



Abstract
Polycyclic aromatic hydrocarbons (PAHs), comprised of fused benzene (C6H6) rings, emit infrared radiation (3–12 μm) due to the vibrational transitions of the C–H bonds of the aromatic rings. The 3.3 μm aromatic band is generally accompanied by the band at 3.4 μm assigned to the vibration of aliphatic C–H bonds of compounds such as PAHs with an excess of peripheral H atoms (Hn–PAHs). Herein we study the stability of fully hydrogenated benzene (or cyclohexane, C6H12) under the impact of stellar radiation in the photodissociation region (PDR) of NGC 7027. Using synchrotron radiation and time-of-flight mass spectrometry, we investigated the ionization and dissociation processes at energy ranges of UV (10–200 eV) and soft X-rays (280–310 eV). Density Functional Theory (DFT) calculations were used to determine the most stable structures and the relevant low-lying isomers of singly charged C6H12 ions. Partial Ion Yield (PIY) analysis gives evidence of the higher tendency toward dissociation of cyclohexane in comparison to benzene. However, because of the high photoabsorption cross-section of benzene at the C1s resonance edge, its photodissociation and photoionization cross-sections are enhanced, leading to a higher efficiency of dissociation of benzene in the PDR of NGC 7027. We suggest that a similar effect is experienced by PAHs in X-ray photon-rich environments, which ultimately acts as an auxiliary protection mechanism of super-hydrogenated polycyclic hydrocarbons. Finally, we propose that the single photoionization of cyclohexane could enhance the abundance of branched molecules in interstellar and circumstellar media.
Export citation and abstract BibTeX RIS
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) are molecular archetypes that can be classified according to the number of fused benzene (C6H6) rings that make up the base structure (Scott 2015). These molecules are identified by spectral features due to the stretching and bending vibrations of aromatic species at 3.3, 6.2, 7.7, 8.6, and 11.3 μm (Allamandola et al. 1989; Peeters et al. 2002; Yang et al. 2017). Frequently, such features are accompanied by bands at 3.4 and 6.9 μm (Zhang & Kwok 2014), characteristic of C–H and C–C aliphatic bonds. These bands are emitted by hydrogenated PAHs (Hn–PAHs) and by species of PAHs with aliphatic side groups observed in many sources, such as H ii regions, interstellar (ISM) and circumstellar media (CSM), planetary nebulae (PNe), and galaxies (Tielens 2008; Li & Draine 2012; Sandford et al. 2013; Hsia et al. 2016; Simonian & Martini 2017; Yang et al. 2017). For example, in the infrared spectra of the well-studied planetary nebula NGC 7027, the 3.3 μm emission feature is much more intense than the 3.4 μm one. The difference between the profile of both bands can be attributed to distinct factors, such as the relative abundances of the aromatic and aliphatic compounds (Tielens 2008), differences in the oscillator strength values of each transition (Yang et al. 2017), the average size of the PAH carbon backbone (Le Page et al. 2003; Montillaud et al. 2013), and the evolution stage of the object (Sandford et al. 2013; Steglich et al. 2013).
Currently, the stability of Hn–PAHs and their role as catalysts in the formation of H2 in different astrophysical environments are of special interest in astrophysics and astrochemistry, and the subject of an intense debate (Reitsma et al. 2014; Gatchell et al. 2015; Cazaux et al. 2016; Wolf et al. 2016). Le Page et al. (2003) studied the hydrogenation and charge state of PAHs in diffuse clouds, and concluded that the size of the PAH influences the stability of highly hydrogenated species. More recently, it was shown that the hydrogenation of the coronene cation,  , follows a site-selective sequence, leading to the appearance of magic numbers of attached hydrogen atoms (Cazaux et al. 2016). Further, Reitsma et al. (2014) verified that although the carbon backbone of a super-hydrogenated PAH is locally weakened, its de-excitation by H loss protects the PAH from fragmentation. These findings suggest that the addition of peripheral hydrogen atoms to PAHs could impart greater stability to these molecules in interstellar and circumstellar photodissociation regions (PDRs). Gatchell et al. (2015) and Wolf et al. (2016), however, have shown that carbon backbone fragmentation was actually increased in collision and photoinduced experiments with super- and fully hydrogenated pyrene cations (
, follows a site-selective sequence, leading to the appearance of magic numbers of attached hydrogen atoms (Cazaux et al. 2016). Further, Reitsma et al. (2014) verified that although the carbon backbone of a super-hydrogenated PAH is locally weakened, its de-excitation by H loss protects the PAH from fragmentation. These findings suggest that the addition of peripheral hydrogen atoms to PAHs could impart greater stability to these molecules in interstellar and circumstellar photodissociation regions (PDRs). Gatchell et al. (2015) and Wolf et al. (2016), however, have shown that carbon backbone fragmentation was actually increased in collision and photoinduced experiments with super- and fully hydrogenated pyrene cations ( ). Their results, therefore, point to a failure of the hydrogenation protection mechanism, at least for small Hn–PAHs.
). Their results, therefore, point to a failure of the hydrogenation protection mechanism, at least for small Hn–PAHs.
It should be clear that there is no consensus in the literature about the role of peripheral H atoms to the photostability of PAHs, aside from some evidence that it should be dependent on the size of the carbon backbone. On the other hand, some of the electronic and structural properties of such macromolecules are expected to be already present in their smallest units. Benzene, the basic building block of a PAH, is composed of an aromatic ring with sp2-type carbon atoms occupying the vertices of a regular hexagon (Martín & Scott 2015). It is one of the most studied systems in chemistry, due to its several remarkable structural, electronic, and reactivity properties (Cardozo et al. 2014; Papadakis & Ottosson 2015). In opposition to alkenes and acyclic polyenes, the first hydrogenation step of benzene in its ground state is endothermic, which is attributed to aromaticity loss (Papadakis et al. 2016). In fact, the partial hydrogenation of benzene is not favored from the thermodynamic point of view, and the development of catalysts and processes to account for this transformation is still the focus of scientific inquiry (Foppa & Dupont 2015). The complete hydrogenation of C6H6 (Figure 1) is accompanied by a change in the type of C atoms (from sp2 to sp3 type), as all π electrons of the valence space are now being used to form the new C–H σ bonds. This process is exothermic by 208 kJ mol−1 (Carey & Sundberg 2007), and ultimately leads to the formation of the cyclohexane (C6H12) molecule.
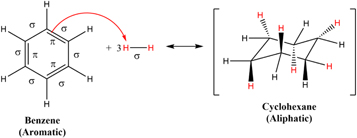
Figure 1. Schematic diagram of the complete hydrogenation reaction of benzene.
Download figure:
Standard image High-resolution imageThe fully hydrogenated counterpart of benzene, cyclohexane, is an aliphatic organic molecule of the cycloalkane group. It is formed by covalent σ bonds of six sp3-type C atoms and 12 peripheral H atoms. Due to the nature of the sp3-type orbitals, C6H12 does not form a planar hexagonal structure. The three-dimensional potential energy surface and the conformational analysis of the neutral C6H12 molecule have been the focus of several studies, such as the ones developed by Leventis et al. (1997) and Kakhiani et al. (2009). To summarize, there are two potential energy minima for the most stable conformer, which is the chair structure of D3d symmetry (see Figure 1, right panel). These minima can interconvert through six different metastable twist-boat structures of D2 symmetry. The interconversion between one of the chair conformers and each of the twist-boat structures follow through either of two transition states: one of C2 symmetry and the other of C1 symmetry, usually identified as a half-chair conformer. Finally, two twist-boat conformers are connected via a boat transition-state structure of C2v symmetry.
Herein, we study the stability of cyclohexane to ionizing and dissociative effects of UV and X-ray radiation. From the experimental data, we determined the main fragmentation pathways, as well as the photoionization and photodissociation cross-sections of C6H12. From computational calculations, the geometries of the main photoionization products are described. Finally, from the photon flux in the PDR of the planetary nebula NGC 7027, we contrast the stability and survival rates of C6H6 and C6H12 in this carbon-rich object (Bernard-Salas et al. 2001; Bernard-Salas & Tielens 2005; Wesson et al. 2010). Whenever possible, we extrapolate our results to the chemistry of PAHs and Hn–PAHs in circumstellar environments. It is worth mentioning that bands related to the mentioned polycyclic molecules have already been detected in NGC 7027 (Bernard-Salas et al. 2001; Goicoechea et al. 2004; Tielens 2008; Boersma et al. 2009; Lau et al. 2016), thus providing an appropriate astrophysical environment for such analysis.
