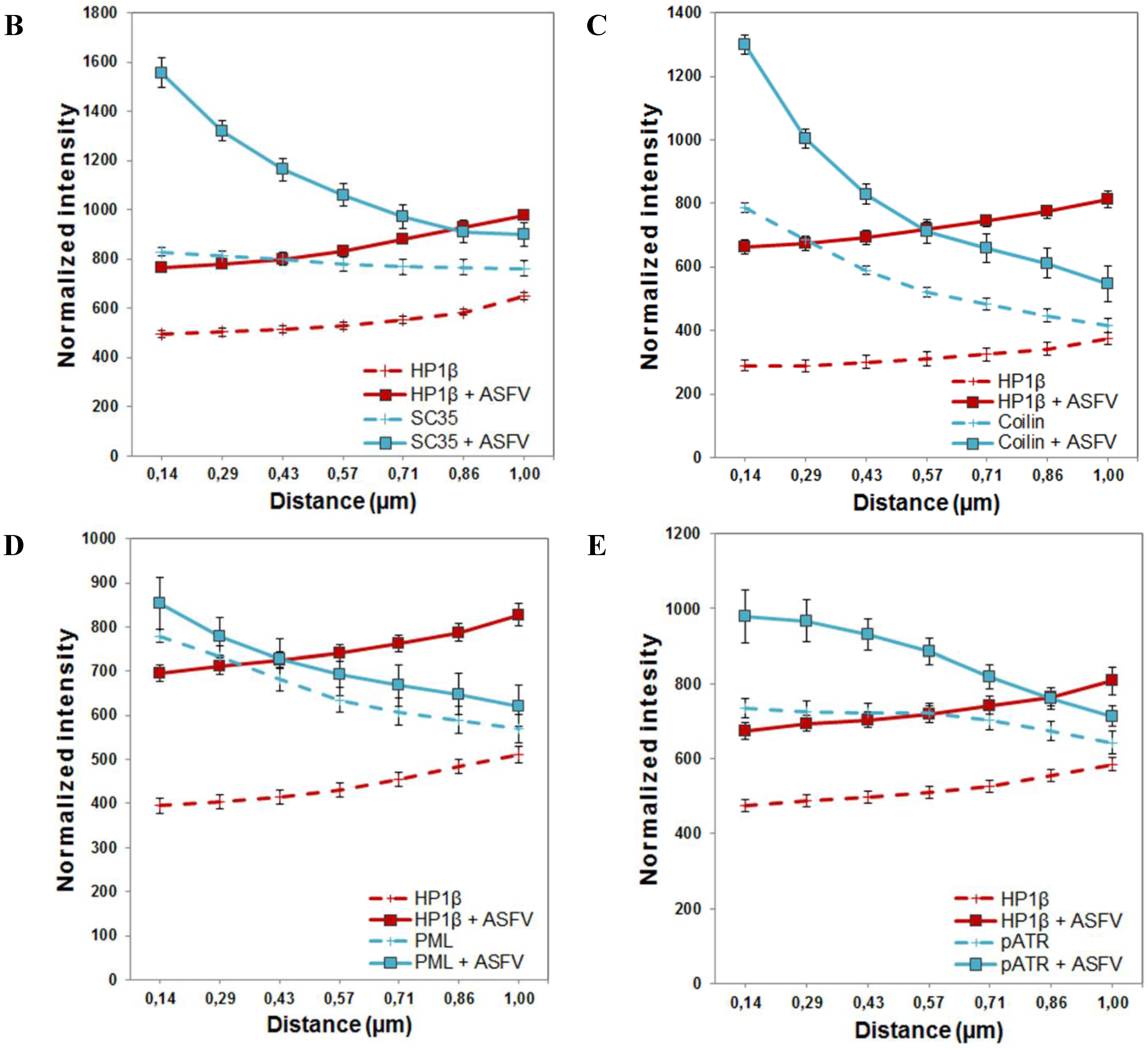
2.5. Subnuclear Domains and ATR Loci Accumulate nearby Heterochromatic Regions during ASFV Infection
3. Discussion



4. Materials and Methods
4.1. Vero Cell Culture and Lentiviral Infection of shRNA
4.2. Virus and Infections
4.3. Antibodies
4.4. Immunofluorescence Studies
4.5. Microscopy and Image Processing
4.6. Radial Analysis of Site Images
4.7. Western Blotting Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sánchez-Vizcaíno, J.M.; Mur, L.; Martínez-López, B. African swine fever (ASF): Five years around Europe. Vet. Microbiol. 2013, 165, 45–50. [Google Scholar] [CrossRef] [PubMed]
- European Commission. African Swine Fever. EFSA Panel on Animal Health and Welfare (AHAW). EFSA J. 2015, 13. [Google Scholar] [CrossRef]
- King, A.M.Q.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Ninth report of the International Committee on Taxonomy of Viruses. In Virus Taxonomy; King, A.M.Q., Ed.; Elsevier: London, UK, 2011; pp. 153–162. [Google Scholar]
- Sánchez, E.G.; Quintas, A.; Nogal, M.; Castelló, A.; Revilla, Y. African swine fever virus controls the host transcription and cellular machinery of protein synthesis. Virus Res. 2013, 173, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Ballester, M.; Rodríguez-Cariño, C.; Pérez, M.; Gallardo, C.; Rodríguez, J.M.; Salas, M.L.; Rodriguez, F. Disruption of nuclear organization during the initial phase of African swine fever virus infection. J. Virol. 2011, 85, 8263–8269. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.; Martins, C.; Ferreira, F. Host DNA damage response facilitates African swine fever virus infection. Vet. Microbiol. 2013, 165, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Granja, A.G.; Nogal, M.L.; Hurtado, C.; Salas, J.; Salas, M.L.; Carrascosa, A.L.; Revilla, Y. Modulation of p53 cellular function and cell death by African swine fever virus. J. Virol. 2004, 78, 7165–7174. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.; Martins, C.L.; Ferreira, F. Early intranuclear replication of African swine fever virus genome modifies the landscape of the host cell nucleus. Virus Res. 2015, 210, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Chromatin regulation of virus infection. Trends Microbiol. 2006, 14, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Jung, J.U. The chromatin landscape of Kaposi’s sarcoma-associated herpesvirus. Viruses 2013, 5, 1346–1373. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 2001, 20, 7266–7273. [Google Scholar] [CrossRef] [PubMed]
- Ihalainen, T.O.; Niskanen, E.A.; Jylhävä, J.; Paloheimo, O.; Dross, N.; Smolander, H.; Langowski, J.; Timonen, J.; Vihinen-Ranta, M. Parvovirus induced alterations in nuclear architecture and dynamics. PLoS ONE 2009, 4, e5948. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Godinez, W.J.; Kim, I.-H.H.; Tektonidis, M.; De Lanerolle, P.; Eils, R.; Rohr, K.; Knipe, D.M. Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, E136–144. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Dauber, B.; Melén, K.; Julkunen, I.; Wolff, T. Analysis of influenza B Virus NS1 protein trafficking reveals a novel interaction with nuclear speckle domains. J. Virol. 2008, 83, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Gama-Carvalho, M.; Condado, I.; Carmo-Fonseca, M. Regulation of adenovirus alternative RNA splicing correlates with a reorganization of splicing factors in the nucleus. Exp. Cell Res. 2003, 289, 77–85. [Google Scholar] [CrossRef]
