Immunogenic SARS-CoV-2 Epitopes: In Silico Study Towards Better Understanding of COVID-19 Disease—Paving the Way for Vaccine Development

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Source of Sequences
2.2. MHC Class I Epitope Prediction
2.3. Comparison of Predicted and Experimentally Known Epitopes
2.4. Epitopes Physicochemical Properties and eMHC-I Complex Stability
2.5. Docking and Structural Analyses
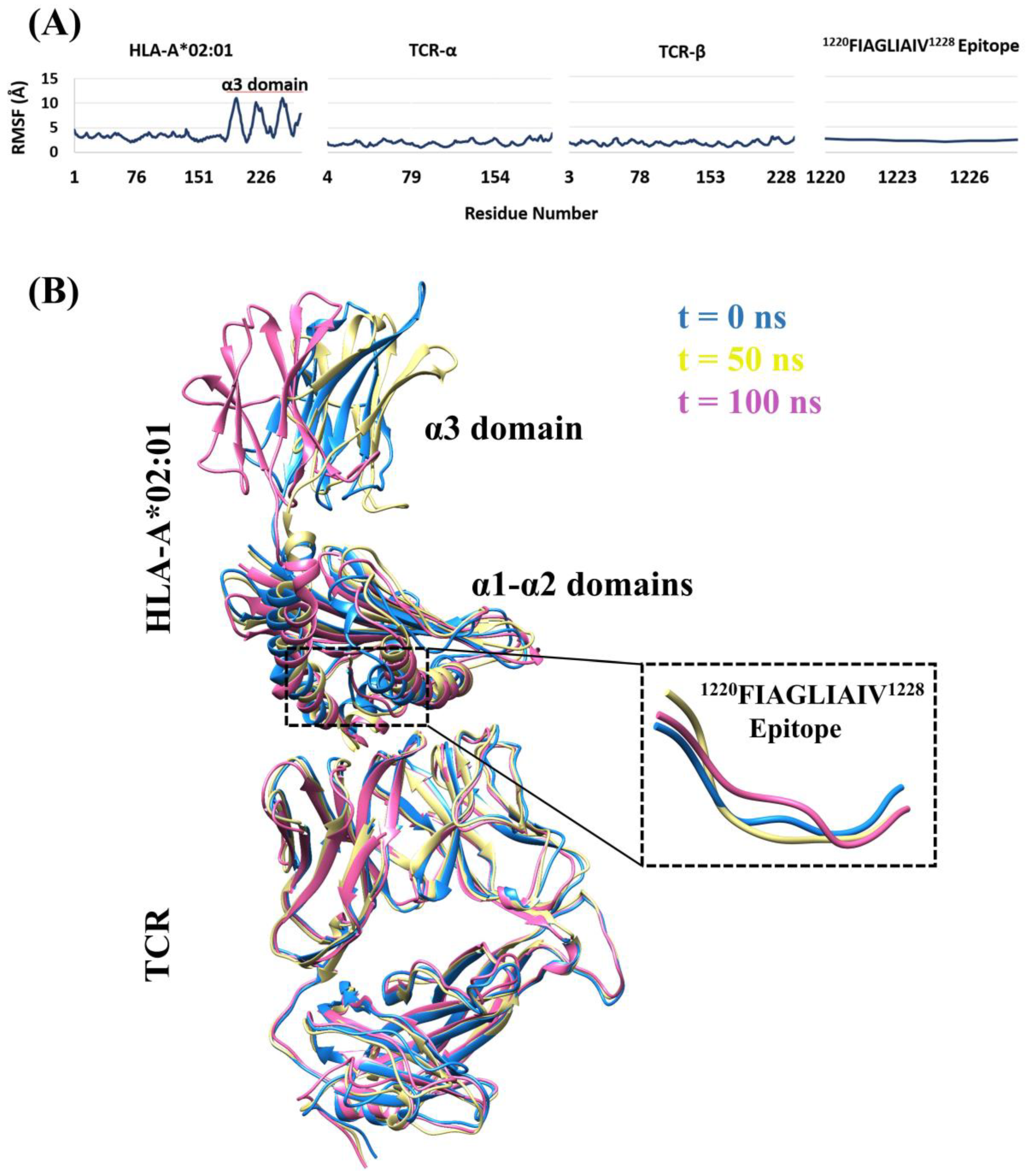
2.6. Molecular Dynamics Simulations
3. Results
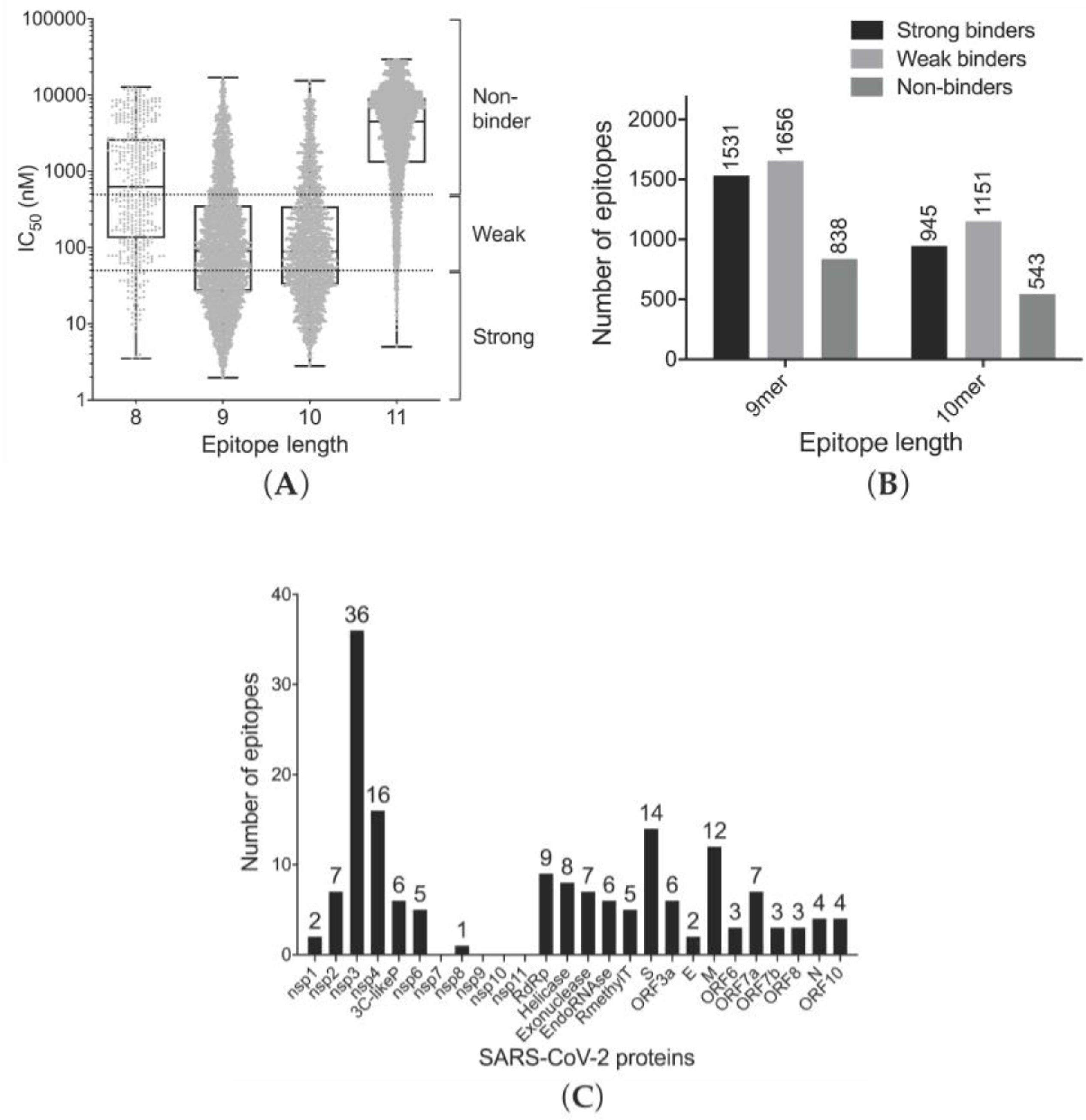
3.1. Prediction of Binding of SARS-CoV-2-Derived Peptides to MHC Class I Receptors
3.2. Analysis of Correlation between in Silico Identified SARS-CoV-2 (This Study) and Experimentally Validated SARS-CoV (from IEDB) Epitopes
3.3. Efficiency of Epitope Presentation to Stimulate an Immune Response
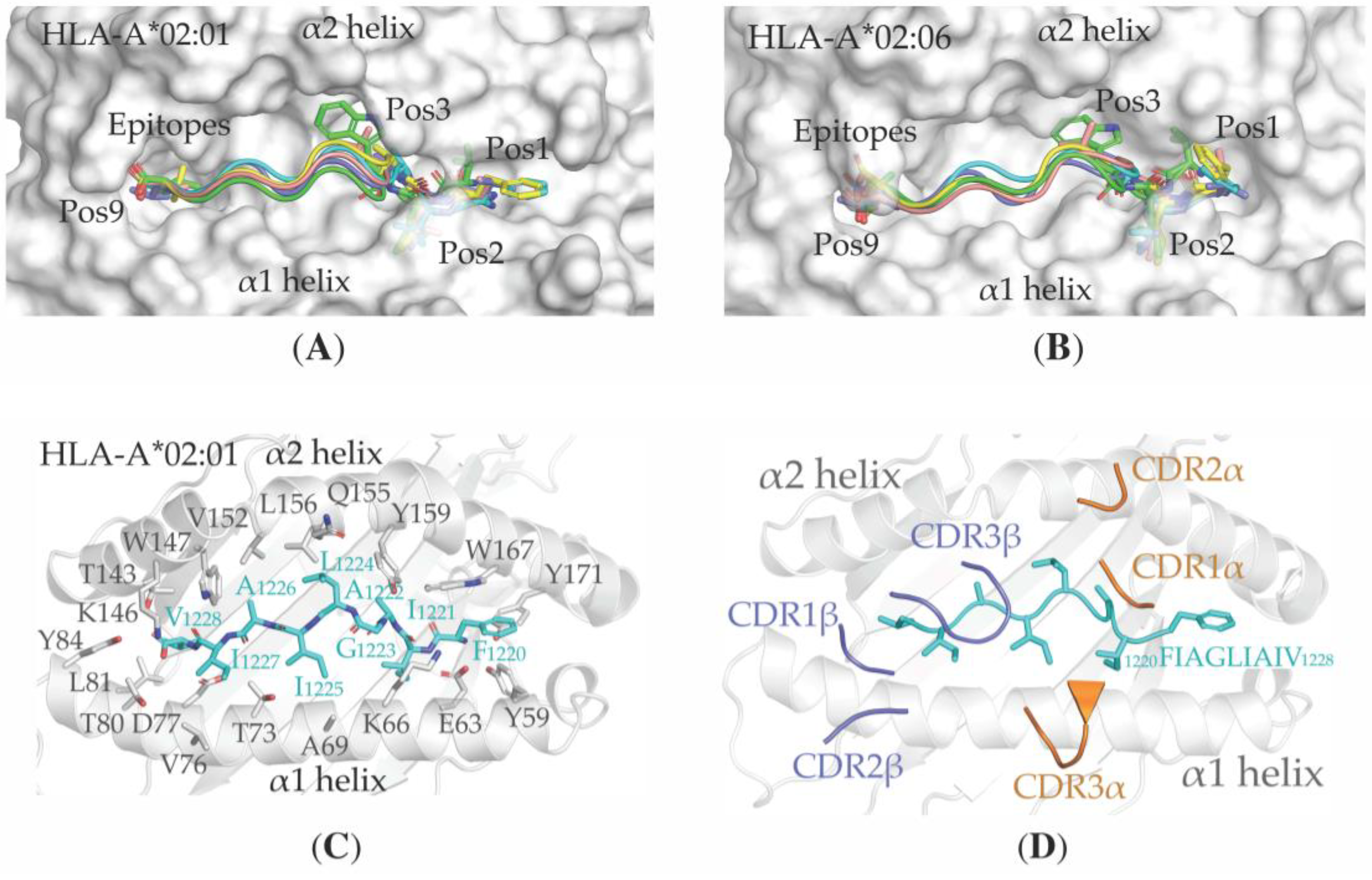
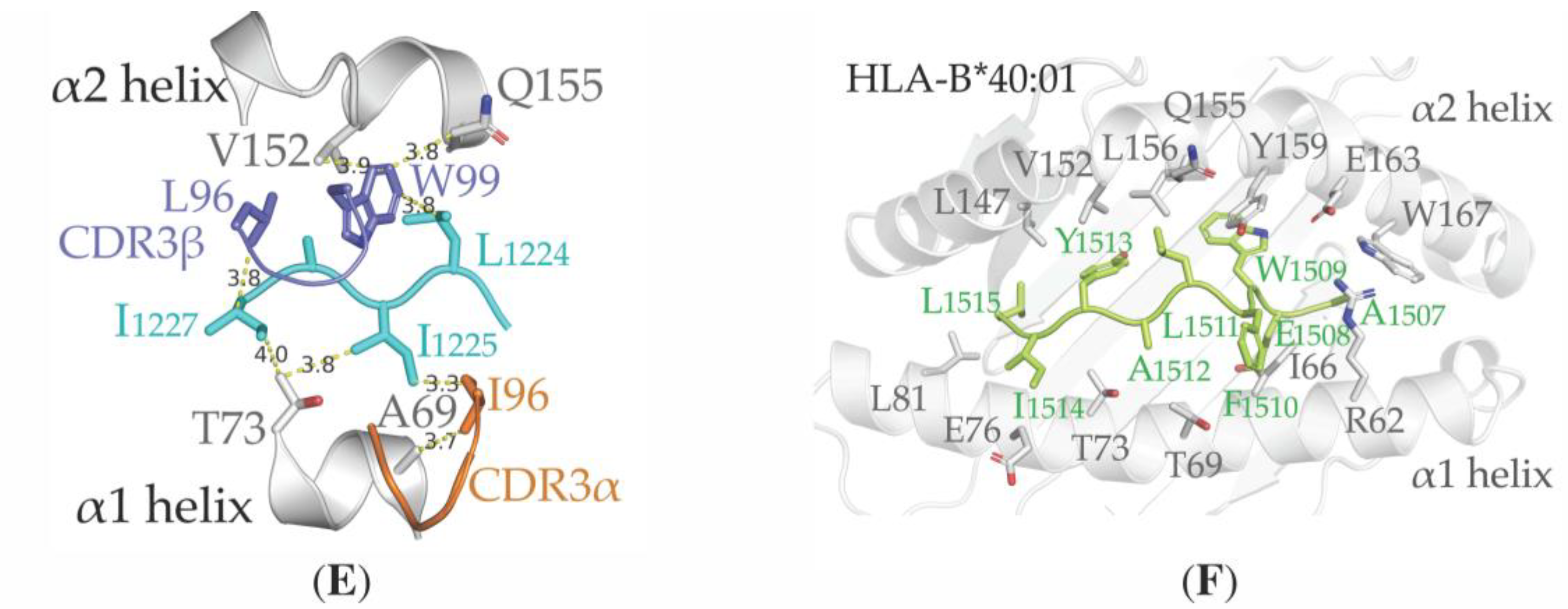
3.4. Structural Properties of the Peptide-HLA-A*02:01-Complexes Defining T Cell Receptor (TCR) Recognition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. Corrigendum: “World Health Organization declares Global Emergency: A review of the 2019 Novel Coronavirus (COVID-19)”. Int. J. Surg. 2020. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymann, D.L. Data sharing and outbreaks: Best practice exemplified. Lancet 2020, 395, 469–470. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, X.J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J. Genet. Genomics 2020, 47, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Singhal, T. A Review of Coronavirus Disease-2019 (COVID-19). Indian J. Pediatr. 2020, 87, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute respiratory distress syndrome advances in diagnosis and treatment. JAMA J. Am. Med. Assoc. 2018, 319, 698–710. [Google Scholar] [CrossRef]