2. Methods
2.1. Experimental Methods
The measurements were performed at the Brazilian Synchrotron Light Source Laboratory (LNLS) using UV and soft X-ray photons selected by a Toroidal Grating Monochromator (TGM) at energies from 10.8 to 307.0 eV. Photons in this energy range cause ionizations from the valence orbitals as well as from the K inner shell of the carbon atom. The experimental setup has been described in detail elsewhere (Lago et al. 2004; Marinho et al. 2006; de Souza et al. 2007). Briefly, UV and X-ray photons, produced at a rate of ∼1012 s−1, perpendicularly intersect the cyclohexane gas sample inside an ultrahigh vacuum chamber. The gaseous sample was injected through a capillary with an internal diameter of 0.8 mm. During the experiments, the pressure inside the chamber with the gas injection was raised and kept constant at approximately 10−6 Torr, ensuring the single-collision regime for the photon–molecule interactions. The injection of cyclohexane in gas phase was made by simple sublimation, without heating, avoiding degradation of the sample, and controlled by an ultrahigh vacuum leak valve. A schematic diagram of the time-of-flight spectrometer inside the experimental vacuum chamber is shown in Figure 2.
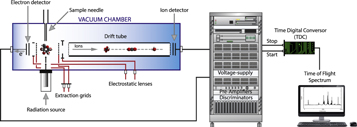
Figure 2. Schematic diagram of the time-of-flight mass spectrometer (TOF-MS) inside the experimental vacuum chamber and the associated electronic components.
Download figure:
Standard image High-resolution imageA static uniform electric field of 485 V cm−1 was used to extract in opposite directions the photoelectrons (PEs) and photoions (PIs) produced by the interaction of the photon beam with the sample. The ionic positively charged species were accelerated and focused into a field-free drift time-of-flight tube with 297 mm length toward a microchannel plate (MCP) detector in a chevron configuration. The detection of one of the PEs by a second MCP detector provides the start signal, while the stop signals are given by the PIs to a multihit fast time to digital converter unit. Thus, standard time-of-flight spectra were obtained using the PE–PI coincidence (PEPICO) technique.
In order to derive astrochemical information, it is necessary to obtain the absolute ionization and dissociation cross-sections. False coincidences coming from aborted double and triple ionization events were subtracted during the data reduction procedure (Simon et al. 1991). Subsequently, the contributions of all singly charged fragments were summed up and normalized to the photoabsorption cross-sections measured by Hitchcock et al. (1986). Assuming that the fluorescence yield is negligible due to the low carbon atomic number (Chen et al. 1981), all absorbed photons should lead to ion formation. The photoionization process that follows inner shell photoabsorption produces instabilities in the molecular structure (nuclear rearrangements), leading to a metastable transient molecule that can break down into several ionic and neutral fragments following different dissociation pathways.
2.2. Computational Details
Density Functional Theory (DFT; Becke 2014) calculations were used to search for the most stable structure and the relevant low-lying isomers of the singly charged C6H12 systems. Around 100 different input molecular structures were initially constructed, taking into account different geometrical features and bonding motifs. Among them, the presence of rings, the ring conformations (chair-like, boat-like, or twist-like), the number of atoms in the ring (from three to six), the type of chain (linear, straight, or branched), and the presence of carbene-like (CR2), methylium-like ( ), and methanium-like (
), and methanium-like ( ) units were considered. After performing a geometry optimization procedure for each of the input molecules, harmonic frequency calculations were performed in order to obtain the respective zeropoint energies and to characterize the nature of the optimized stationary points. Different spin multiplicities (doublet and quartet) were investigated. However, all relevant structures for this work were doublet (
) units were considered. After performing a geometry optimization procedure for each of the input molecules, harmonic frequency calculations were performed in order to obtain the respective zeropoint energies and to characterize the nature of the optimized stationary points. Different spin multiplicities (doublet and quartet) were investigated. However, all relevant structures for this work were doublet ( ). The + · notation is used to indicate that the single ionization of cyclohexane leads to a radical cation species, with an odd number of electrons. The geometries of minimum energy were classified in ascending order of enthalpy values at 298 K (H298). A simple notation (aB+·) was used to label the different isomers found in this work. In this notation, the first superscript (a) represents the multiplicity of the system, and the bold script (B) represents the isomer stability in an enthalpy scale ranging from the most stable (larger negative enthalpy) to the less stable one. The enthalpy values are shown relative to the structure that more closely resembles the neutral molecular geometry (isomer 25+·). The optimized bond distances and the atomic charges obtained by fitting the electrostatic potential (ESP) were used to assist the determination of the Lewis structure of the species. All calculations were performed with the Jaguar 7.9 program (Bochevarov et al. 2013).
). The + · notation is used to indicate that the single ionization of cyclohexane leads to a radical cation species, with an odd number of electrons. The geometries of minimum energy were classified in ascending order of enthalpy values at 298 K (H298). A simple notation (aB+·) was used to label the different isomers found in this work. In this notation, the first superscript (a) represents the multiplicity of the system, and the bold script (B) represents the isomer stability in an enthalpy scale ranging from the most stable (larger negative enthalpy) to the less stable one. The enthalpy values are shown relative to the structure that more closely resembles the neutral molecular geometry (isomer 25+·). The optimized bond distances and the atomic charges obtained by fitting the electrostatic potential (ESP) were used to assist the determination of the Lewis structure of the species. All calculations were performed with the Jaguar 7.9 program (Bochevarov et al. 2013).
3. Results and Discussion
Section 3.1 shows the experimental results concerning the production of ionized species, from the parent ion ( ) to its photodissociation products. Section 3.2 shows the theoretical results for the most stable structures of the
) to its photodissociation products. Section 3.2 shows the theoretical results for the most stable structures of the  species. In Section 3.3, the absolute photoionization and photodissociation cross-sections are calculated. Finally, in Section 3.4, the astrochemical implications of the results obtained in the previous sections are applied to the chemistry of cyclohexane and Hn–PAHs in interstellar and circumstellar photon-rich environments.
species. In Section 3.3, the absolute photoionization and photodissociation cross-sections are calculated. Finally, in Section 3.4, the astrochemical implications of the results obtained in the previous sections are applied to the chemistry of cyclohexane and Hn–PAHs in interstellar and circumstellar photon-rich environments.
3.1. Experimental Results
Mass spectra obtained by the technique of simple coincidences of monochromatic photon beam bombardment on cyclohexane at selected UV and X-ray energies can be seen in Figure 3. The production of ionic fragments are initially determined by means of multiple-Gaussian-function fitting to obtain the area of the peaks and the Partial Ion Yield, PIY(%), of each fragment ion obtained as
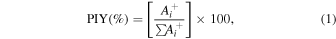
where  is the area of each ion i and
is the area of each ion i and  is the sum of the areas of all peaks in the spectrum. The uncertainties of the partial yields are estimated to be around 10%. Only fragments with PIY greater than 1%, together with the
is the sum of the areas of all peaks in the spectrum. The uncertainties of the partial yields are estimated to be around 10%. Only fragments with PIY greater than 1%, together with the  series and the parent ion—also designated by M+—are shown. The PIYs for all measured energies are listed in Tables 1 and 2.
series and the parent ion—also designated by M+—are shown. The PIYs for all measured energies are listed in Tables 1 and 2.
