
Estimation of Hydrazone Derivative Compounds as Anti-Bacterial Agents Against Staphylococcus aureous Through Molecular Docking and Self Consisted Field
DOI:
https://doi.org/10.48048/tis.2023.5593Keywords:
Anti-bacterial, Hydrazone, Docking, SCF, Staphylococcus aureus, Anti-biotic, Molecular dynamicAbstract
Molecular docking is an important computational method for drug design. It can be used to predict the binding interaction of receptor with the ligand. Hydrazone were reported as chemical compounds with =CH-N-NH group. In addition, hydrazone derivative compounds were also reported have various activities, such as antibacterial agents against Staphylococcus aureus. S. aureus is a very serious problem because this bacteria is increasingly resistant to various types of antibiotics (multidrug resistance). S. aureus is also having extraordinary adaptability thus, it can be resistant to many antibiotics. The purpose of this study is to predict whether the hydrazone compounds were active as anti-bacterial agents and also to ensure that the binding interaction is stable before and after docking calculation. Molecular docking research was conducted using the protein target 1N67 (PDB ID), which was derived from the crystallographic structure of hydrazone derivative chemicals. Four hydrazone derivative substances were docked to the protein in this investigation with grid boxes along x, y, and z radii of 67, 86 and 66, respectively. The positive control was chloramphenicol. Self-Consisted Field (SCF) was calculated using AM1 as basis set and molecular dynamic simulation was performed using CHARMM27 force field. The results showed that from 4 tested compounds, only compound 3 with the binding free energy of −6.535 kcal/mol was estimated as active agent against S. aureus. This compound showed very good potential to be an anti-bacterial with the lowest binding free energy value with the highest stability level.
HIGHLIGHTS
- Due to in silico study such as molecular docking, molecular dynamic simulation is an important computational method for drug design. It can be used to predict the binding orientation of small molecule drug candidates to their protein targets. In addition, docking is also can be applied to predict small molecule affinity and activity. Thus, molecular docking has an important role in drug discovery and rational design
- The ligand interaction with the receptor was more stable due to the decreased binding free energy value
- The lowest binding free energy and lowest RMSD value of this molecule, which may presumably cause compound 3 to has better binding affinity compare to other compounds
GRAPHICAL ABSTRACT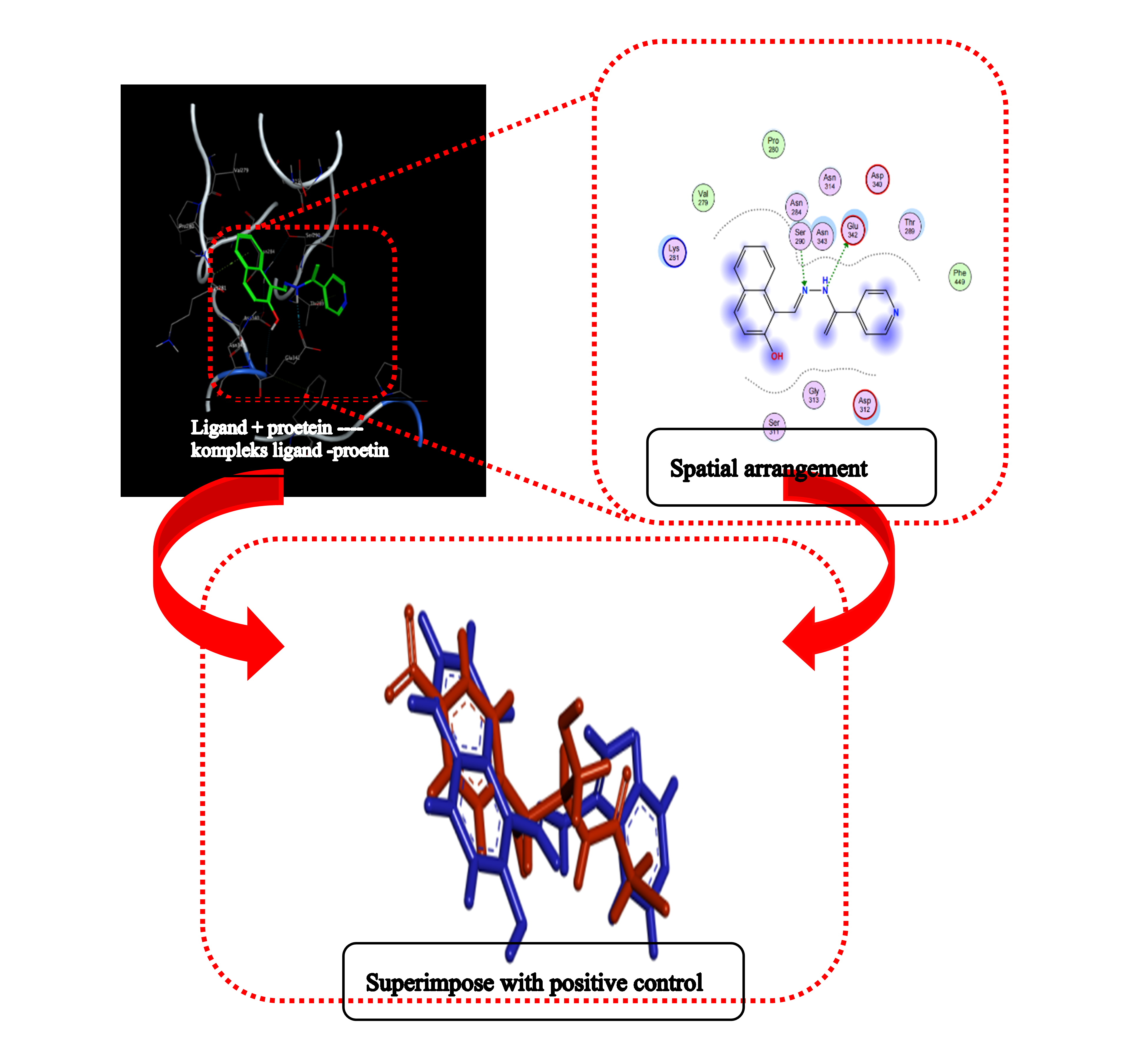
Downloads
Metrics
References
CD Vincy, JDD Tarika, XDD Dexlin, A Rathika and TJ Beaula. Exploring the antibacterial activity of 1, 2 diaminoethane hexanedionic acid by spectroscopic, electronic, ELF, LOL, RDG analysis and molecular docking studies using DFT method. J. Mol. Struct. 2022; 1247, 131388.