- Verdoni, L.; Mazza, A.; Gervasoni, A.; Martelli, L.; Ruggeri, M.; Ciuffreda, M.; Bonanomi, E.; D’Antiga, L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet 2020, 395, 1771–1778. [Google Scholar] [CrossRef]
- Al Awaidy, S.T.; Khamis, F. Middle east respiratory syndrome coronavirus (Mers-cov) in Oman: Current situation and going forward. Oman Med. J. 2019, 34, 181–183. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Srinivasan, S.; Cui, H.; Gao, Z.; Liu, M.; Lu, S.; Mkandawire, W.; Narykov, O.; Sun, M.; Korkin, D. Structural genomics of SARS-COV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses 2020, 12, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denesyuk, A.I.; Johnson, M.S.; Salo-Ahen, O.M.H.; Uversky, V.N.; Denessiouk, K. NBCZone: Universal three-dimensional construction of eleven amino acids near the catalytic nucleophile and base in the superfamily of (chymo)trypsin-like serine fold proteases. Int. J. Biol. Macromol. 2020, 153, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Crux, N.B.; Elahi, S. Human Leukocyte Antigen (HLA) and immune regulation: How do classical and non-classical HLA alleles modulate immune response to human immunodeficiency virus and hepatitis C virus infections? Front. Immunol. 2017, 8, 1–26. [Google Scholar] [CrossRef]
- Lin, M.; Tseng, H.K.; Trejaut, J.A.; Lee, H.L.; Loo, J.H.; Chu, C.C.; Chen, P.J.; Su, Y.W.; Lim, K.H.; Tsai, Z.U.; et al. Association of HLA class I with severe acute respiratory syndrome coronavirus infection. BMC Med. Genet. 2003, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ng, O.W.; Chia, A.; Tan, A.T.; Jadi, R.S.; Leong, H.N.; Bertoletti, A.; Tan, Y.J. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine 2016, 34, 2008–2014. [Google Scholar] [CrossRef]