Table 1. Partial Ion Yield, PIY (%), as a Function of the Photon Energy in the UV and Soft X-Ray Regions
| m/q | Attribution | PIY (≥1%), Per Energy (eV) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10.8 | 11.4 | 12.4 | 13.0 | 14.0 | 16.0 | 20.0 | 50.0 | 80.0 | 100.0 | 146.0 | 200.0 | ||
| 1 | H+ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 4.2 | 4.7 | 5.0 | 3.9 | 4.4 |
| 2 |
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 0.2 | 0.3 | 0.4 | 0.3 | 0.4 |
| 3 |
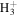
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 0.0 | 0.1 | 0.0 | 0.1 |
| 15 |
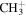
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 6.0 | 5.9 | 5.9 | 4.1 | 4.2 |
| 26 |
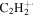
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 1.9 | 2.7 | 4.1 | 4.7 |
| 27 |

|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 10.8 | 10.9 | 12.9 | 14.0 | 12.7 |
| 28 |
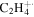
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 1.4 | 2.7 | 7.0 | 5.0 | 6.5 | 4.0 | 3.8 |
| 29 |
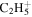
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 1.6 | 3.6 | 4.2 | 5.1 | 3.2 | 4.2 |
| 38 |
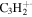
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 2.4 | 2.2 |
| 39 |
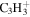
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 8.9 | 9.0 | 7.6 | 11.5 | 12.9 |
| 40 |
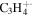
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 1.9 | 1.6 | 2.1 | 2.2 | 2.9 |
| 41 |
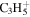
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 10.9 | 15.1 | 15.5 | 16.4 | 12.7 | 14.4 | 11.5 |
| 42 |
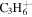
|
⋯ | ⋯ | 4.5 | 6.3 | 9.1 | 12.7 | 9.4 | 4.8 | 4.3 | 6.4 | 3.7 | 3.1 |
| 43 |
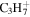
|
⋯ | ⋯ | 1.9 | 3.2 | 5.1 | 5.2 | 4.7 | 2.1 | 1.9 | 2.7 | 1.6 | 1.2 |
| 50 |
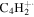
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 1.4 | 1.2 |
| 51 |

|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 1.6 | 1.6 |
| 53 |
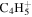
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 1.5 | 1.3 | 1.1 | 1.7 | 1.8 |
| 54 |
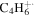
|
⋯ | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ | 1.3 | 1.4 | 1.1 | 1.1 | 1.4 | 1.4 |
| 55 |

|
⋯ | ⋯ | 3.0 | 4.0 | 5.4 | 9.0 | 9.6 | 5.5 | 4.9 | 4.2 | 3.6 | 3.4 |
| 56 |
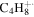
|
⋯ | ⋯ | 22.7 | 29.5 | 34.1 | 29.8 | 26.8 | 10.0 | 11.2 | 9.8 | 5.5 | 6.0 |
| 57 |
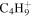
|
⋯ | ⋯ | 1.9 | 1.9 | 2.2 | 1.9 | 1.7 | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ |
| 69 |
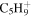
|
⋯ | ⋯ | 5.9 | 6.7 | 9.0 | 5.1 | 4.0 | 1.7 | 1.1 | 1.3 | ⋯ | ⋯ |
| 83 |
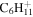
|
⋯ | ⋯ | ⋯ | 2.2 | 1.7 | 1.6 | 1.4 | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ |
| 84 |
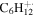
|
93.8 | 93.9 | 53.4 | 41.9 | 27.2 | 18.4 | 17.4 | 7.1 | 5.3 | 5.5 | 4.6 | 4.5 |
| 85 | 13C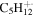
|
6.2 | 6.1 | 2.8 | 2.4 | 1.5 | 1.2 | 1.0 | ⋯ | ⋯ | ⋯ | ⋯ | ⋯ |
Note. Only the fragments with intensity greater than 1% were shown, except the ions  and
and  .
.
Download table as: ASCIITypeset image
Table 2. Partial Ion Yield, PIY (%), as a Function of the Photon Energy around C1s Resonance in the Soft X-Ray Region
| m/q | Attribution | PIY (≥1%), Per Energy (eV) | ||||
|---|---|---|---|---|---|---|
| 284.8 | 287.7 | 300.0 | 301.0 | 307.0 | ||
| 1 | H+ | 10.0 | 8.2 | 9.2 | 9.5 | 9.3 |
| 2 |
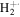
|
0.8 | 0.7 | 0.7 | 0.7 | 0.6 |
| 3 |

|
0.1 | 0.1 | 0.1 | 0.1 | 0.1 |
| 14 |

|
1.2 | 1.0 | 1.4 | 1.2 | 1.2 |
| 15 |
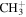
|
8.5 | 5.3 | 8.2 | 7.8 | 7.5 |
| 26 |
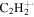
|
7.4 | 6.5 | 8.3 | 8.5 | 8.0 |
| 27 |
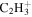
|
18.6 | 17.4 | 17.8 | 17.5 | 16.1 |
| 28 |
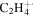
|
4.2 | 3.9 | 4.8 | 4.2 | 3.9 |
| 29 |
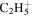
|
2.9 | 2.3 | 2.1 | 2.5 | 2.1 |
| 37 | C3H+ | 2.4 | 2.9 | 2.7 | 2.4 | 3.4 |
| 38 |
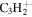
|
5.3 | 3.4 | 3.5 | 3.1 | 4.2 |
| 39 |
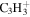
|
12.4 | 14.8 | 12.8 | 14.2 | 12.5 |
| 40 |
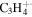
|
2.9 | 2.4 | 1.8 | 1.6 | 2.0 |
| 41 |
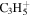
|
5.7 | 6.4 | 6.2 | 6.7 | 5.2 |
| 42 |
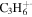
|
⋯ | 1.5 | 1.1 | 1.1 | 1.2 |
| 50 |
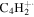
|
2.5 | 3.4 | 2.6 | 2.4 | 3.4 |
| 51 |
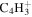
|
1.8 | 2.7 | 2.2 | 2.4 | 2.3 |
| 53 |
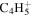
|
1.1 | 1.3 | 1.2 | 1.4 | ⋯ |
| 63 |
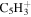
|
⋯ | ⋯ | 1.0 | 1.0 | 1.5 |
| 84 |
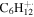
|
0.9 | 1.4 | 0.8 | 0.9 | 0.6 |
Note. Only the ionic fragments with intensity greater than 1% were shown, except the  ,
,  and parent ion
and parent ion  .
.
Download table as: ASCIITypeset image
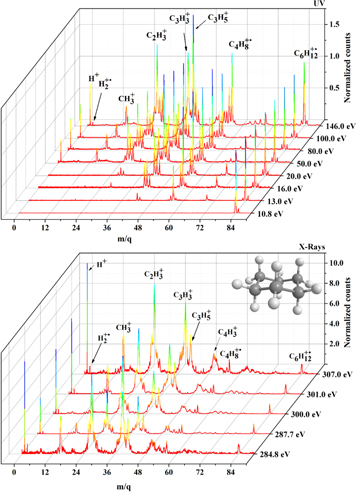
Figure 3. Selected mass spectra of the cyclohexane molecule recorded at UV (10–200 eV) and soft X-ray photons (280–310 eV). The color variation indicates the differences in the intensity of the peaks. For each energy, the counts are normalized with respect to those of the parent ion,  . The PIY values of all measured energies are listed in Tables 1 and 2. See details in the text.
. The PIY values of all measured energies are listed in Tables 1 and 2. See details in the text.
Download figure:
Standard image High-resolution image3.1.1. Ultraviolet (UV)
Figure 4, top panel, shows the production of  as a function of the photon energy in the UV range (10.8 eV to 200.0 eV). For energies close to the first Ionization Potential (IP) of cyclohexane (9.8 eV, as determined by Dewar & Worley 1969), the photodissociation process is poorly activated. The PIY value of the parent ion decreases by a factor of almost 9 in the UV range, remaining at only 4.5% at 200.0 eV.
as a function of the photon energy in the UV range (10.8 eV to 200.0 eV). For energies close to the first Ionization Potential (IP) of cyclohexane (9.8 eV, as determined by Dewar & Worley 1969), the photodissociation process is poorly activated. The PIY value of the parent ion decreases by a factor of almost 9 in the UV range, remaining at only 4.5% at 200.0 eV.
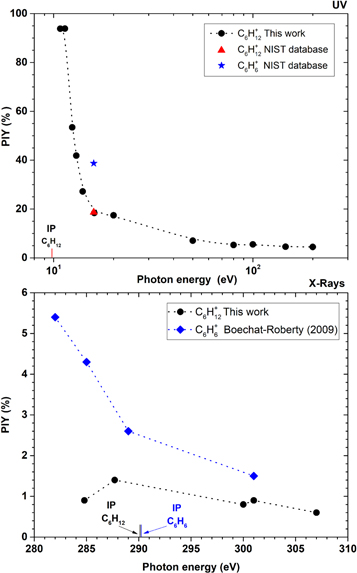
Figure 4. Production (Partial Ion Yield, PIY) of  as a function of the photon energy. Top panel, in the UV range: the red triangle and the blue star indicate the values of
as a function of the photon energy. Top panel, in the UV range: the red triangle and the blue star indicate the values of  and
and  , respectively, both obtained from the NIST database (Johnson 2013). Bottom, in the X-ray range: comparison of the yields of ionized cyclohexane with ionized benzene (blue dots taken from Boechat-Roberty et al. 2009). The ionization energies of cyclohexane (290.12 eV) and benzene (290.24 eV) are also indicated (Kolczewski et al. 2006).
, respectively, both obtained from the NIST database (Johnson 2013). Bottom, in the X-ray range: comparison of the yields of ionized cyclohexane with ionized benzene (blue dots taken from Boechat-Roberty et al. 2009). The ionization energies of cyclohexane (290.12 eV) and benzene (290.24 eV) are also indicated (Kolczewski et al. 2006).