- James, N.J.; Howell, G.J.; Walker, J.H.; Blair, G.E. The role of Cajal bodies in the expression of late phase adenovirus proteins. Virology 2010, 399, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Zimmerman, N.; Chen, T.; Domagala, M.; Frappier, L. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog. 2008, 4, e1000100. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Ryabov, E.V.; Kalinina, N.O.; Rakitina, D.V; Gillespie, T.; MacFarlane, S.; Haupt, S.; Brown, J.W.S.; Taliansky, M. Cajal bodies and the nucleolus are required for a plant virus systemic infection. EMBO J. 2007, 26, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Möller, A.; Schmitz, M.L. Viruses as hijackers of PML nuclear bodies. Arch. Immunol. Ther. Exp. (Warsz). 2003, 51, 295–300. [Google Scholar] [PubMed]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Sarkari, F.; Wang, X.; Nguyen, T.; Frappier, L. The Herpesvirus Associated Ubiquitin Specific Protease, USP7, Is a Negative Regulator of PML Proteins and PML Nuclear Bodies. PLoS ONE 2011, 6, e16598. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Papior, P.; Rechter, S.; Stamminger, T. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J. Virol. 2008, 82, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Lieberman, P.M.; Jung, J.U.; McBride, A.A.; Morris, K.V.; Ott, M.; Margolis, D.; Nieto, A.; Nevels, M.; Parks, R.J.; Kristie, T.M. Snapshots: Chromatin control of viral infection. Virology 2013, 435, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Genomes in conflict: Maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 2010, 64, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Fillingham, J.; Greenblatt, J.F. A histone code for chromatin assembly. Cell 2008, 134, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Polo, S.E.; Almouzni, G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol. Cell 2012, 46, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.I.S.; Jia, S. Heterochromatin revisited. Nat. Rev. Genet. 2007, 8, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Ball, A.R.; Yokomori, K. HP1: Heterochromatin binding proteins working the genome. Epigenetics Off. J. DNA Methylation Soc. 2010, 5, 287–292. [Google Scholar] [CrossRef]
- Eskeland, R.; Eberharter, A.; Imhof, A. HP1 binding to chromatin methylated at H3K9 is enhanced by auxiliary factors. Mol. Cell. Biol. 2007, 27, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Luijsterburg, M.S.; Dinant, C.; Lans, H.; Stap, J.; Wiernasz, E.; Lagerwerf, S.; Warmerdam, D.O.; Lindh, M.; Brink, M.C.; Dobrucki, J.W.; et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol. 2009, 185, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Moudry, P.; Hodny, Z.; Lukas, J.; Rajpert-De Meyts, E.; Bartek, J. Heterochromatin marks HP1γ, HP1α and H3K9me3, and DNA damage response activation in human testis development and germ cell tumours. Int. J. Androl. 2011, 34, e103–e113. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.L.; Kristie, T.M. The dynamics of HCF-1 modulation of herpes simplex virus chromatin during initiation of infection. Viruses 2013, 5, 1272–1291. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, V.; Everett, R.D. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol. 2010, 84, 4026–4040. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Maul, G.G. Mouse cytomegalovirus immediate-early protein 1 binds with host cell repressors to relieve suppressive effects on viral transcription and replication during lytic infection. J. Virol. 2003, 77, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Zakaryan, H.; Stamminger, T. Nuclear remodelling during viral infections. Cell. Microbiol. 2011, 13, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Lamond, A.I.; Spector, D.L. Nuclear speckles: A model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 2003, 4, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.X.; Imperiale, M.J. Design stars: How small DNA viruses remodel the host nucleus. Future Virol. 2012, 7, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Salomoni, P.; Luo, J.; Shih, A.; Zhong, S.; Gu, W.; Pandolfi, P.P. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2000, 2, 730–736. [Google Scholar] [PubMed]
- De Stanchina, E.; Querido, E.; Narita, M.; Davuluri, R.V.; Pandolfi, P.P.; Ferbeyre, G.; Lowe, S.W. PML is a direct p53 target that modulates p53 effector functions. Mol. Cell 2004, 13, 523–535. [Google Scholar] [CrossRef]
- Pampin, M.; Simonin, Y.; Blondel, B.; Percherancier, Y.; Chelbi-Alix, M.K. Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. J. Virol. 2006, 80, 8582–8592. [Google Scholar] [CrossRef] [PubMed]
- Dellaire, G.; Ching, R.W.; Ahmed, K.; Jalali, F.; Tse, K.C.K.; Bristow, R.G.; Bazett-Jones, D.P. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR. J. Cell Biol. 2006, 175, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Vink, E.I.; Zheng, Y.; Yeasmin, R.; Stamminger, T.; Krug, L.T.; Hearing, P. Impact of Adenovirus E4-ORF3 oligomerization and protein localization on cellular gene expression. Viruses 2015, 7, 2428–2449. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-E.; Lee, J.-H.; Kim, E.T.; Shin, H.J.; Gu, S.Y.; Seol, H.S.; Ling, P.; Lee, C.H.; Ahn, J.-H. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J. Virol. 2011, 85, 11928–11937. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates, J.R.; Gray, N.S.; Lee, J.-D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010, 18, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, A.L.; Bustos, M.J.; Leon, P. Methods for growing and titrating African swine fever virus: field and laboratory samples. Curr. Protoc. Cell Biol. 2011, 53, 26.14.1–26.14.25. [Google Scholar]