- Tsao, Y.P.; Lin, J.Y.; Jan, J.T.; Leng, C.H.; Chu, C.C.; Yang, Y.C.; Chen, S.L. HLA-A*0201 T-cell epitopes in severe acute respiratory syndrome (SARS) coronavirus nucleocapsid and spike proteins. Biochem. Biophys. Res. Commun. 2006, 344, 63–71. [Google Scholar] [CrossRef]
- Kohyama, S.; Ohno, S.; Suda, T.; Taneichi, M.; Yokoyama, S.; Mori, M.; Kobayashi, A.; Hayashi, H.; Uchida, T.; Matsui, M. Efficient induction of cytotoxic T lymphocytes specific for severe acute respiratory syndrome (SARS)-associated coronavirus by immunization with surface-linked liposomal peptides derived from a non-structural polyprotein 1a. Antivir. Res. 2009, 84, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B. Presence of SARS-CoV-2-reactive T cells in COVID-19 patients and healthy donors. medRxiv 2020. [Google Scholar] [CrossRef]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [Green Version]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI viral Genomes resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [Green Version]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lund, O.; Nielsen, M.; Kesmir, C.; Petersen, A.G.; Lundegaard, C.; Worning, P.; Sylvester-Hvid, C.; Lamberth, K.; Røder, G.; Justesen, S.; et al. Definition of supertypes for HLA molecules using clustering of specificity matrices. Immunogenetics 2004, 55, 797–810. [Google Scholar] [CrossRef]
- Sidney, J.; Peters, B.; Frahm, N.; Brander, C.; Sette, A. HLA class I supertypes: A revised and updated classification. BMC Immunol. 2008, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.; Fenoy, E.; Harndahl, M.; Kristensen, A.B.; Nielsen, I.K.; Nielsen, M.; Buus, S. Pan-Specific Prediction of Peptide–MHC Class I Complex Stability, a Correlate of T Cell Immunogenicity. J. Immunol. 