Download figure:
Standard image High-resolution imageA significant production of  is observed when the production of
is observed when the production of  starts to drop fast at around 12.4 eV (see Figure 3). That is, the original molecular ion breaks up through the loss of a neutral ethylene (C2H4) unit. The loss of only hydrogen atoms from the parent ion (
starts to drop fast at around 12.4 eV (see Figure 3). That is, the original molecular ion breaks up through the loss of a neutral ethylene (C2H4) unit. The loss of only hydrogen atoms from the parent ion ( , n = 1–8) as well the loss of one carbon atom (
, n = 1–8) as well the loss of one carbon atom ( , n = 1–8) are inhibited, as observed by their low relative amounts (see Table 1). The production of
, n = 1–8) are inhibited, as observed by their low relative amounts (see Table 1). The production of  reaches a maximum value (34.1%) at 14.0 eV and then it starts to decrease, reaching 26.8% at 20.0 eV. In this energy range, the production of
reaches a maximum value (34.1%) at 14.0 eV and then it starts to decrease, reaching 26.8% at 20.0 eV. In this energy range, the production of  more than doubles, from 4% to 9%. This indicates that part of the closed-shell
more than doubles, from 4% to 9%. This indicates that part of the closed-shell  production comes through the loss of a hydrogen atom from the open-shell
production comes through the loss of a hydrogen atom from the open-shell  radical cation system. Other significant ions produced in the 14.0 eV to 20.0 eV energy range are the
radical cation system. Other significant ions produced in the 14.0 eV to 20.0 eV energy range are the  (M+–
(M+– ),
),  (M+–C3H6), and
(M+–C3H6), and  species.
species.
At 100.0 eV, the production of ions from the ethyl ( ), methyl (
), methyl ( ), and hydrogen (
), and hydrogen ( ) groups begins to increase. The most produced ion is the
) groups begins to increase. The most produced ion is the  species, followed by
species, followed by  (M+–
(M+– ) and
) and  . The production of the cyclopropenyl molecule
. The production of the cyclopropenyl molecule  becomes significant at 100.0 eV, and at 200.0 eV, this species is the most produced one.
becomes significant at 100.0 eV, and at 200.0 eV, this species is the most produced one.
By comparing the present results with the PIY of the literature data of the National Institute of Standards and Technology (NIST; Johnson 2013), we verified that the fragmentation pattern at 70 eV electron energy already resembles the one induced by 16 eV photons. It is also interesting to note that at the electron impact of 70 eV (or photon impact of 16 eV), the abundance of the aromatic ring parent ion is twice that of the cyclohexane parent ion.
3.1.2. Soft X-Rays
The fragmentation pattern observed in the soft X-ray region around the C1s resonance energy is significantly different from that measured in the UV region (see Figure 3). From 284.8 eV to 307.0 eV, more than 90% of the ion yield comes from the light fragment ion contribution, from the  (and
(and  ) to
) to  groups. The parent ion production (see Figure 4, bottom panel) ranges from merely 0.6% (307.0 eV) to 1.4% at the C1s resonance energy (287.7 eV). This profile contrasts reasonably with the one presented by the parent ion of benzene, for which a production of 5.4% was observed at 282.4 eV, a value more than five times larger than the average production of the parent ion of cyclohexane at the C1s edge. These values reveal a higher propensity of cyclohexane to photodissociate after absorbing a soft X-ray photon in the C1s edge. This tendency is related to the higher molecular rigidity of benzene and its parent ion, in comparison to cyclohexane and its ionization product. By far, the most relevant fragments produced by the photodissociation of cyclohexane are
groups. The parent ion production (see Figure 4, bottom panel) ranges from merely 0.6% (307.0 eV) to 1.4% at the C1s resonance energy (287.7 eV). This profile contrasts reasonably with the one presented by the parent ion of benzene, for which a production of 5.4% was observed at 282.4 eV, a value more than five times larger than the average production of the parent ion of cyclohexane at the C1s edge. These values reveal a higher propensity of cyclohexane to photodissociate after absorbing a soft X-ray photon in the C1s edge. This tendency is related to the higher molecular rigidity of benzene and its parent ion, in comparison to cyclohexane and its ionization product. By far, the most relevant fragments produced by the photodissociation of cyclohexane are  and
and  , which are related to cyclopropenyl cation (Zhao et al. 2014) and protonated acetylene (Glassgold et al. 1992), respectively.
, which are related to cyclopropenyl cation (Zhao et al. 2014) and protonated acetylene (Glassgold et al. 1992), respectively.
Figure 5 shows the comparison of the different ions resulting from the fragmentation of benzene and cyclohexane at the C1s resonance energy. By analyzing the production of the parent ion and other  species, it is possible to see that the main difference between benzene and cyclohexane is that the backbone fragmentation is significantly more pronounced in the latter. This result gives evidence that the hydrogenated benzene molecule shows, after X-ray photoionization, a higher tendency for dissociation than its aromatic counterpart. Consequently, a high production of ions pertaining to the
species, it is possible to see that the main difference between benzene and cyclohexane is that the backbone fragmentation is significantly more pronounced in the latter. This result gives evidence that the hydrogenated benzene molecule shows, after X-ray photoionization, a higher tendency for dissociation than its aromatic counterpart. Consequently, a high production of ions pertaining to the  ,
,  , and
, and  groups is observed for cyclohexane. In fact, the formation of
groups is observed for cyclohexane. In fact, the formation of  ,
,  , and
, and  is particularly high for C6H12 compared to aromatic systems, such as benzene (Boechat-Roberty et al. 2009) and toluene (C7H8; Monfredini et al. 2016). On the other hand, the fragmentation of benzene into the ionic species
is particularly high for C6H12 compared to aromatic systems, such as benzene (Boechat-Roberty et al. 2009) and toluene (C7H8; Monfredini et al. 2016). On the other hand, the fragmentation of benzene into the ionic species  to
to  is significantly more pronounced. Concerning the Hn series, the H+ production is relatively the same for both species. In turn,
is significantly more pronounced. Concerning the Hn series, the H+ production is relatively the same for both species. In turn,  and
and  are much more efficiently produced from the break up of the aliphatic structure than from benzene.
are much more efficiently produced from the break up of the aliphatic structure than from benzene.

Figure 5. Top: comparison between the mass spectra of cyclohexane (red) and benzene (blue) at the respective C1s resonance energies. Bottom: normalized abundances of the  group produced by the fragmentation of cyclohexane and benzene at 301.0 eV.
group produced by the fragmentation of cyclohexane and benzene at 301.0 eV.
Download figure:
Standard image High-resolution imageA comparison between the fragmentation pattern of benzene and cyclohexane around the parent ion m/q value at 301 eV photon impact is shown in Figure 5, bottom panel. For benzene, all C6Hn cations were observed, and the most abundant ions are C6H+,  ,
,  , and
, and  . For cyclohexane, the results are quite different. Several of the C6Hn cations were absent, such as
. For cyclohexane, the results are quite different. Several of the C6Hn cations were absent, such as  ,
,  ,
,  ,
,  , and
, and  . These results indicate that stable C6Hn ionic structures are not directly achievable from the molecular rearrangement of cyclohexane after ionization. The most relevant C6Hn ions produced by the photodissociation of C6H12 are
. These results indicate that stable C6Hn ionic structures are not directly achievable from the molecular rearrangement of cyclohexane after ionization. The most relevant C6Hn ions produced by the photodissociation of C6H12 are  and
and  and
and  .
.
3.2. Theoretical Results
The structural and spectroscopic properties of the singly charged cyclohexane radical cation,  , and similar cycloalkane cations have been exhaustively studied by both Ion Cyclotron Resonance (ICR) techniques (Dunbar 1984) and collisional activation measurements (Borchers et al. 1977). Dunbar (1976) has shown that strong optical absorptions in the visible region could be observed for gas-phase radical cations of saturated hydrocarbons. In contrast, radical cations that originated from linear alkenes present weak visible absorptions and strong UV peaks. For cyclohexane and larger cycloalkane rings, the optical spectrum is similar to that of n-alkanes, which indicates that they retain their ring structures upon ionization. A profile similar to that of alkenes is observed for cyclopentane (C5H10) and smaller cycloalkane rings, indicating that a ring-opening isomerization occurs in these radical ions prior to fragmentation (Benz & Dunbar 1979).
, and similar cycloalkane cations have been exhaustively studied by both Ion Cyclotron Resonance (ICR) techniques (Dunbar 1984) and collisional activation measurements (Borchers et al. 1977). Dunbar (1976) has shown that strong optical absorptions in the visible region could be observed for gas-phase radical cations of saturated hydrocarbons. In contrast, radical cations that originated from linear alkenes present weak visible absorptions and strong UV peaks. For cyclohexane and larger cycloalkane rings, the optical spectrum is similar to that of n-alkanes, which indicates that they retain their ring structures upon ionization. A profile similar to that of alkenes is observed for cyclopentane (C5H10) and smaller cycloalkane rings, indicating that a ring-opening isomerization occurs in these radical ions prior to fragmentation (Benz & Dunbar 1979).