- Fodor, B.D.; Shukeir, N.; Reuter, G.; Jenuwein, T. Mammalian Su(var) genes in chromatin control. Annu. Rev. Cell Dev. Biol. 2010, 26, 471–501. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; Shilatifard, A.; Workman, J.; Zhang, Y. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Workman, J.L. The heterochromatin protein 1 (HP1) family: put away a bias toward HP1. Mol. Cells 2008, 26, 217–227. [Google Scholar] [PubMed]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases (HDACs) in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin. Epigenetics 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Aygün, O.; Mehta, S.; Grewal, S.I.S. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat. Struct. Mol. Biol. 2013, 20, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Chaurushiya, M.S.; Weitzman, M.D. Chromatin at the intersection of viral infection and DNA damage. Biochim. Biophys. Acta 2010, 1799, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Molina, Y.; Martínez, F.; Tang, Q. Nuclear domain 10 of the viral aspect. World J. Virol. 2013, 2, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Netherton, C.L.; Wileman, T.E. African swine fever virus organelle rearrangements. Virus Res. 2013, 173, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Stamminger, T. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta 2008, 1783, 2207–2221. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, E.; Van Ostade, X. Crosstalk between viruses and PML nuclear bodies: A network-based approach. Front. Biosci. 2011, 16, 2910–2920. [Google Scholar] [CrossRef]
- Wang, S.; Long, J.; Zheng, C. The potential link between PML NBs and ICP0 in regulating lytic and latent infection of HSV-1. Protein Cell 2012, 3, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Hu, L.; Makielski, K.; Pandolfi, P.P.; Gjoerup, O.V. Functional connection between Rad51 and PML in homology-directed repair. PLoS ONE 2011, 6, e25814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, C.L.; Ramalho, M.; Nadkarni, N.; May Kong, W.; Higgins, C.F.; Krauzewicz, N. Role for centromeric heterochromatin and PML nuclear bodies in the cellular response to foreign DNA. Mol. Cell. Biol. 2006, 26, 2583–2594. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Murray, J.; Orr, A.; Preston, C.M. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 2007, 81, 10991–11004. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Workman, J.L. The changing faces of HP1: From heterochromatin formation and gene silencing to euchromatic gene expression: HP1 acts as a positive regulator of transcription. BioEssays News Rev. Mol. Cell. Dev. Biol. 2011, 33, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; Dal Zuffo, R.; Matti, V.; D’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Porwal, M.; Cohen, S.; Snoussi, K.; Popa-Wagner, R.; Anderson, F.; Dugot-Senant, N.; Wodrich, H.; Dinsart, C.; Kleinschmidt, J.A.; Panté, N.; Kann, M. Parvoviruses Cause Nuclear Envelope Breakdown by Activating Key Enzymes of Mitosis. PLoS Pathog. 2013, 9, e1003671. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.; Cliffe, A.; Chang, L.; Knipe, D.M. Role for A-Type Lamins in Herpesviral DNA Targeting and Heterochromatin Modulation. PLoS Pathog. 2008, 4, e1000071. [Google Scholar] [CrossRef] [PubMed]