2016, 197, 1517–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Chen, G.; Weng, N.-P.; Mariuzza, R.A. Structural basis for clonal diversity of the human T-cell response to a dominant influenza virus epitope. J. Biol. Chem. 2017, 292, 18618–18627. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, K.Y.; Ren, E.C. Structural insights into the binding of hepatitis B virus core peptide to HLA-A2 alleles: Towards designing better vaccines. Eur. J. Immunol. 2011, 41, 2097–2106. [Google Scholar] [CrossRef]
- Rose, P.W.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dimitropoulos, D.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Prlić, A.; Quesada, M.; et al. The RCSB Protein Data Bank: New resources for research and education. Nucleic Acids Res. 2013, 41, 475–482. [Google Scholar] [CrossRef]
- London, N.; Raveh, B.; Cohen, E.; Fathi, G.; Schueler-Furman, O. Rosetta FlexPepDock web server—High resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011, 39, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Tamirat, M.Z.; Kurppa, K.J.; Elenius, K.; Johnson, M.S. Deciphering the structural effects of activating EGFR somatic mutations with molecular dynamics simulation. J. Vis. Exp. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K. Amber 2018; University of California, San Francisco: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zipeto, D.; Beretta, A. HLA-C and HIV-1: Friends or foes? Retrovirology 2012, 9, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Sher, X. Systematically benchmarking peptide-MHC binding predictors: From synthetic to naturally processed epitopes. PLoS Comput. Biol. 2018, 14, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, 5–9. [Google Scholar] [CrossRef]