The ring-opening mechanisms for the cyclopentane and cyclohexane radical cations have been studied by van der Hart (2001) using ab initio calculations. The author showed that the ring-opening barrier heights in both cases are comparable, being significantly lower than the ionization energies. Although these results could not explain the ICR experimental results, they suggest that open-chain radical cations are accessible by the photoionization of neutral cyclohexane. Moreover, it was also shown by van der Hart (2001) that some of the acyclic  radical cations are thermodynamically more stable than six- or five-membered rings. Since there is still no information about which isomer represents the ground state of the parent ion of cyclohexane, a careful computational analysis has been made in order to identify the most stable isomers. Some of the optimized
radical cations are thermodynamically more stable than six- or five-membered rings. Since there is still no information about which isomer represents the ground state of the parent ion of cyclohexane, a careful computational analysis has been made in order to identify the most stable isomers. Some of the optimized  radical cations are shown in Figure 6.
radical cations are shown in Figure 6.

Figure 6. Minimum energy geometries of the singly charged C6H12 ions. The enthalpy values at 298 K, relative to the 25+· isomer (the cyclic chair singly charged cyclohexane), are shown in parentheses in kcal mol−1. The isomer notation is described in Section 2.2. The symbols (⊕) and (·) indicate the positions of the charge and the open-shell electron in the molecules, respectively. The bold lines in 25+· and 28+· indicate the front view of the molecule. The dashed lines in 23+·, 26+·, and 27+· indicate one-electron σ bonds. See the text for details.
Download figure:
Standard image High-resolution imageThe global minimum structure of the  system is the 2,3-dimethyl-2-butene (or tetramethyl-ethylene) radical cation (21+·), 25.4 kcal mol−1 more stable than the cyclic chair singly charged cyclohexane species, 25+·. The central C–C bond length is 1.420 Å, a value between the C=C bond in ethylene (1.3305 Å; Craig et al. 2006) and the C–C bond in ethane (1.522 Å; Harmony 1990). A torsional angle of 13° is observed between two adjacent methyl groups. These CH3 substituents act as stabilizing agents for both radicals and carbocations, which could explain the high stability of such a structure in comparison to 25+·. The energy difference between the neutral chair cyclohexane and 21+· is 8.6 eV, and is 9.7 eV between the former and 25+·. The photon energies used in this work are significantly higher than the mentioned energy differences, the experimental ionization potential and the isomerization barriers calculated by van der Hart (2001). It is expected, therefore, that 21+· is being produced in our experiments as the main
system is the 2,3-dimethyl-2-butene (or tetramethyl-ethylene) radical cation (21+·), 25.4 kcal mol−1 more stable than the cyclic chair singly charged cyclohexane species, 25+·. The central C–C bond length is 1.420 Å, a value between the C=C bond in ethylene (1.3305 Å; Craig et al. 2006) and the C–C bond in ethane (1.522 Å; Harmony 1990). A torsional angle of 13° is observed between two adjacent methyl groups. These CH3 substituents act as stabilizing agents for both radicals and carbocations, which could explain the high stability of such a structure in comparison to 25+·. The energy difference between the neutral chair cyclohexane and 21+· is 8.6 eV, and is 9.7 eV between the former and 25+·. The photon energies used in this work are significantly higher than the mentioned energy differences, the experimental ionization potential and the isomerization barriers calculated by van der Hart (2001). It is expected, therefore, that 21+· is being produced in our experiments as the main  species.
species.
Structure 22+· is the 3-methyl-2-pentene radical cation, another branched singly charged molecule. It is 9.2 kcal mol−1 less stable than 21+· and 16.2 kcal mol−1 more stable than the chair structure. The smaller branch degree of 22+· in comparison to 21+· is responsible for its lower thermodynamic stability. This trend is also observed for the non-branched linear 2-hexene radical cation (24+·), which is 10.3 kcal mol−1 less stable than 22+·.
Structures 23+· and 25+· to 28+· are cyclic, with the rings presenting from three to six carbon atoms. The most stable cyclic  is the 1,1,2-trimethyl-cyclopropane radical cation (23+·). The C1–C2 bond length is 1.906 Å, which suggests that it is composed of a two-center–one-electron σ bond. This bonding pattern has been observed for some neutral (Hübner et al. 2014) and radical (Hoefelmeyer & Gabbaï 2000; Moret et al. 2013) boron-containing compounds, and represents a challenge for chemical bond models (de Sousa & Nascimento 2017; Fantuzzi et al. 2017b).
is the 1,1,2-trimethyl-cyclopropane radical cation (23+·). The C1–C2 bond length is 1.906 Å, which suggests that it is composed of a two-center–one-electron σ bond. This bonding pattern has been observed for some neutral (Hübner et al. 2014) and radical (Hoefelmeyer & Gabbaï 2000; Moret et al. 2013) boron-containing compounds, and represents a challenge for chemical bond models (de Sousa & Nascimento 2017; Fantuzzi et al. 2017b).
The next cyclic structure is the chair cyclohexane radical cation (25+·). The neutral cyclohexane molecule in the chair conformation has D3d symmetry, and the cation can distort to two different conformations of C2h symmetry because of Jahn–Teller instability. In addition to these conformations, an unsymmetrical chair structure with Cs symmetry was also characterized in the literature. Lunell et al. (1985) has shown that the relaxation of the inversion symmetry leads to a stabilization of ∼7 kcal mol−1. In our calculations, we did not impose any symmetry constraint for optimizing 25+· or, analogously, 28+·, which naturally converged to a boat-like structure with C2 symmetry. The energy difference between both optimized cyclohexane radical cations is 6.5 kcal mol−1.
Finally, the isomers 26+· and 27+· are the 1,1,2-trimethyl-cyclobutane and methyl-cyclopentane radical cations, respectively. The presence of an elongated carbon–carbon bond (1.964 Å for 26+· and 2.150 Å) suggests, as for structure 23+·, the formation of a two-center–one-electron σ bond. The implications of these results for the chemistry in the PDR of NGC 7027 are discussed in Section 3.4.
3.3. Absolute Photoionization and Photodissociation Cross-sections
Before going into the details of the determination of the photoionization and photodissociation cross-sections, it is interesting to compare the absolute X-ray photoabsorption cross-sections of benzene and its hydrogenated analogues, as shown in Figure 7. A strong photoabsorption peak is observed for benzene at 285.2 eV, with a maximum value of 2.6 × 10−17 cm2. This feature is attributed to a K-shell transition from a C1s orbital to the lowest unoccupied π* orbital of e2u symmetry (Horsley et al. 1985). The shape of this transition, together with additional features in the K-shell spectra related to the splitting of π* and σ* resonances, has been used to prove the aromatic character of heterocyclic rings (Stöhr 1992), such as borazine (Doering et al. 1986). A second peak (287.2 eV, 4.4 × 10−18 cm2) is also observed for benzene in Figure 7, which is assigned to a transition to a 3p Rydberg orbital (Horsley et al. 1985).
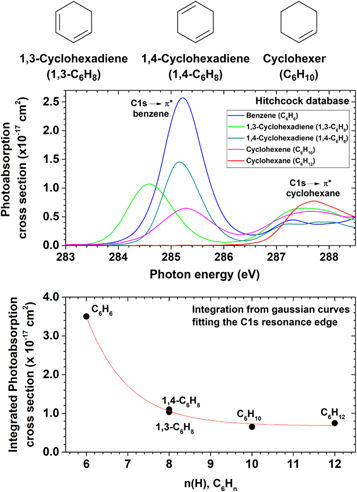
Figure 7. Absolute photoabsorption cross-sections as a function of the number of H atoms. Top: benzene (C6H6, blue), 1,3-cyclohexadiene (C6H8, green), 1,4-cyclohexadiene (C6H8, dark cyan), cyclohexene (C6H10, pink), and cyclohexane (C6H12, red), adapted from the Hitchcock database (Hitchcock et al. 1986, 1987; Hitchcock & Rühl 1989; Hitchcock 1994). Bottom: comparison between the areas of the C1s  * resonance bands after a Gaussian-fitting procedure. The red line is an adjusted exponential decay curve, with a coefficient of determination R2 = 0.9978.
* resonance bands after a Gaussian-fitting procedure. The red line is an adjusted exponential decay curve, with a coefficient of determination R2 = 0.9978.