A Afifurrahman, K Samadin and S Aziz. Pola kepekaan bakteri Staphylococcus Aureus terhadap antibiotik vancomycin di RSUP Dr. Mohammad Hoesin Palembang. Majalah Kedokteran Sriwijaya 2014; 46, 266-70.
MA Brockhurst, F Harrison, JW Veening, E Harrison, G Blackwell, Z Iqbal and C Maclean. Assessing evolutionary risks of resistance for new antimicrobial therapies. Nat. Ecol. Evol. 2019; 3, 515-7.
V Cattoir and B Felden. Future antibacterial strategies: From basic concepts to clinical challenges. J. Infect. Dis. 2019; 220, 350-60.
RJ Fair and Y Tor. Antibiotics and bacterial resistance in the 21st century. Perspect. Med. Chem. 2014; 6, 25-64.
VL Simpkin, MJ Renwick, R Kelly and E Mossialos. Incentivising innovation in antibiotic drug discovery and development: Progress, challenges and next steps. J. Antibiot. 2017; 70, 1087-96.
NP Belskaya, W Daehan, VA Bakulev, NP Belskaya, W Daehan and VA Bakulev. Synthesis and properties of hydrazone bearing amide, thioamide, and amidine function. Arkivoc 2010; 1, 275-332.
A Patel, S Bari, G Talete, J Patel and M Sarangpani. Synthesis and antimicrobial activity of some new 63 isatin derivatives. Iranian J. Pharmaceut. Sci. 2006; 4, 249-54.
A Zamri, N Frimayanti and HY Teruna. Docking and molecular dynamic simulations: Study of 1,3,4-oxadiazolechalcone hybrid derivatives to search new active anticancer agents. Thai J. Pharmaceut. Sci. 2016; 40, 179-84.
N Frimayanti, I Ikhtiarudin, R Dona, TT Agustini, F Murdiya and A Zamri. A computational approach to drug discovery: Search for chalcone analogues as the potential candidates for anti colorectal cancer (HT29). Walailak J. Sci. Tech. 2020; 17, 64-74.
B Lakhani, KM Thayer, E Black and DL Beveridge. Sppectral analysis of molecular dynamic simulations on PDZ: MD sector. J. Biomol. Struct. Dynam. 2019; 38, 781-90.
N Frimayanti, M Yaeghoobi, H Namavar, I Ikhtiarudin and M Afzali. In silico studies and biological evaluation of chalcone-based 1,5-benzothiazepines as new potential H1N1 neuraminidase inhibitors. J. Appl. Pharmaceut. Sci. 2020; 10, 86-94.
P Fong, CH Hao and LR Meng. In silico and in vitro anti-Helicobacter pylori effects of combinations of phytochemicals and antibiotics. Molecules 2019; 24, 3608-11.
N Frimayanti, M Yaeghoobi, I Ikhtiarudin, DRW Putri, H Namavar and FS Bitaraf. Insight on the In silico study and biological activity assay of chalcone-based 1, 5-Benzothiazepines as potential inhibitor for breast cancer MCF7. Chiang Mai Univ. J. Nat. Sci. 2021; 20, e2021019.
W Brutting. Physics of organic semiconductor. WILEY-VCH Verlag, Weinheim, Germany, 2005.
Y Ma, Y Tao, H Qu, C Wang, F Yan, X Gao and M Zhang. Exploration of plant-derived natural polyphenols toward COVID-19 main protease inhibitors: DFT, molecular docking approach, and molecular dynamics simulations. RSC Adv. 2022; 12, 5357-68.
Z Lian-Gang, L Wei and Y Zhi-Xiang. A frontier molecular orbital theory approach to understanding the mayr equation and to quantifying nucleophilicity and electrophilicity by using HOMO and LUMO energies. Asian J. Org. Chem. 2012; 1, 336-45.