- Harndahl, M.; Rasmussen, M.; Roder, G.; Dalgaard Pedersen, I.; Sørensen, M.; Nielsen, M.; Buus, S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur. J. Immunol. 2012, 42, 1405–1416. [Google Scholar] [CrossRef]
- Colleton, B.A.; Huang, X.-L.; Melhem, N.M.; Fan, Z.; Borowski, L.; Rappocciolo, G.; Rinaldo, C.R. Primary Human Immunodeficiency Virus Type 1-Specific CD8+ T-Cell Responses Induced by Myeloid Dendritic Cells. J. Virol. 2009, 83, 6288–6299. [Google Scholar] [CrossRef] [Green Version]
- Bettencourt, P.; Müller, J.; Nicastri, A.; Cantillon, D.; Madhavan, M.; Charles, P.D.; Fotso, C.B.; Wittenberg, R.; Bull, N.; Pinpathomrat, N.; et al. Identification of antigens presented by MHC for vaccines against tuberculosis. npj Vaccines 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Chowell, D.; Krishna, S.; Becker, P.D.; Cocita, C.; Shu, J.; Tan, X.; Greenberg, P.D.; Klavinskis, L.S.; Blattman, J.N.; Anderson, K.S. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Natl. Acad. Sci. USA 2015, 112, E1754–E1762. [Google Scholar] [CrossRef] [Green Version]
- Anft, M.; Paniskaki, K.; Blazquez-Navarro, A.; Doevelaar, A.A.N.; Seibert, F.; Hoelzer, B.; Skrzypczyk, S.; Kohut, E.; Kurek, J.; Zapka, J.; et al. COVID-19 progression is potentially driven by T cell immunopathogenesis. medRxiv 2020. [Google Scholar] [CrossRef]
- Grifoni, A.; Sidney, J.; Zhang, Y.; Scheuermann, R.H.; Peters, B.; Sette, A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 2020, 27, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Schmitz, K.S.; Raadsen, M.P.; Grifoni, A.; Okba, N.M.A.; Endeman, H.; van den Akker, J.P.; Molenkamp, R.; Koopmans, M.P.G.; van Gorp, E.C.M.; et al. Phenotype of SARS-CoV-2-specific T-cells in COVID-19 patients with acute respiratory distress syndrome. medRxiv 2020. [Google Scholar] [CrossRef]