Download figure:
Standard image High-resolution imageThe increase in the number of hydrogen atoms from benzene (C6H6) to the isomers 1,3- and 1,4-hexadiene (C6H8) is followed by a significant decrease in the photoabsorption cross-section values at the respective C1s  * resonance energies. This trend is also observed for the more hydrogenated six-membered ring molecules, cyclohexene (C6H10) and cyclohexane (C6H12). In the case of the latter, besides the reduction in the cross-section value, there is also a significant displacement of the resonance energy to a higher photon energy value from 285.2 eV to 287.7 eV (Figure 7, top panel). The absence of a peak around 285 eV comes from the fact that there is no sp2-like atom in cyclohexane, and the feature in 287.7 eV is attributed to a transition to the 4a1π* (CH2) orbital (Hitchcock et al. 1986), whose maximum value is 7.7 × 10−18 cm2. By fitting the C1s
* resonance energies. This trend is also observed for the more hydrogenated six-membered ring molecules, cyclohexene (C6H10) and cyclohexane (C6H12). In the case of the latter, besides the reduction in the cross-section value, there is also a significant displacement of the resonance energy to a higher photon energy value from 285.2 eV to 287.7 eV (Figure 7, top panel). The absence of a peak around 285 eV comes from the fact that there is no sp2-like atom in cyclohexane, and the feature in 287.7 eV is attributed to a transition to the 4a1π* (CH2) orbital (Hitchcock et al. 1986), whose maximum value is 7.7 × 10−18 cm2. By fitting the C1s  * resonance bands with Gaussian functions and integrating, an exponential decay profile between the integrated photoabsorption cross-sections and the number of hydrogen atoms in the C6Hn molecules is observed. The fluorescence yield can be assumed to be negligible, due to the low C atomic number (Chen et al. 1981). Moreover, since typical K-shell core hole lifetimes of light elements are in the femtosecond range (Drescher et al. 2002), and therefore some orders of magnitude faster than a vibrational period, it is possible to conclude that the photorelaxation of the neutral molecule by internal conversion and energy redistribution into vibrational modes, such as the one described by the cationic excited states of PAHs (Marciniak et al. 2015), is also negligible. Finally, given the fact that these core hole states are embedded in the electronic continua of ionic states (Carravetta et al. 1988), we can conclude that the only relaxation channels of such metastable highly excited states are the photoionization and photodissociation processes. Therefore, the high photoabsorption cross-section of benzene in comparison to cyclohexane could also impart a higher photodissociation rate, depending on the intensity of the photon flux at the C1s resonance edge of benzene.
* resonance bands with Gaussian functions and integrating, an exponential decay profile between the integrated photoabsorption cross-sections and the number of hydrogen atoms in the C6Hn molecules is observed. The fluorescence yield can be assumed to be negligible, due to the low C atomic number (Chen et al. 1981). Moreover, since typical K-shell core hole lifetimes of light elements are in the femtosecond range (Drescher et al. 2002), and therefore some orders of magnitude faster than a vibrational period, it is possible to conclude that the photorelaxation of the neutral molecule by internal conversion and energy redistribution into vibrational modes, such as the one described by the cationic excited states of PAHs (Marciniak et al. 2015), is also negligible. Finally, given the fact that these core hole states are embedded in the electronic continua of ionic states (Carravetta et al. 1988), we can conclude that the only relaxation channels of such metastable highly excited states are the photoionization and photodissociation processes. Therefore, the high photoabsorption cross-section of benzene in comparison to cyclohexane could also impart a higher photodissociation rate, depending on the intensity of the photon flux at the C1s resonance edge of benzene.
The full description of the determination of absolute cross-sections can be found elsewhere (Pilling et al. 2006; Fantuzzi et al. 2011). Briefly, the non-dissociative single ionization (photoionization) cross-section σphi and the dissociative single ionization (photodissociation) cross-section σphd of molecules are obtained from

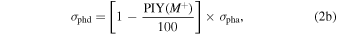
where σpha is the photoabsorption cross-section (Hitchcock et al. 1986) and M+ represents the parent ion. The PIY values of benzene were taken from Boechat-Roberty et al. (2009).
The absolute cross-sections of cyclohexane and benzene at the C1s edge are shown in Figure 8 and Table 3. The photoabsorption cross-section value of cyclohexane at the C1s resonance energy, 7.7 × 10−18 cm2 (Hitchcock et al. 1986), is about half of the benzene one, which is 2.6 × 10−17 cm2 (Hitchcock et al. 1987). Since a stronger photoabsorption leads to higher photoionization and photodissociation cross-sections, the addition of peripheral hydrogen atoms to the aromatic moiety could afford greater stability against X-ray dissociation. These values are used to determine the survival rates of both molecules in the PDR of the planetary nebula NGC 7027, which are discussed in the following section.
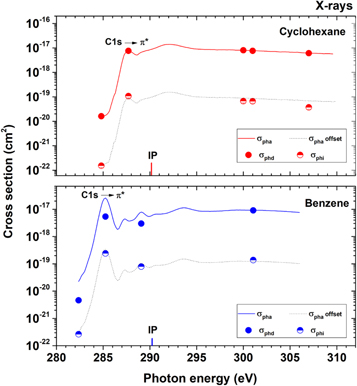
Figure 8. Dissociative ionization (photodissociation) σphd (filled circle), non-dissociative single ionization (photoionization) σphi (half-filled circle), and absolute photoabsorption σpha (solid line) cross-sections as a function of the X-rays energies. Top: cyclohexane (red); bottom: benzene (blue). The dotted lines are an offset of the photoabsorption cross-section; they are only shown to guide the eyes. The photoabsorption cross-section values are obtained from Hitchcock et al. (1987) and Hitchcock et al. (1986) for benzene and cyclohexane, respectively. See details in text.
Download figure:
Standard image High-resolution imageTable 3. Absolute Photoabsorption (σpha), Photodissociation (σphd), and Photoionization (σphi) Cross-sections of Cyclohexane and Benzene, as well as Their Respective Rates kphd and kphi, and the Half-lives, t1/2, under X-Ray Photon Fluxes, FX (E), in NGC 7027
| Energy (eV) | σpha (cm2) | σphd (cm2) | σphi (cm2) | FX(E) (cm−2 s−1) | kphd (s−1) | kphi (s−1) | t1/2 (years) |
|---|---|---|---|---|---|---|---|
| Cyclohexane | |||||||
| 284.8 | 1.6 × 10−20 | 1.6 × 10−20 | 1.5 × 10−22 | 7.9 × 105 | 1.3 × 10−14 | 1.2 × 10−16 | 1.7 × 106 |
| 287.7† | 7.7 × 10−18 | 7.6 × 10−18 | 1.1 × 10−19 | 8.3 × 105 | 6.3 × 10−12 | 8.9 × 10−14 | 3.5 × 103 |
| 300.0 | 8.1 × 10−18 | 8.0 × 10−18 | 6.6 × 10−20 | 1.0 × 106 | 8.1 × 10−12 | 6.7 × 10−14 | 2.7 × 103 |
| 301.0 | 7.7 × 10−18 | 7.6 × 10−18 | 6.6 × 10−20 | 1.0 × 106 | 7.8 × 10−12 | 6.8 × 10−14 | 2.8 × 103 |
| Benzene | |||||||
| 282.4 | 2.3 × 10−20 | 2.2 × 10−20 | 1.2 × 10−21 | 7.5 × 105 | 1.6 × 10−14 | 9.2 × 10−16 | 1.4 × 106 |
| 285.2† | 2.6 × 10−17 | 2.5 × 10−17 | 1.1 × 10−18 | 7.9 × 105 | 1.9 × 10−11 | 8.7 × 10−13 | 1.1 × 103 |
| 289.1 | 7.8 × 10−18 | 7.6 × 10−18 | 2.0 × 10−19 | 8.5 × 105 | 6.5 × 10−12 | 1.7 × 10−13 | 3.4 × 103 |
| 301.0 | 9.2 × 10−18 | 9.0 × 10−18 | 1.4 × 10−19 | 1.0 × 106 | 9.3 × 10−12 | 1.4 × 10−13 | 2.4 × 103 |
Note. The σpha values are obtained from Hitchcock et al. (1986, 1987). The † symbol indicates the C1s resonance energies.
Download table as: ASCIITypeset image
3.4. Astrochemical Implications
3.4.1. Survival of Cyclohexane and Benzene in the Photodissociation Region of the Planetary Nebula NGC 7027
NGC 7027 is a young carbon-rich planetary nebula located in the Cygnus constellation at a distance of 880 pc (Masson 1986; Latter et al. 2000; Bernard-Salas et al. 2001; Hasegawa & Kwok 2001, 2003; Kastner et al. 2001; Wesson et al. 2010). This nebula has a distinct structure, with an ionized elliptical envelope lying at the center of an extended molecular envelope. Intense lines of highly ionized Ne atom were observed in infrared spectra obtained by ISO-SWS of NGC 7027 (Bernard-Salas et al. 2001) and in X-ray spectra taken by the Chandra X-ray Observatory (Kastner et al. 2001). At the interface between the cold molecular region and the ionized front, there is the PDR where the chemistry is controlled by penetrating UV and X-ray photons from the central star (Lau et al. 2016; Latter et al. 2000). The morphology of NGC 7027 presents three outflows in the H ii region interacting with the outermost regions (Cox et al. 1997; Santander-García et al. 2012; Lau et al. 2016). The variations in the radiative flux of the object provide a non-equivalent mixture of chemical species within the outer layers, thus providing environments with different characteristics within the same nebula (Arnoult et al. 2000). Consequently, it stimulates the formation and destruction of a wide variety of molecular species detected in both neutral and ionized states, such as H2, CO,  ,
,  , CH+, c-C3H2, PAHs, and aliphatic hydrocarbons (Hasegawa et al. 2000; Hasegawa & Kwok 2001; Lau et al. 2016). The physical properties of the central star, a very hot white dwarf with a temperature around 2 × 105 K and an estimated luminosity of 7700 L⊙ (Latter et al. 2000), promote a chemically rich medium. This implies that different reaction mechanisms could be taking place, both in the gas phase and on the surface of grains. Ultimately, such an environment could uphold the formation of complex ions and organic molecules, among them PAHs (Herbst & van Dishoeck 2009).