- Cibulka, J.; Fraiberk, M.; Forstova, J. Nuclear Actin and Lamins in Viral Infections. Viruses 2012, 4, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Snoussi, K.; Kann, M. Interaction of parvoviruses with the nuclear envelope. Adv. Biol. Regul. 2014, 54, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Marchion, D.C.; Bicaku, E.; Turner, J.G.; Schmitt, M.L.; And, D.R.M.; Munster, P.N. HDAC2 regulates chromatin plasticity and enhances DNA vulnerability. Mol. Cancer Ther. 2009, 8, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Wendling, D. Histone deacetylases in viral infections. Clin. Epigenetics 2010, 1, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Moreno, R.; Barrado-Gil, L.; Galindo, I.; Alonso, C. Analysis of HDAC6 and BAG3-aggresome pathways in African swine fever viral factory formation. Viruses 2015, 7, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Adhya, D.; Dutta, K.; Kundu, K.; Basu, A. Histone deacetylase inhibition by Japanese encephalitis virus in monocyte/macrophages: a novel viral immune evasion strategy. Immunobiology 2013, 218, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Siddiquey, M.; Nakagawa, H.; Iwata, S.; Kanazawa, T.; Suzuki, M.; Imadome, K.-I.; Fujiwara, S.; Goshima, F.; Murata, T.; Kimura, H. Anti-tumor effects of suberoylanilide hydroxamicacid on Epstein-Barr virus-associated T cell andnatural killer cell lymphoma. Cancer Sci. 2014, 105, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Conn, K.; Schang, L. Chromatin dynamics during lytic infection with herpes simplex virus 1. Viruses 2013, 5, 1758–1786. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Studach, L.L.; Andrisani, O.M. Proteins ZNF198 and SUZ12 are down-regulated in hepatitis B virus (HBV) X protein-mediated hepatocyte transformation and in HBV replication. Hepatology 2011, 53, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Rabellino, A.; Carter, B.J.; Konstantinidou, G.; Shwu-Yuan, W.; Rimessi, A.; Byers, L.A.; Heymach, J.V.; Girard, L.; Chiang, C.-M.; Teruya-Feldstein, J.; Scaglioni, P.P. The SUMO E3-ligase PIAS1 regulates the tumor suppressor PML and its oncogenic counterpart PML-RARA. Cancer Res. 2012, 72, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.; Scholl, T.; Mebus, C.A.; Fisch, H.; Lawman, M. Modulation of porcine peripheral blood-derived macrophage functions by in vitro infection with African swine fever virus (ASFV) isolates of different virulence. Viral Immunol. 1987, 1, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Spiluttini, B.; Gu, B.; Belagal, P.; Smirnova, A.S.; Nguyen, V.T.; Hébert, C.; Schmidt, U.; Bertrand, E.; Darzacq, X.; Bensaude, O. Splicing-independent recruitment of U1 snRNP to a transcription unit in living cells. J. Cell Sci. 2010, 123, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simões, M.; Rino, J.; Pinheiro, I.; Martins, C.; Ferreira, F. Alterations of Nuclear Architecture and Epigenetic Signatures during African Swine Fever Virus Infection. Viruses 2015, 7, 4978-4996. https://doi.org/10.3390/v7092858
Simões M, Rino J, Pinheiro I, Martins C, Ferreira F. Alterations of Nuclear Architecture and Epigenetic Signatures during African Swine Fever Virus Infection. Viruses. 2015; 7(9):4978-4996. https://doi.org/10.3390/v7092858
Chicago/Turabian StyleSimões, Margarida, José Rino, Inês Pinheiro, Carlos Martins, and Fernando Ferreira. 2015. "Alterations of Nuclear Architecture and Epigenetic Signatures during African Swine Fever Virus Infection" Viruses 7, no. 9: 4978-4996. https://doi.org/10.3390/v7092858