- Funke, J.; Dürr, R.; Dietrich, U.; Koch, J. Key words Natural Killer Cells in HIV-1 Infection: A Double-Edged Sword. AIDS Rev. 2011, 13, 67–76. [Google Scholar]
- Piai, A.; Fu, Q.; Cai, Y.; Ghantous, F.; Xiao, T.; Shaik, M.M.; Peng, H.; Rits-Volloch, S.; Chen, W.; Seaman, M.S.; et al. Structural basis of transmembrane coupling of the HIV-1 envelope glycoprotein. Nat. Commun. 2020, 11, 2317. [Google Scholar] [CrossRef]
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human leukocyte antigen susceptibility map for SARS-CoV-2. J. Virol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.; Barker, D.J.; Georgiou, X.; Cooper, M.A.; Flicek, P.; Marsh, S.G.E. IPD-IMGT/HLA Database. Nucleic Acids Res. 2020, 48, D948–D955. [Google Scholar] [CrossRef]
- Boyle, L.H.; Goodall, J.C.; Gaston, J.S.H. Major histocompatibility complex class I-restricted alloreactive CD4 + T cells. Immunology 2004, 112, 54–63. [Google Scholar] [CrossRef]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the N- and O- glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 1–20. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific analysis of the SARS-CoV-2 glycan shield. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, W.; Mao, Y.; Wang, S. Site-specific N-glycosylation Characterization of Recombinant. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Ng, M.H.L.; Cheng, S.H.; Lau, K.M.; Leung, G.M.; Khoo, U.S.; Zee, B.C.W.; Sung, J.J.Y. Immunogenetics in SARS: A casecontrol study. Hong Kong Med. J. 2010, 16, 29–33. [Google Scholar] [PubMed]
- Sanchez-Mazas, A. HLA studies in the context of coronavirus outbreaks. Swiss Med. Wkly. 2020, 150, w20248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Epitopes Described in—Immune Epitope Database (IEDB). Available online: http://www.iedb.org/details_v3.php?type=reference&id=1000425 (accessed on 15 April 2020).