, CH+, c-C3H2, PAHs, and aliphatic hydrocarbons (Hasegawa et al. 2000; Hasegawa & Kwok 2001; Lau et al. 2016). The physical properties of the central star, a very hot white dwarf with a temperature around 2 × 105 K and an estimated luminosity of 7700 L⊙ (Latter et al. 2000), promote a chemically rich medium. This implies that different reaction mechanisms could be taking place, both in the gas phase and on the surface of grains. Ultimately, such an environment could uphold the formation of complex ions and organic molecules, among them PAHs (Herbst & van Dishoeck 2009).
The determination of molecular σphd values is of significant importance for estimating the molecular abundance in both interstellar and circumstellar environments. The decreasing abundance of a given molecule subjected to a radiation field in the photon energy range E2 − E1 inside a gaseous dusty cloud can be written as (Cottin et al. 2003; Boechat-Roberty et al. 2009)
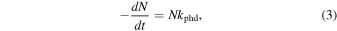
where N is the column density (cm−2) and kphd is the photodissociation rate (s−1), given by
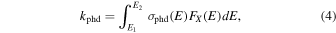
where  is the photodissociation cross-section (cm2) and FX(E) is the photon flux (photons cm−2 eV−1 s−1), both as a function of the photon energy E = hν.
is the photodissociation cross-section (cm2) and FX(E) is the photon flux (photons cm−2 eV−1 s−1), both as a function of the photon energy E = hν.
Therefore, it is possible to determine the half-life of a given molecule with the following equation (Andrade et al. 2010):
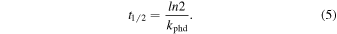
In addition, we can also determine the photoionization rate, kphi, given by
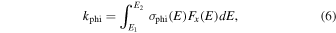
where σphi(E) (cm2) is the photoionization cross-section.
In order to know the X-ray photon flux (FX) values in the PDR of NGC 7027, we used the equation
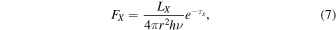
where Lx = 1.3 × 1032 ergs s−1 is the integrated X-ray luminosity from 0.2 to 2.5 keV, reported by Kastner et al. (2001), r = 5.21 × 1016 cm is the distance from the central star to a position inside the PDR (Agúndez et al. 2010), and τX is the X-ray optical depth, given by Deguchi et al. (1990):

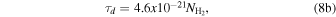
which accounts for the absorption due to materials on the envelope along a radius. In Equations (8(a)) and (8(b)), τd is the UV optical depth of dust grains at 1000 Å, proposed by Morris & Jura (1983), the factor α = 0.054 for energies below 0.6 keV, and  is the column density of H2, for which we adopted the value of 1.3 × 1021 cm−2 obtained by Agúndez et al. (2010). The energy 0.6 keV corresponds to the oxygen O1s absorption edge (Deguchi et al. 1990).
is the column density of H2, for which we adopted the value of 1.3 × 1021 cm−2 obtained by Agúndez et al. (2010). The energy 0.6 keV corresponds to the oxygen O1s absorption edge (Deguchi et al. 1990).
Figure 9 shows the X-ray optical depth in the PDR of NGC 7027, its photon flux with and without the attenuation, and the estimated half-lives of cyclohexane and benzene in this object. The X-ray optical depth ranges from 2.4 (282.0 eV) to 0.3 (600.0 eV), which shows that the object is essentially transparent for energies above the O1s resonance energy. In the vicinity of the C1s resonance energies, the attenuated photon flux values range from 7.4 × 105 cm−2 s−1 eV−1 to 1.1 × 106 cm−2 s−1 eV−1. Although the maximum X-ray attenuated photon flux is not located on the C1s edge, the photoabsorption cross-sections significantly decay after the inner shell excitation energies (Sakamoto et al. 2010). Therefore, the half-lives obtained in this work are a good estimation of the X-ray survival rates of the molecules in NGC 7027.

Figure 9. Top: X-ray optical depth, τX, in the photon energy range from 280 to 600 eV. Middle: X-ray photon flux, FX(E), with and without attenuation in the same photon energy range, arriving at the PDR of the planetary nebula NGC 7027 at a position r from the central star of 5.21 × 1016 cm. The pink circles indicate the energies studied herein. Bottom: half-lives, t1/2, of C6H12 (red circles) and C6H6 (blue triangles) at energies around the C1s resonance of each molecule (lines are included to guide the eye).
Download figure:
Standard image High-resolution imageThe photoionization and photodissociation rates, as well as the estimated t1/2 values of C6H12 and C6H6, are shown in Table 3. We estimate that the half-life of cyclohexane is 3.5 × 103 yr in the C1s resonance energy (287.7 eV), more than three times the half-life of benzene at 285.2 eV (1.1 × 103 years). This large distinction is directly related to differences in the photoabsorption cross-sections of both molecules at the mentioned energies (7.7 × 10−18 cm2 for cyclohexane and 2.6 × 10−17 cm2 for benzene). On the other hand, in the vicinity of the C1s resonance energy, the survival of both molecules is practically the same. By integrating the half-life curves of both molecules and comparing the areas, it is possible to see that the survival of cyclohexane on the C1s edge is ∼20% higher than that of benzene. Since the abundances of interstellar molecules depend on both their formation and destruction rates, we suggest that the richness of interstellar benzene in comparison to its fully hydrogenated counterpart comes from more effective mechanisms of formation, such as the one described by Jones et al. (2011). This result indicates, therefore, that the addition of peripheral atoms in the basic PAH unit could impart greater X-ray stability in photodissociation regions.
At this point, it is important to make a comparative analysis of the findings discussed in the last paragraph with the ones presented in Figure 5, as they could be naïvely interpreted as contradictory. From Figure 5, it is shown that the backbone fragmentation of cyclohexane is significantly more pronounced than that of benzene. This means that once photoabsorption has taken place, the fully hydrogenated molecule is more likely to photodetach its carbon skeleton. In other words, the survival probability of cyclohexane is smaller than that of benzene after the photoabsorption process. However, the benzene molecule has a strong absorption feature at the C1s edge as shown in Figure 7. This feature disappears entirely when additional hydrogen atoms are inserted into the carbon backbone. Consequently, the photoabsorption cross-section of benzene is larger than that of cyclohexane, which ultimately leads to a higher efficiency of dissociation for the aromatic molecule. The protective effect of additional hydrogen atoms is, therefore, related to a damping out process of the strong absorption feature of benzene. The extrapolation of such a finding to PAHs of high molecular mass will be discussed in the next subsection.
A final remark should be made with regard to the survival of cyclohexane in PDR regions, such as the one in NGC 7027. As shown in Section 3.2, the global minimum of the C6H12 radical cation (21+·) is a highly branched structure. The first alkyl branched molecule detected in the interstellar medium was the isopropyl-cyanide (i-C3H7CN) species, in a recent study developed by Belloche et al. (2014). The present results suggest that, in a PDR region, the photoionization of C6H12 could induce the formation of the mentioned highly branched radical cation. Since methyl substituents help in the stabilization of carbocations and radical centers, it is expected that the molecular rearrangement that follows the photoionization process of both cyclic and open-chain hydrocarbons could lead to an enhancement of branched molecules in PDR regions.
3.4.2. Stability of Super-hydrogenated PAHs in Photodissociation Regions
In this last subsection, the consequences of the results presented herein on the stability of PAHs and Hn–PAHs in X-ray photon-rich environments are examined and discussed in the context of previous studies (Reitsma et al. 2014; Gatchell et al. 2015; Wolf et al. 2016).