Y Huang, C Rong, R Zhang and S Liu. Evaluating frontier orbital energy and HOMO/LUMO gap with descriptors from density functional reactivity theory. J. Mol. Model. 2017; 23, 3175-82.
F Murdiya, N Frimayanti and M Yaeghoobi. Application of molecular dynamics study and homo lumo calculation on the ionized air for high voltage engineering. In: Proceedings of the 2018 2nd International Conference on Electrical Engineering and Informatics, Batam, Indonesia. 2018, p. 171-74.
J Jasril, I Ikhtiarudin, A Zamri, HY Teruna and N Frimayanti. New fluorinated chalcone and pyrazoline analogs: Synthesis, docking and molecular dynamic studies as anticancer agents. Thai J. Pharmaceut. Sci. 2017; 41, 93-8.
T Sakano, MI Mahamood, T Yamashita and H Fujitani. Molecular dynamics analysis to evaluate docking pose prediction. Biophys. Physicobiology 2016; 13, 181-94.
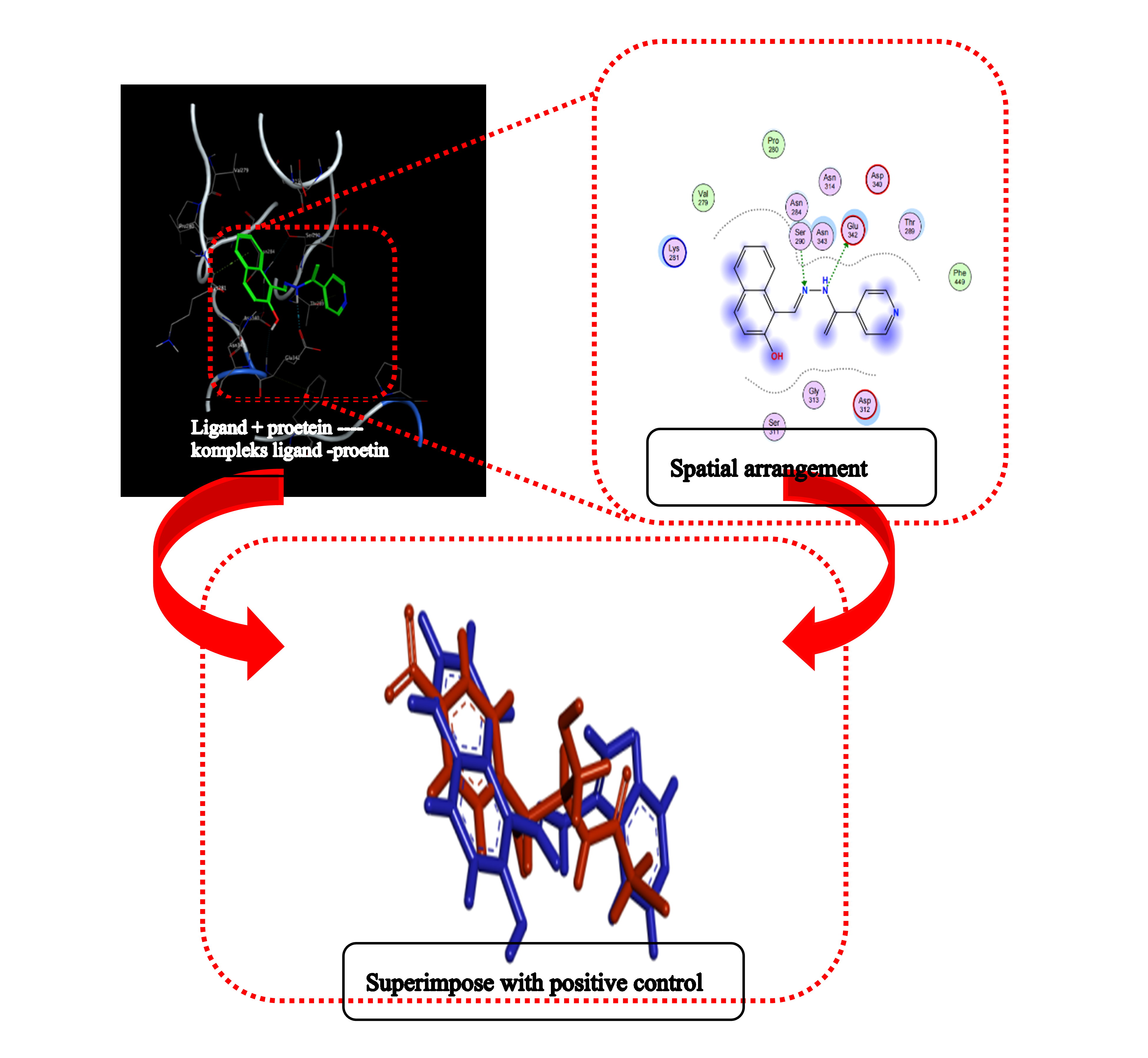
Downloads
Published
How to Cite
Issue
Section
License
Copyright (c) 2022 Walailak University

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.