- Ishizuka, J.; Grebe, K.; Shenderov, E.; Peters, B.; Chen, Q.; Peng, Y.; Wang, L.; Dong, T.; Pasquetto, V.; Oseroff, C.; et al. Quantitating T Cell Cross-Reactivity for Unrelated Peptide Antigens. J. Immunol. 2009, 183, 4337–4345. [Google Scholar] [CrossRef]
- Negahdaripour, M.; Golkar, N.; Hajighahramani, N.; Kianpour, S.; Nezafat, N.; Ghasemi, Y. Harnessing self-assembled peptide nanoparticles in epitope vaccine design. Biotechnol. Adv. 2017, 35, 575–596. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Length (aa) | NCBI RefSeq Accession ID |
|---|---|---|
| nsp1 | 180 | YP_009725297.1 |
| nsp2 | 638 | YP_009725298.1 |
| nsp3 | 1945 | YP_009725299.1 |
| nsp4 | 500 | YP_009725300.1 |
| 3C-like proteinase (3CLpro) | 306 | YP_009725301.1 |
| nsp6 | 290 | YP_009725302.1 |
| nsp7 | 83 | YP_009725303.1 |
| nsp8 | 198 | YP_009725304.1 |
| nsp9 | 113 | YP_009725305.1 |
| nsp10 | 139 | YP_009725306.1 |
| nsp11 | 13 | YP_009725312.1 |
| RNA-dependent RNA polymerase (RdRp) | 932 | YP_009725307.1 |
| Helicase | 601 | YP_009725308.1 |
| 3′-to-5′ exonuclease (35EXO) | 527 | YP_009725309.1 |
| Endo RNAse (EndoR) | 346 | YP_009725310.1 |
| 2′-O-ribose methyltransferase | 298 | YP_009725311.1 |
| Surface glycoprotein (S) | 1273 | YP_009724390.1 |
| ORF3a | 275 | YP_009724391.1 |
| Envelope protein (E) | 75 | YP_009724392.1 |
| Membrane glycoprotein (M) | 222 | YP_009724393.1 |
| ORF6 | 61 | YP_009724394.1 |
| ORF7a | 121 | YP_009724395.1 |
| ORF7b | 43 | YP_009725318.1 |
| ORF8 | 121 | YP_009724396.1 |
| Nucleocapsid phosphoprotein (N) | 419 | YP_009724397.2 |
| ORF10 | 38 | YP_009725255.1 |
| Epitopes | Protein | Allotype | Supertype | Combined Score | Predicted IC50 (nM) |
|---|---|---|---|---|---|
| 738DTDFVNEFY746 | RdRp | A*01:01 | A01 | 3.619 | 2.83 |
| 1505LVAEWFLAY1513 | nsp3 | A*29:02 | A01 | 2.748 | 3.02 |
| 289SHFAIGLAL297 | Helicase | B*39:01 | B39 | 2.168 | 4.55 |
| 1507AEWFLAYIL1515 | nsp3 | B*40:01 | B44 | 2.036 | 4.88 |
| 1505LVAEWFLAY1513 | nsp3 | B*35:01 | A01 | 2.748 | 5.66 |
| 1507AEWFLAYIL1515 | nsp3 | B*40:02 | B44 | 2.036 | 7.64 |
| 217AMDEFIERY225 | EndoR | A*01:01 | A01 | 3.138 | 10.47 |
| 1505LVAEWFLAY1513 | nsp3 | B*15:01 | A01 | 2.748 | 11.16 |
| 1505LVAEWFLAY1513 | nsp3 | A*26:01 | A01 | 2.748 | 18.88 |
| Epitopes | Protein | Epitope Mutation | Combined Score | Allotypes | Predicted IC50 (nM) | Experimental IC50 (nM) |
|---|---|---|---|---|---|---|
| 1220FIAGLIAIV1228 | S | No | 1.212 | A*02:01 A*02:06 A*68:02 | 10.29 11.13 8.32 | 1.48 2.8 0.54 |
| 17VLLFLAFVV25 | E | No | 1.213 | A*02:01 A*02:06 | 21.72 107.83 | 5.62 12.6 |
| 20FLAFVVFLL28 | E | No | 1.440 | A*02:01 A*02:06 | 5.26 51.99 | 0.23 2.57 |
| 204VLAWLYAAV212 | 3CLpro | No | 1.173 | A*02:01 A*02:06 | 13.40 29.50 | 0.435 8.79 |