In Section 3.1, we have shown that, after an X-ray photoabsorption process, the cyclohexane molecule is almost entirely dissociated, as the average partial ion yield of  is around 1%. The small production of cyclohexane's parent ion is due to the weak carbon backbone rigidity of the fully hydrogenated six-membered ring, whose high energetic chair-like structure of the C6H12 monocation (25+·, see Section 3.2) will break away to form more stable acyclic ions, such as 21+·, or dissociate into smaller fragments. The carbon backbone rigidity of benzene, on the other hand, is responsible for keeping the PIY values around 5% at the C1s resonance energy. In fact, only after a double ionization process is the six-membered ring surpassed by a different structure as the global minimum. In this case, it acquires a pentagonal–pyramidal carbon arrangement (Jašík et al. 2014; Fantuzzi et al. 2017a).
is around 1%. The small production of cyclohexane's parent ion is due to the weak carbon backbone rigidity of the fully hydrogenated six-membered ring, whose high energetic chair-like structure of the C6H12 monocation (25+·, see Section 3.2) will break away to form more stable acyclic ions, such as 21+·, or dissociate into smaller fragments. The carbon backbone rigidity of benzene, on the other hand, is responsible for keeping the PIY values around 5% at the C1s resonance energy. In fact, only after a double ionization process is the six-membered ring surpassed by a different structure as the global minimum. In this case, it acquires a pentagonal–pyramidal carbon arrangement (Jašík et al. 2014; Fantuzzi et al. 2017a).
As the carbon backbone size increases, the resistance of super-hydrogenated PAHs toward dissociation is also expected to increase. The fragmentation of perhydropyrene (C16H26) will still be greater than that of pyrene (C16H10), as the collision experiments developed by Gatchell et al. (2015) suggest, but not so pronounced as that of C6H12. However, by increasing even more the number of carbon atoms, the fragmentation of the carbon backbone in a Hn-PAH could be reduced to a secondary process, being exceeded by hydrogen elimination to ultimately form a PAH ion, as suggested by Reitsma et al. (2014) in experiments with coronene (C24H12). In this scenario, the excess of peripheral H atoms acts as a protection mechanism for the PAH structure, as already described earlier in this paper.
Parallel to the mechanism proposed by Reitsma et al. (2014), an auxiliary photostabilization mechanism could be present in super-hydrogenated PAHs. As shown in Section 3.3, the strong photoabsorption cross-section of the C1s  * resonance energy of benzene (2.6 × 10−17 cm2) is more than three times higher than the one of cyclohexane (7.7 × 10−18 cm2). This is a common feature of aromatic molecules, as shown in Figure 10, and is expected to be present in the X-ray photoabsorption spectra of PAHs. In comparison, adamantane (C10H16), a polycyclic fully hydrogenated non-aromatic hydrocarbon, does not have such a feature. In fact, the spectral profile of adamantane shows several resemblances to that of cyclohexane, and as such its dissociative ionization (Candian et al. 2018). In this perspective, we suggest that an auxiliary protection mechanism could exist in super-hydrogenated PAHs. By decreasing the X-ray photoabsorption cross-section, a Hn-PAH molecule will also have a smaller photodissociation cross-section, which ultimately enhances its photostability in PDR regions. This mechanism, to the best of our knowledge, proposed for the first time in the literature, should contribute to explaining the existence of hydrogenated PAHs in interstellar and circumstellar media. The importance of such a mechanism in comparison to the one described by Reitsma et al. (2014) will be the focus of a future study.
* resonance energy of benzene (2.6 × 10−17 cm2) is more than three times higher than the one of cyclohexane (7.7 × 10−18 cm2). This is a common feature of aromatic molecules, as shown in Figure 10, and is expected to be present in the X-ray photoabsorption spectra of PAHs. In comparison, adamantane (C10H16), a polycyclic fully hydrogenated non-aromatic hydrocarbon, does not have such a feature. In fact, the spectral profile of adamantane shows several resemblances to that of cyclohexane, and as such its dissociative ionization (Candian et al. 2018). In this perspective, we suggest that an auxiliary protection mechanism could exist in super-hydrogenated PAHs. By decreasing the X-ray photoabsorption cross-section, a Hn-PAH molecule will also have a smaller photodissociation cross-section, which ultimately enhances its photostability in PDR regions. This mechanism, to the best of our knowledge, proposed for the first time in the literature, should contribute to explaining the existence of hydrogenated PAHs in interstellar and circumstellar media. The importance of such a mechanism in comparison to the one described by Reitsma et al. (2014) will be the focus of a future study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. Absolute photoabsorption cross-sections of benzene (C6H6; blue), cyclohexane (C6H12; red) naphthalene (C10H8; cyan), adamantane (C10H16; wine), phenanthrene (C14H10; pink), and pyrene (C16H10; orange), adapted from the Hitchcock database (Hitchcock 1994).
Download figure:
Standard image High-resolution image{kind=link}
4. Summary
In this work, we probed the stability of the fully hydrogenated benzene molecule, cyclohexane, in circumstellar PDRs. We obtained the fragmentation pattern of C6H12 under interaction with UV and X-ray photons. The production of the radical parent cation, as well as its photodissociation products, was evaluated from 10.8 eV to 307.0 eV photon energy, and their production was directly compared with the previous data of benzene, the basic unit of a PAH molecule. The main geometrical features of the most stable structures of the parent ion were described. We determined the absolute photoionization and photodissociation cross-sections of cyclohexane, and their values were compared to those of benzene. The astrochemical implications of the experimental data and computational results were discussed in the context of the PDR of the carbon-rich planetary nebula NGC 7027, which provided insights into the photostability of super-hydrogenated PAHs in such photon-rich environments.
A decreasing exponential profile is observed for the production of the parent ion of cyclohexane in the range of energies from 10.8 eV to 200.0 eV, and its production is approximately half of benzene's parent ion around 16 eV. From 10.8 eV to 20.0 eV, ions from the  and
and  families are the ones most produced, while in the UV range from 20.0 eV to 100.0 eV, families with lesser amounts of carbon atoms are preferentially formed. For higher UV energies, the production of
families are the ones most produced, while in the UV range from 20.0 eV to 100.0 eV, families with lesser amounts of carbon atoms are preferentially formed. For higher UV energies, the production of  becomes significant, a trend that is also observed in the soft X-ray region. The astrochemically relevant
becomes significant, a trend that is also observed in the soft X-ray region. The astrochemically relevant  and
and  ions were also observed with reasonable abundances, likely formed from H+ migration to a CH2 unit followed by dissociation. Stable C6Hn ions, on the other hand, are not efficiently formed after the molecular rearrangements that follows the X-ray ionization process of neutral cyclohexane.
ions were also observed with reasonable abundances, likely formed from H+ migration to a CH2 unit followed by dissociation. Stable C6Hn ions, on the other hand, are not efficiently formed after the molecular rearrangements that follows the X-ray ionization process of neutral cyclohexane.
The smaller photoabsorption of cyclohexane in comparison to that of benzene is responsible for its lower photoionization and photodissociation cross-sections. By obtaining these absolute values and combining them with the X-ray photon flux in the PDR of NGC 7027, it was possible to evaluate the photoionization and photodissociation rates of cyclohexane, as well as its half-life, in the mentioned astrophysical object. We estimate that cyclohexane has a half-life of 3.5 × 103 yr in the C1s resonance energy of 287.7 eV. On the C1s edge, the survival of cyclohexane is ∼20% higher than that of benzene. Since the strong  * aromatic transition of PAHs is expected to be suppressed as the number of peripheral H atoms increases, such process will ultimately result in an enhancement of the X-ray stability of Hn–PAHs to photodissociation processes, which is described herein as an auxiliary protection mechanism of super-hydrogenated PAHs. A damping out process of the strong absorption feature of aromatic molecules is, thus, related to the protective effect of additional hydrogen atoms in Hn–PAHs. Finally, as the most stable
* aromatic transition of PAHs is expected to be suppressed as the number of peripheral H atoms increases, such process will ultimately result in an enhancement of the X-ray stability of Hn–PAHs to photodissociation processes, which is described herein as an auxiliary protection mechanism of super-hydrogenated PAHs. A damping out process of the strong absorption feature of aromatic molecules is, thus, related to the protective effect of additional hydrogen atoms in Hn–PAHs. Finally, as the most stable  structures are acyclic branched species, we propose that the single photoionization of cyclohexane and other saturated hydrocarbons could enhance the abundance of branched molecules in PDRs.
structures are acyclic branched species, we propose that the single photoionization of cyclohexane and other saturated hydrocarbons could enhance the abundance of branched molecules in PDRs.
The authors would like to thank Dr. H. Luna of the Instituto de Física-UFRJ and the staff of the Brazilian Synchrotron Light Laboratory (LNLS) for their valuable help during the experiments, and LNLS, CNPq, FAPERJ and CAPES for the financial support.
Software: Schrödinger Jaguar (v7.9; Bochevarov et al. 2013).