| 184VLWAHGFEL192 | 35EXO | No | 1.360 | A*02:01 A*02:06 | 5.78 34.55 | 0.40 20.3 |
| 330LLSAGIFGA338 | nsp3 | I335V | 1.217 | A*02:01 A*02:06 | 10.09 14.54 | 8.1 24.6 |
| Epitopes | Allotypes | Half-Life (in Hours) | Secondary Structure | Localization | GRAVY Score |
|---|---|---|---|---|---|
| 1220FIAGLIAIV1228 | A*02:01 A*02:06 A*68:02 | 5.11 5.42 0.63 | helix | transmembrane | 3.056 |
| 17VLLFLAFVV25 | A*02:01 A*02:06 | 4.13 1.90 | helix | transmembrane | 3.489 |
| 20FLAFVVFLL28 | A*02:01 A*02:06 | 11 5.48 | helix | transmembrane | 3.333 |
| 204VLAWLYAAV212 | A*02:01 A*02:06 | 8.13 4.42 | helix | intravirion | 2.133 |
| 184VLWAHGFEL192 | A*02:01 A*02:06 | 6.51 2.26 | strand-coil-helix | intravirion | 0.933 |
| Epitopes | Allotype | Epitope Mutation | Half-Life (in Hours) | Localization | GRAVY Score |
|---|---|---|---|---|---|
| 738DTDFVNEFY746 | A*01:01 | E744D T739I | 2.84 | intravirion | −0.689 |
| 1505LVAEWFLAY1513 | A*29:02 | No | 3.64 | transmembrane | 1.389 |
| 289SHFAIGLAL297 | B*39:01 | H290Y A296S L297F | 2.02 | intravirion | 1.567 |
| 1507AEWFLAYIL1515 | B*40:01 | No | 2.04 | transmembrane | 1.422 |
| 1505LVAEWFLAY1513 | B*35:01 | No | 1.69 | transmembrane | 1.389 |
| 1507AEWFLAYIL1515 | B*40:02 | No | 3.81 | transmembrane | 1.422 |
| 217AMDEFIERY225 | A*01:01 | A217V F221L R224Q | 1.26 | intravirion | −0.589 |
| 1505LVAEWFLAY1513 | B*15:01 | No | 7.51 | transmembrane | 1.389 |
| 1505LVAEWFLAY1513 | A*26:01 | No | 1.33 | transmembrane | 1.389 |
| A*02:01 | Fiagliaiv | Vllflafvv | Flafvvfll | Vlawlyaav | Vlwahgfel | |
|---|---|---|---|---|---|---|
| A*02:06 | ||||||
| Fiagliaiv | 0 | 0.685 | 0.566 | 0.554 | 0.852 | |
| Vllflafvv | 0.940 | 0 | 0.65 | 0.881 | 0.699 | |
| Flafvvfll | 1.002 | 0.636 | 0 | 0.518 | 0.888 | |
| Vlawlyaav | 0.599 | 0.669 | 0.501 | 0 | 1.039 | |
| Vlwahgfel | 1.296 | 1.250 | 1.470 | 1.432 | 0 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranga, V.; Niemelä, E.; Tamirat, M.Z.; Eriksson, J.E.; Airenne, T.T.; Johnson, M.S. Immunogenic SARS-CoV-2 Epitopes: In Silico Study Towards Better Understanding of COVID-19 Disease—Paving the Way for Vaccine Development. Vaccines 2020, 8, 408. https://doi.org/10.3390/vaccines8030408
Ranga V, Niemelä E, Tamirat MZ, Eriksson JE, Airenne TT, Johnson MS. Immunogenic SARS-CoV-2 Epitopes: In Silico Study Towards Better Understanding of COVID-19 Disease—Paving the Way for Vaccine Development. Vaccines. 2020; 8(3):408. https://doi.org/10.3390/vaccines8030408
Chicago/Turabian StyleRanga, Vipin, Erik Niemelä, Mahlet Z. Tamirat, John E. Eriksson, Tomi T. Airenne, and Mark S. Johnson. 2020. "Immunogenic SARS-CoV-2 Epitopes: In Silico Study Towards Better Understanding of COVID-19 Disease—Paving the Way for Vaccine Development" Vaccines 8, no. 3: 408. https://doi.org/10.3390/vaccines8030408